
Hydrogen-Bonding Secondary Coordination Sphere Effect on CO2 Reduction



Abstract
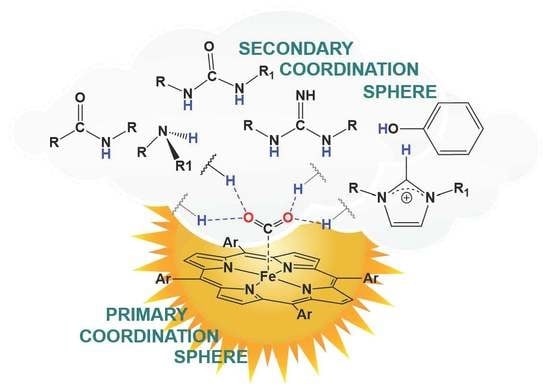
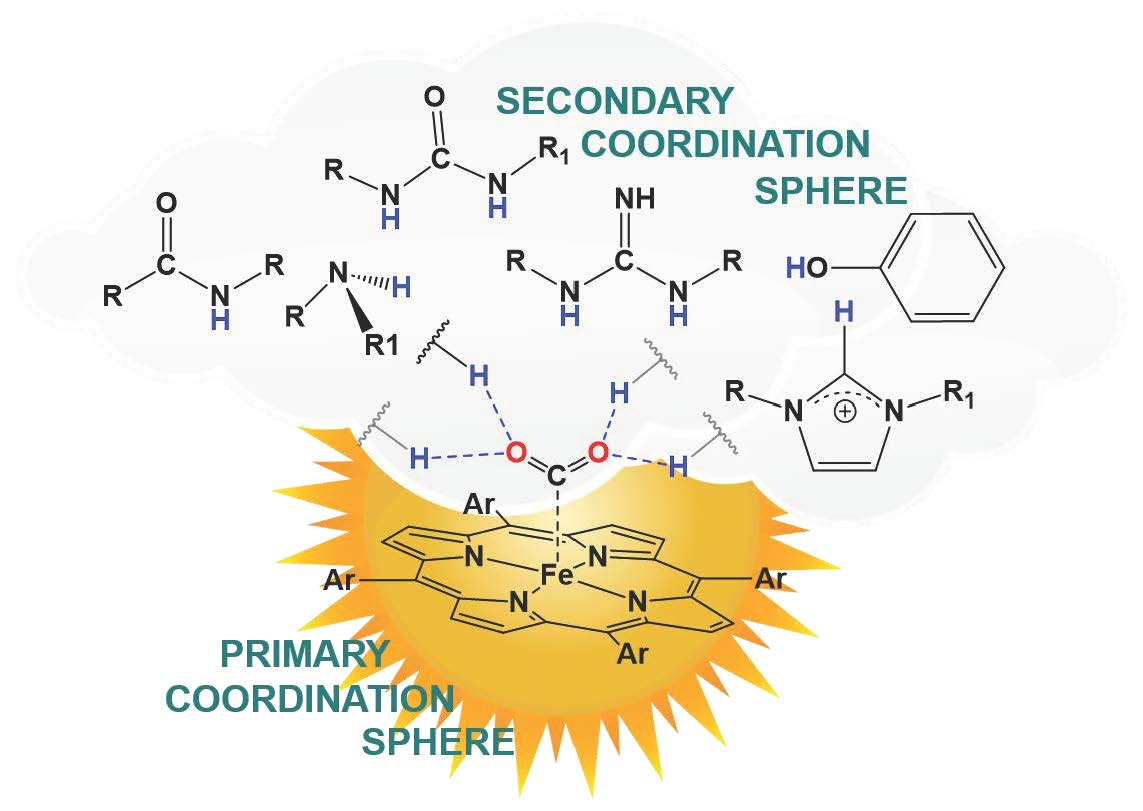
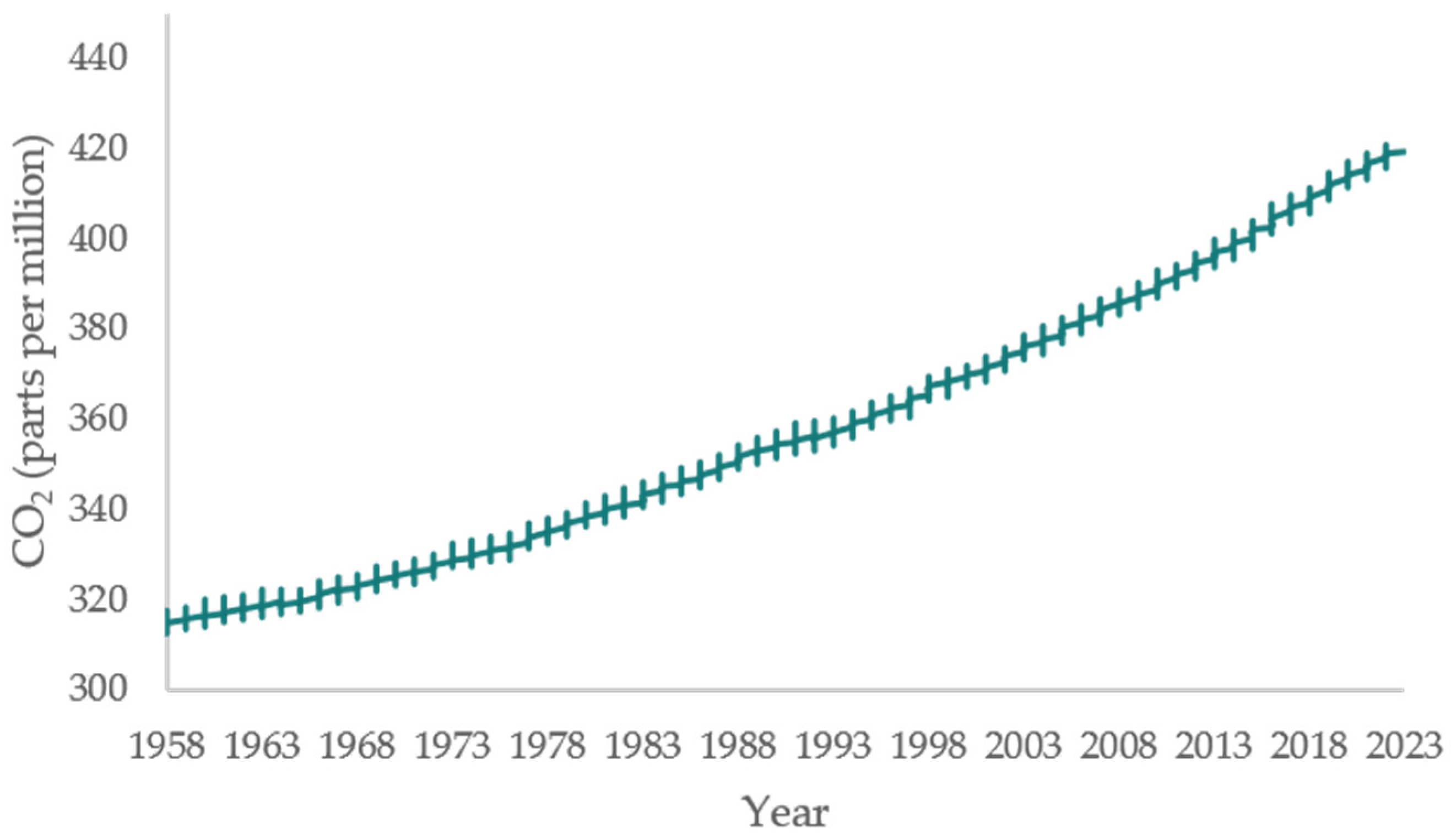
1. Introduction
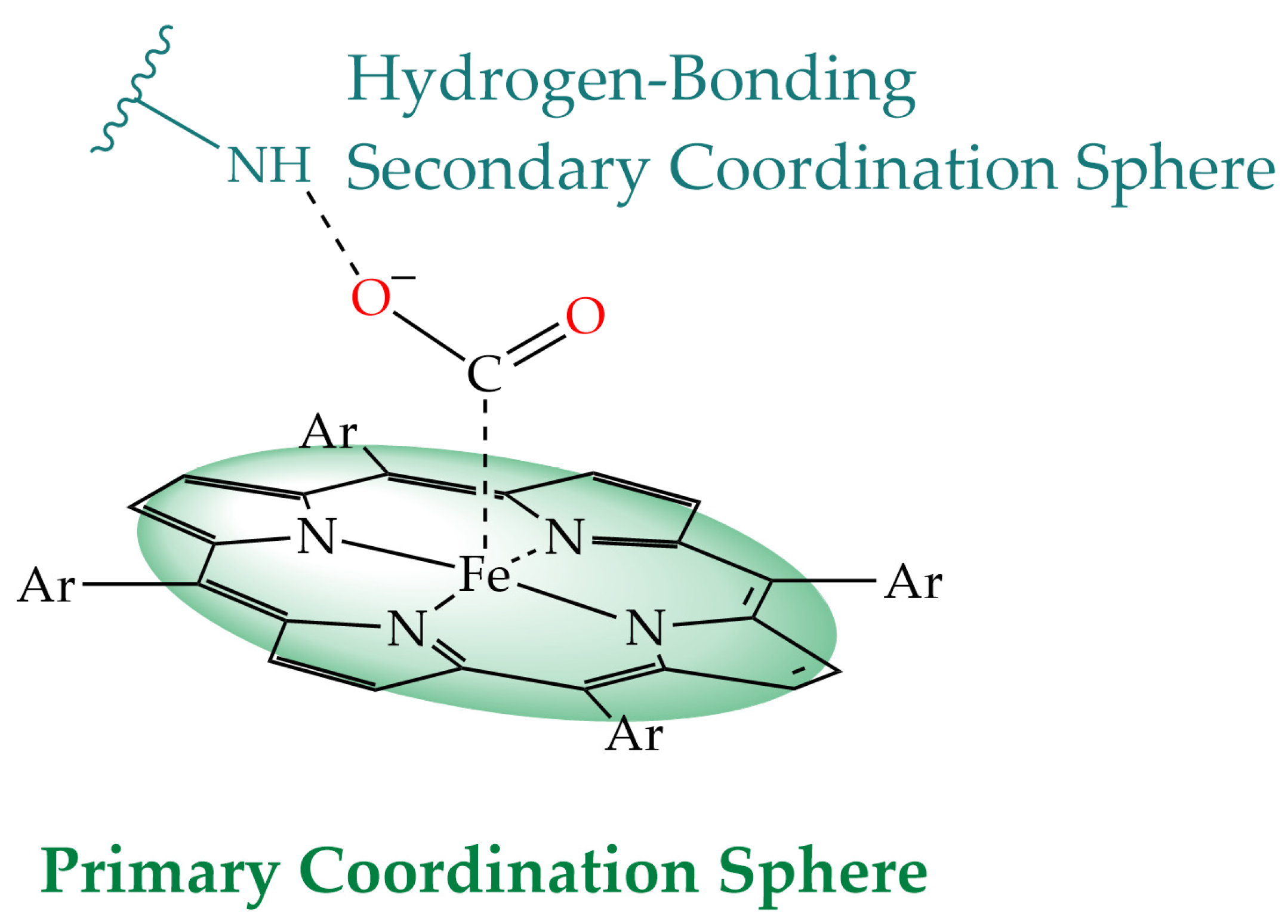
2. Hydrogen-Bonding Secondary Coordination Sphere Effect
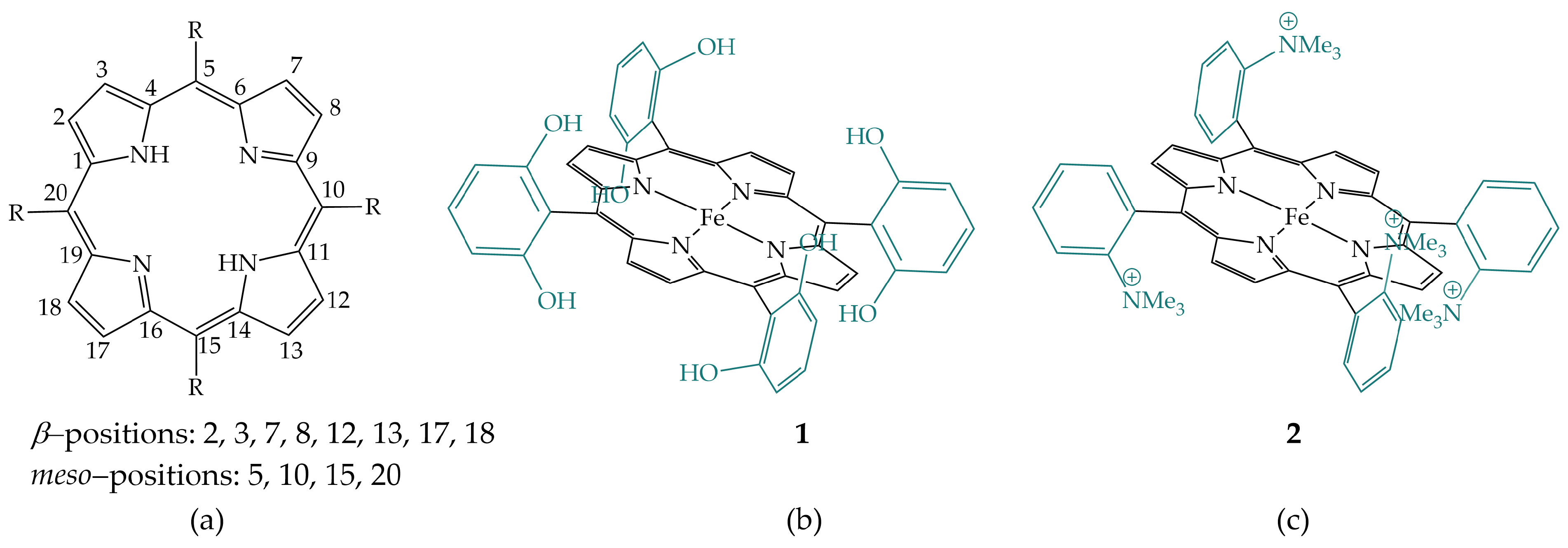
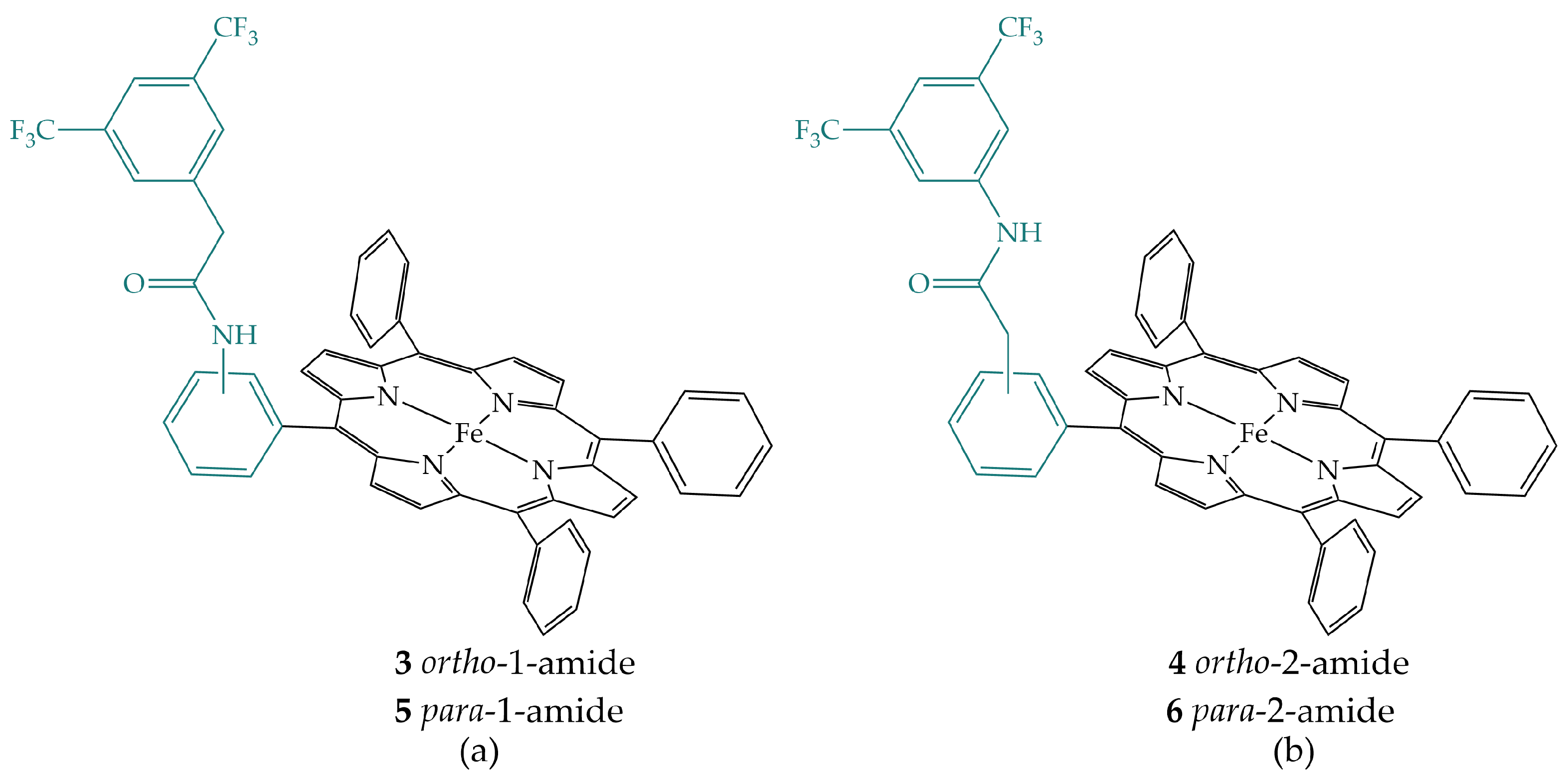
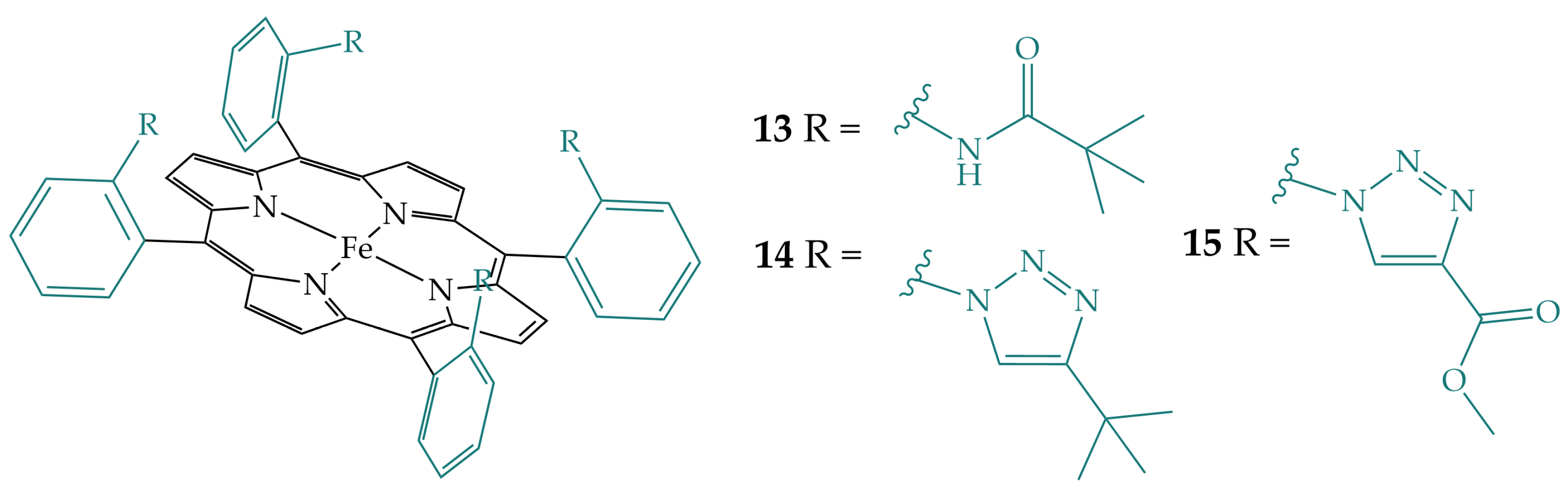
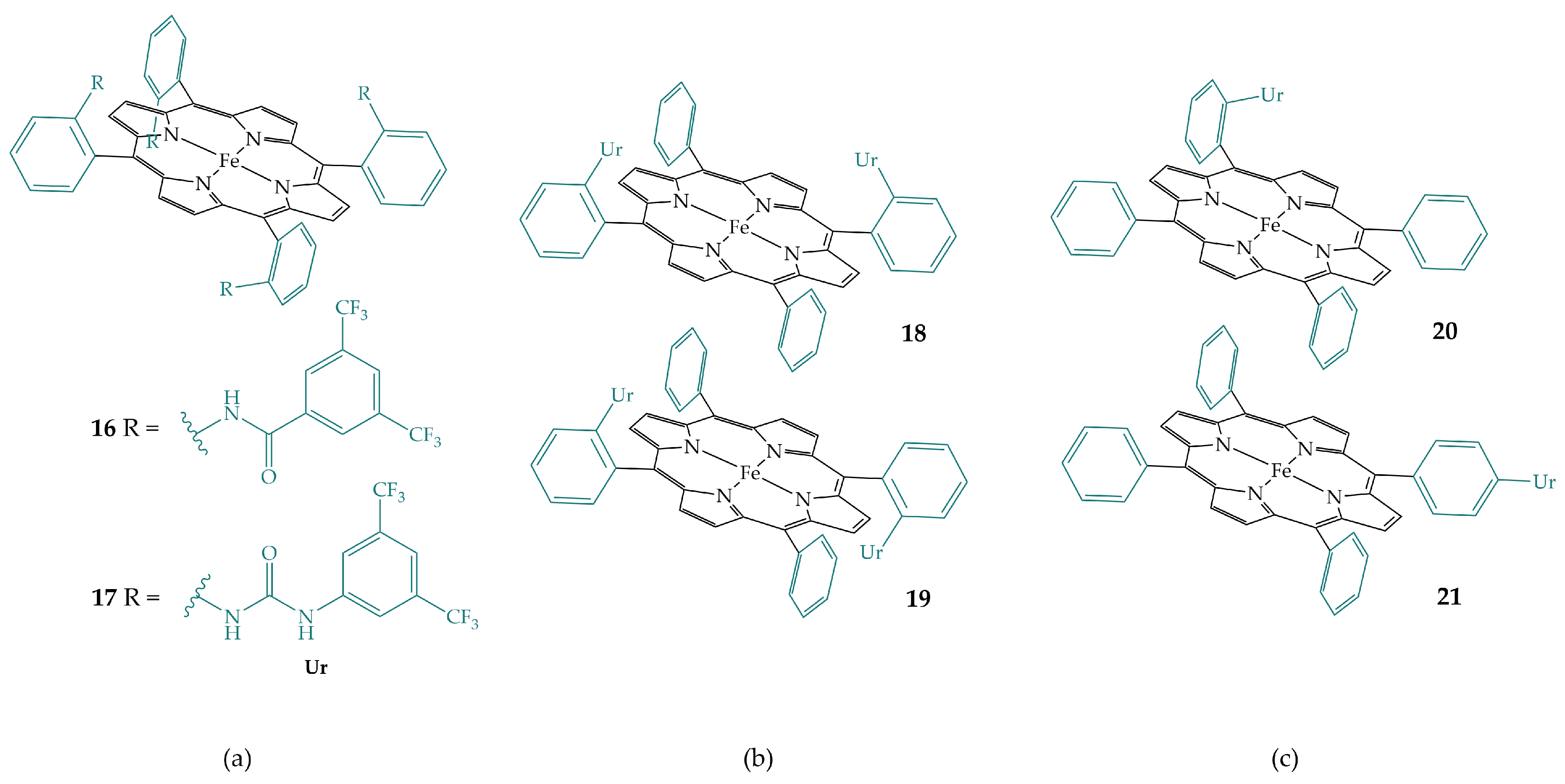
2.1. Amides in the Secondary Coordination Sphere
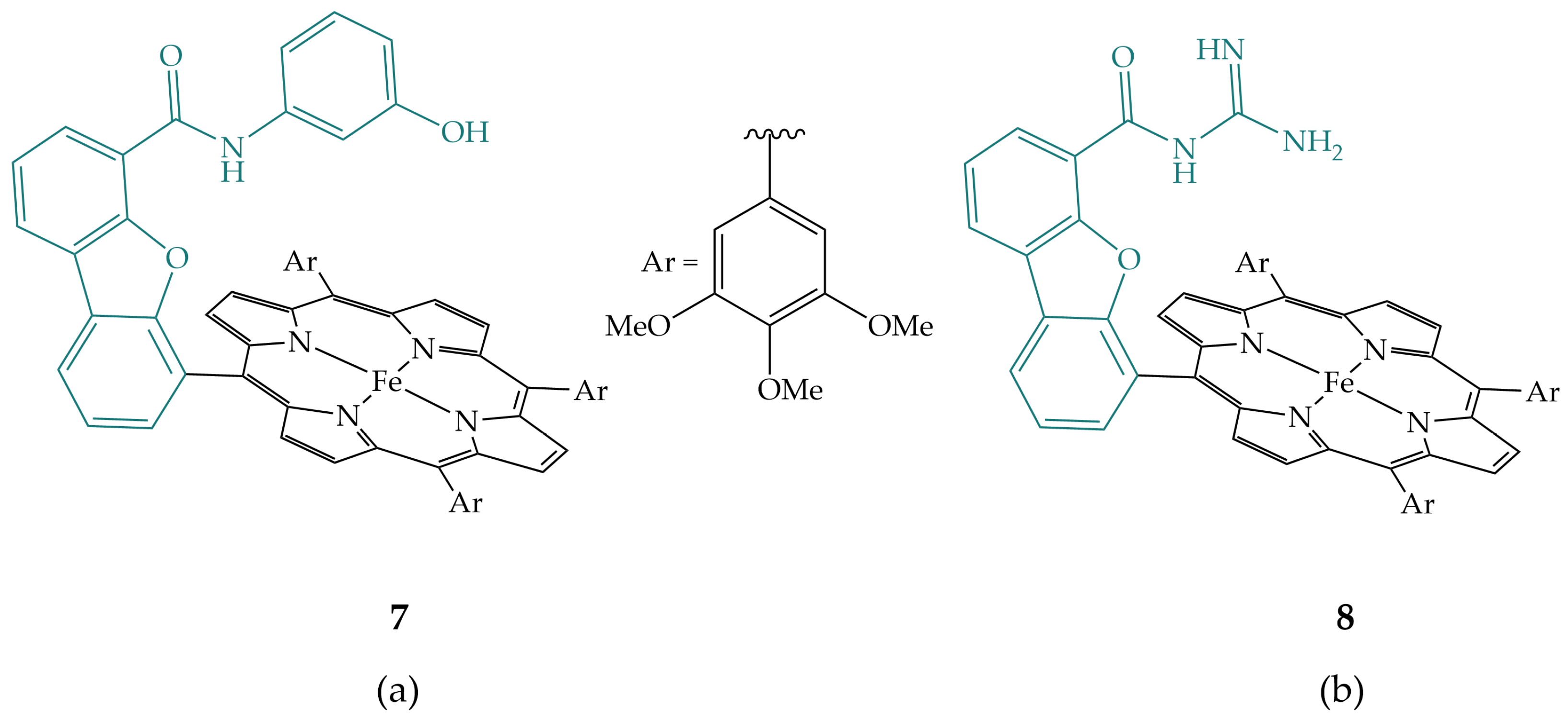
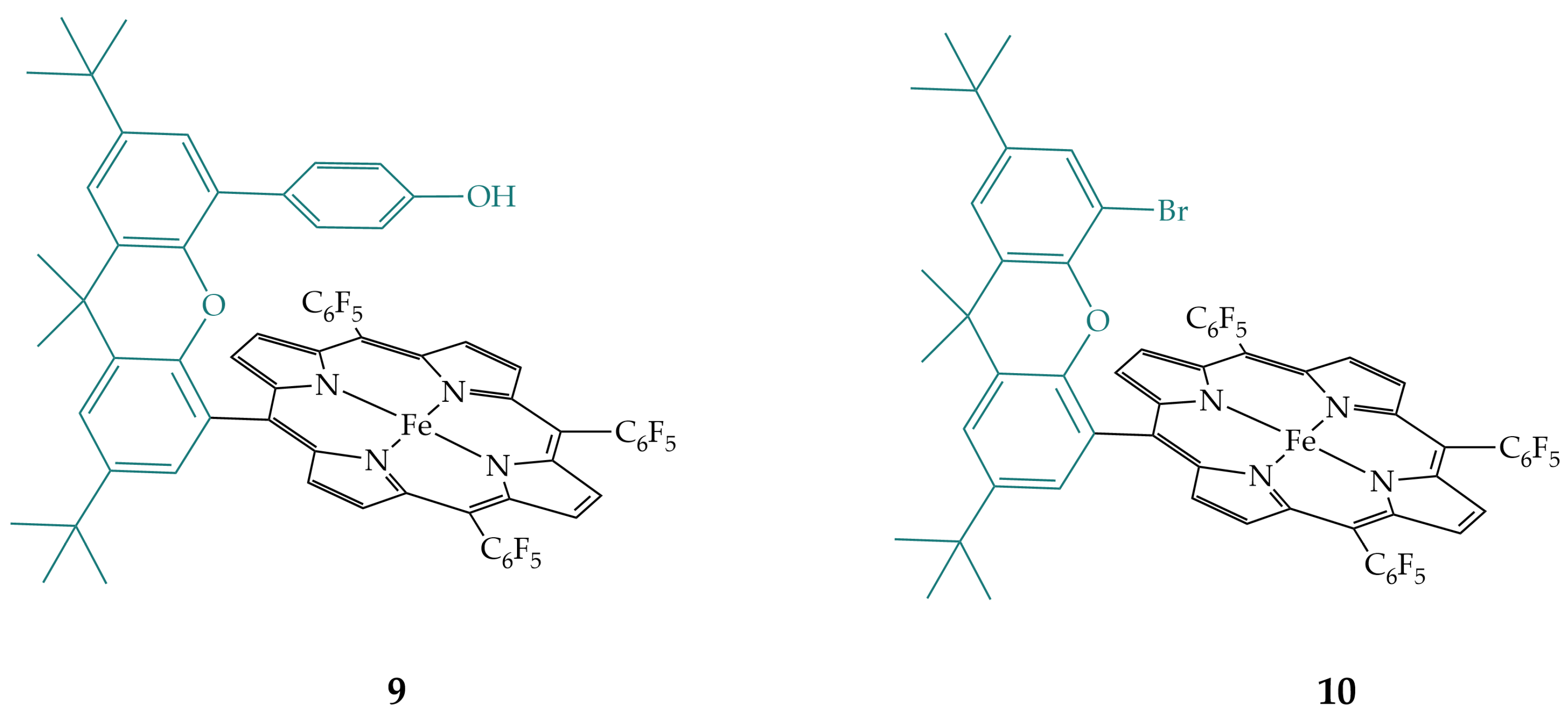
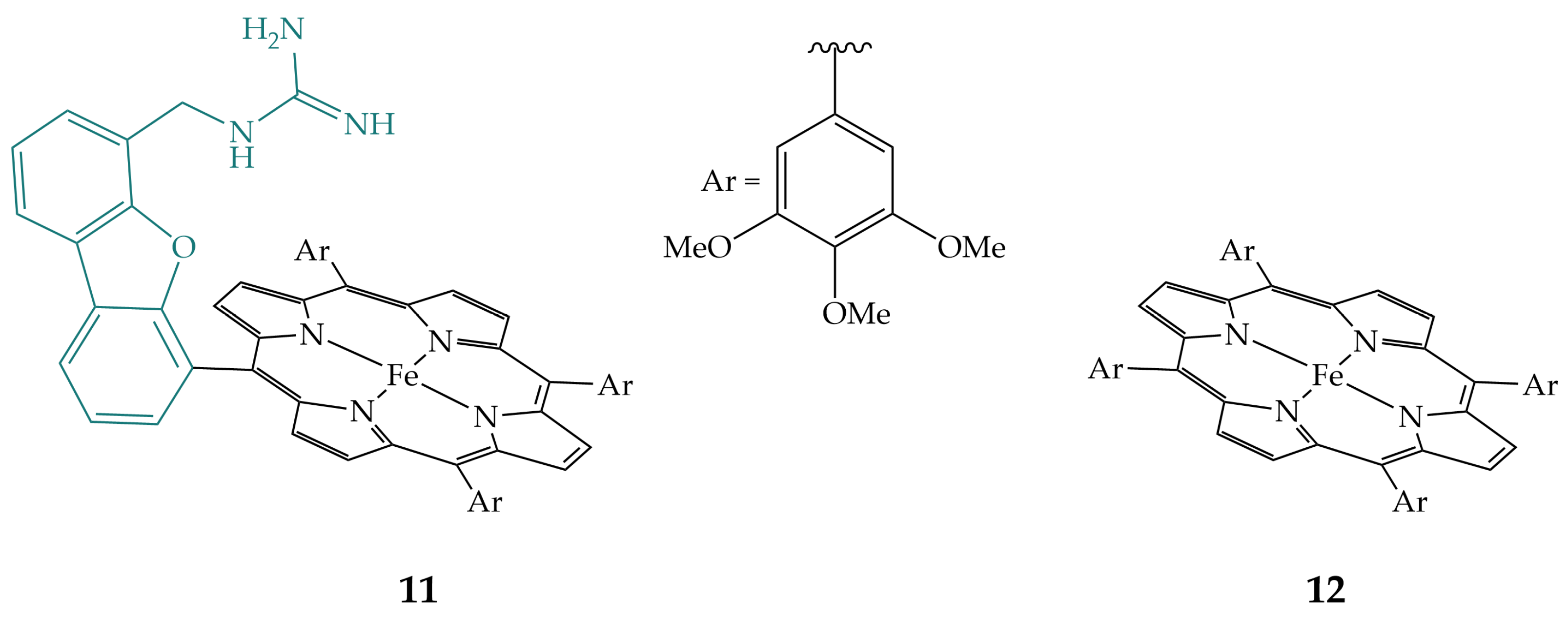
2.2. Phenols and Guanidines in the Secondary Coordination Sphere
2.3. Triazoles in the Secondary Coordination Sphere
2.4. Ureas in the Secondary Coordination Sphere
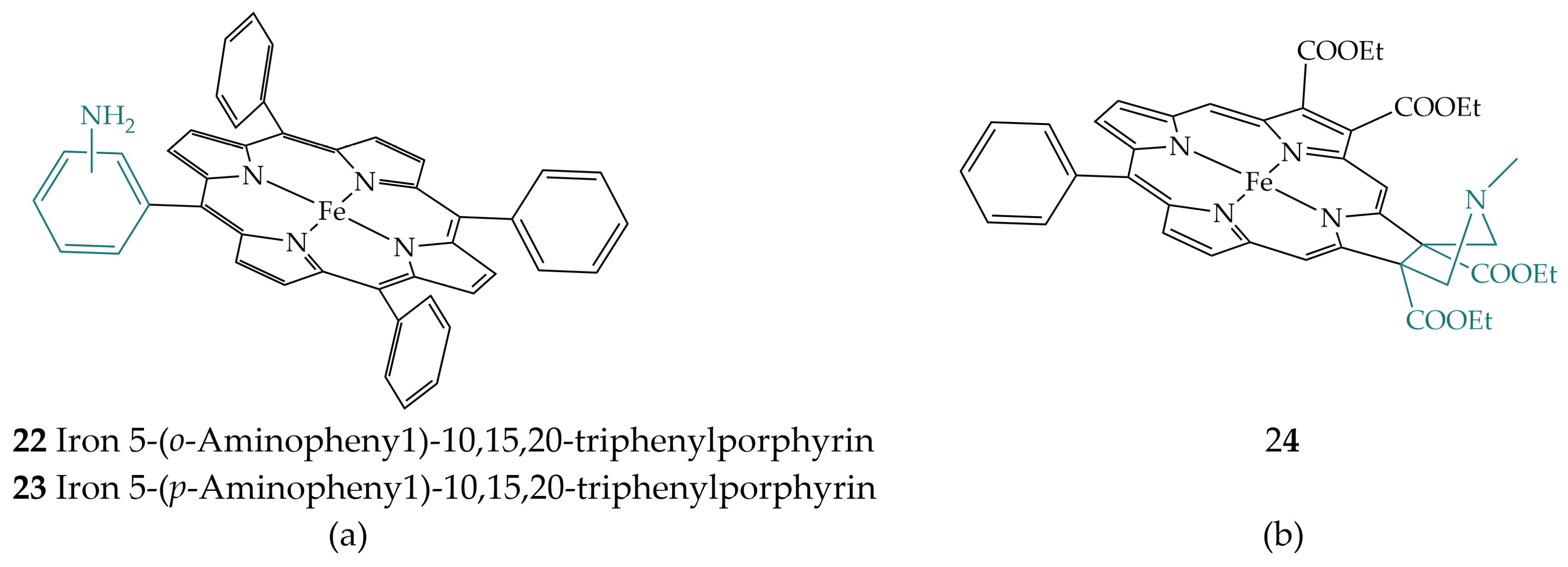
2.5. Amines in the Secondary Coordination Sphere
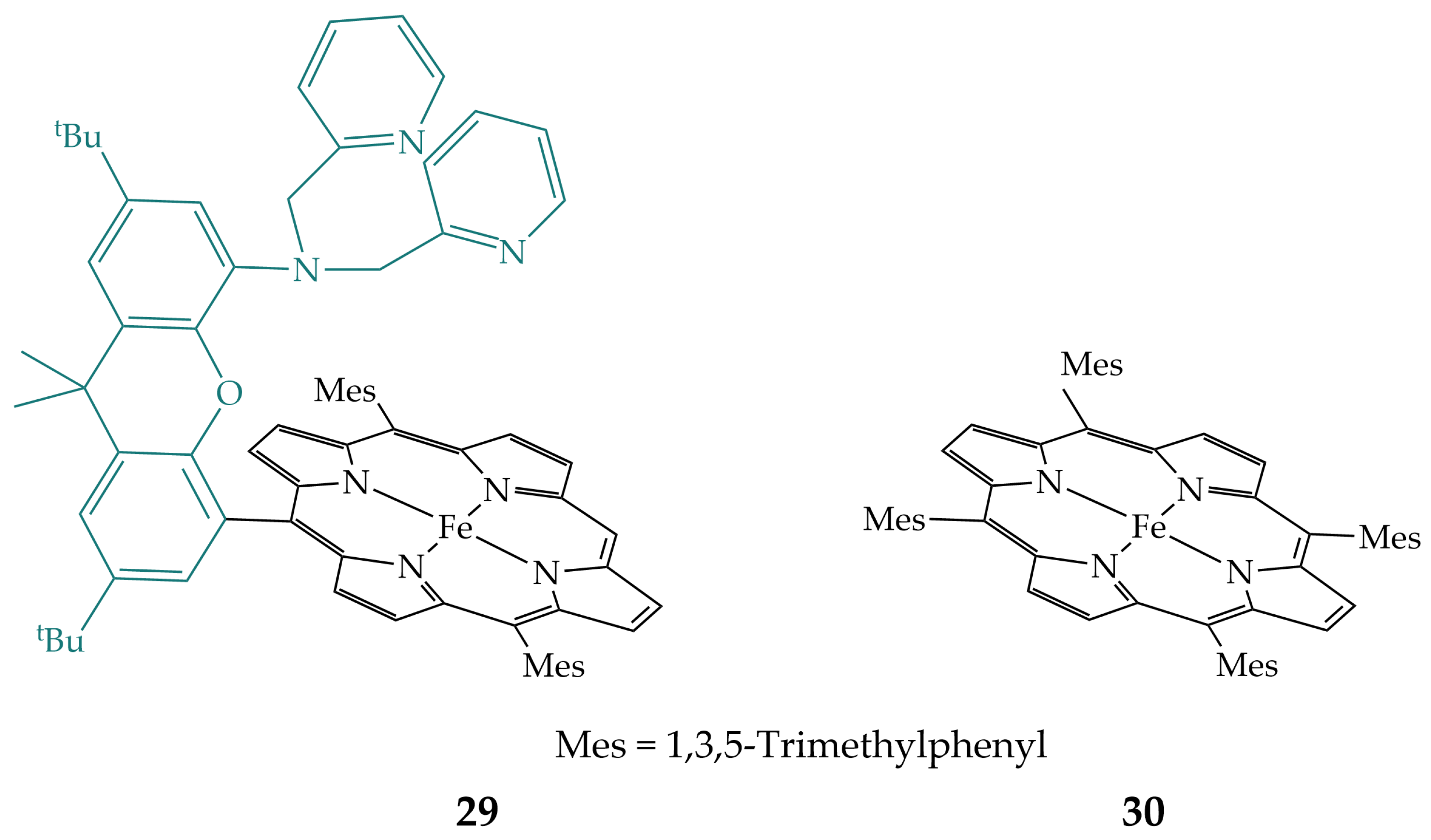
2.6. Pyridines in the Secondary Coordination Sphere
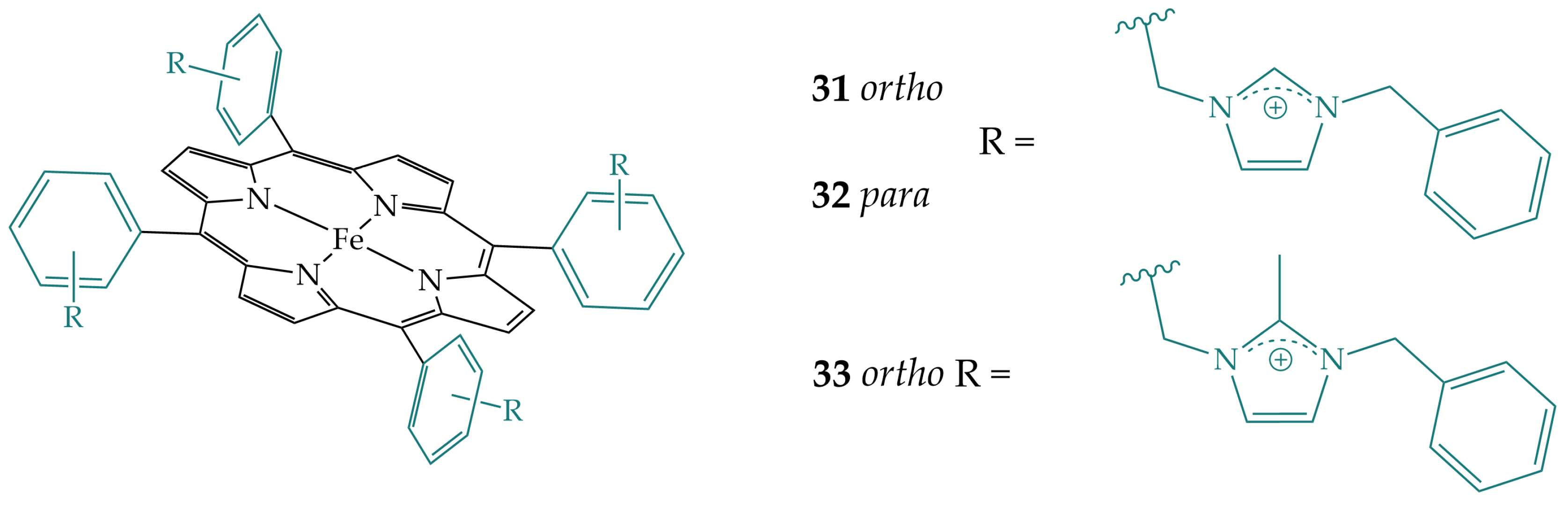
2.7. Imidazoles in the Secondary Coordination Sphere
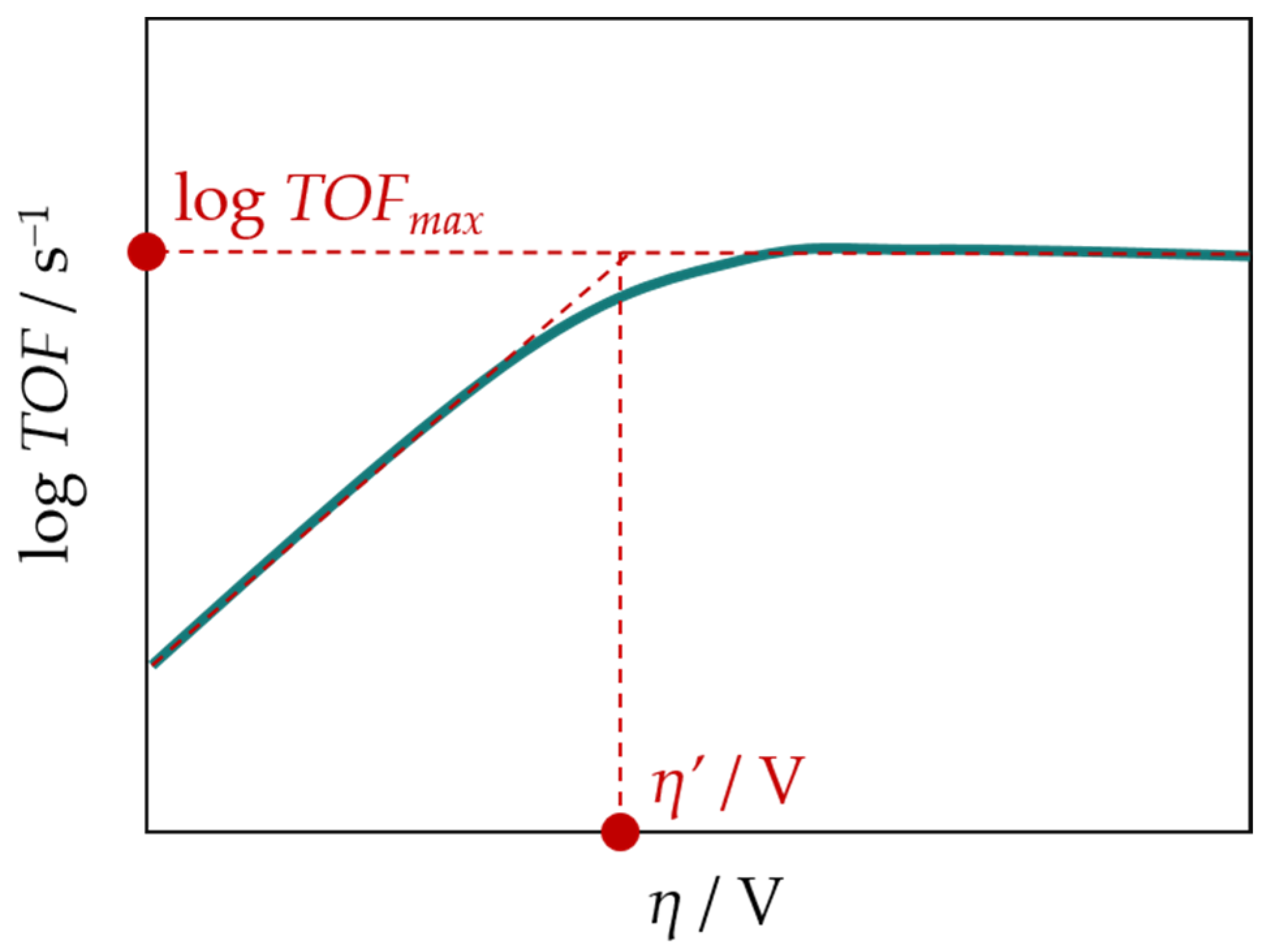
2.8. Summary of Data for Iron Porphyrins
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Carbon Dioxide. National Oceanic and Atmospheric Administration. Available online: https://climate.nasa.gov/ (accessed on 6 March 2023).
- Kinzel, N.W.; Werlé, C.; Leitner, W. Transition Metal Complexes as Catalysts for the Electroconversion of CO2: An Organometallic Perspective. Angew. Chem. Int. Ed. 2021, 60, 11628–11686. [Google Scholar] [CrossRef]
- Pappijn, C.A.R.; Ruitenbeek, M.; Reyniers, M.-F.; Van Geem, K.M. Challenges and Opportunities of Carbon Capture and Utilization: Electrochemical Conversion of CO2 to Ethylene. Front. Energy Res. 2020, 8, 557466. [Google Scholar]
- Francke, R.; Schille, B.; Roemelt, M. Homogeneously Catalyzed Electroreduction of Carbon Dioxide−Methods, Mechanisms, and Catalysts. Chem. Rev. 2018, 118, 4631–4701. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, H.; Xiang, H.; Rasul, S.; Fontmorin, J.-M.; Izadi, P.; Roldan, A.; Taylor, R.; Feng, Y.; Banerji, L.; et al. How to Go Beyond C1 Products with Electrochemical Reduction of CO2. Sustain. Energy Fuels 2021, 5, 5893–5914. [Google Scholar] [CrossRef]
- Zhang, W.; Lai, W.; Cao, R. Energy-Related Small Molecule Activation Reactions: Oxygen Reduction and Hydrogen and Oxygen Evolution Reactions Catalyzed by Porphyrin- and Corrole-Based Systems. Chem. Rev. 2017, 117, 3717–3797. [Google Scholar] [CrossRef]
- Amanullah, S.; Saha, P.; Dey, A. Recent Developments in the Synthesis of Bio-Inspired Iron Porphyrins for Small Molecule Activation. Chem. Commun. 2022, 58, 5808–5828. [Google Scholar] [CrossRef] [PubMed]
- Dedić, D.; Dorniak, A.; Rinner, U.; Schöfberger, W. Recent Progress in (Photo-)-Electrochemical Conversion of CO2 With Metal Porphyrinoid-Systems. Front. Chem. 2021, 9, 685619. [Google Scholar] [CrossRef]
- Bhugun, I.; Lexa, D.; Savéant, J.-M. Catalysis of the Electrochemical Reduction of Carbon Dioxide by Iron(0) Porphyrins. Synergistic Effect of Lewis Acid Cations. J. Phys. Chem. 1996, 100, 19981–19985. [Google Scholar] [CrossRef]
- Lei, K.; Xia, B.Y. Electrocatalytic CO2 Reduction: From Discrete Molecular Catalysts to Their Integrated Catalytic Materials. Chem. Eur. J. 2022, 28, e202200141. [Google Scholar]
- Amanullah, S.; Saha, P.; Nayek, A.; Ahmed, M.E.; Dey, A. Biochemical and Artificial Pathways for the Reduction of Carbon Dioxide, Nitrite and the Competing Proton Reduction: Effect of 2nd Sphere Interactions in Catalysis. Chem. Soc. Rev. 2021, 50, 3755–3823. [Google Scholar] [CrossRef]
- Gotico, P.; Leibl, W.; Halime, Z.; Aukauloo, A. Shaping the Electrocatalytic Performance of Metal Complexes for CO2 Reduction. ChemElectroChem 2021, 8, 3472–3481. [Google Scholar] [CrossRef]
- Nichols, A.W.; Machan, C.W. Secondary-Sphere Effects in Molecular Electrocatalytic CO2 Reduction. Front. Chem. 2019, 7, 397. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338, 90–94. [Google Scholar] [CrossRef]
- Azcarate, I.; Costentin, C.; Robert, M.; Savéant, J.-M. Dissection of Electronic Substituent Effects in Multielectron−Multistep Molecular Catalysis. Electrochemical CO2-to-CO Conversion Catalyzed by Iron Porphyrins. J. Phys. Chem. C 2016, 120, 28951–28960. [Google Scholar] [CrossRef]
- Azcarate, I.; Costentin, C.; Robert, M.; Savéant, J.-M. Through-Space Charge Interaction Substituent Effects in Molecular Catalysis Leading to the Design of the Most Efficient Catalyst of CO2-to-CO Electrochemical Conversion. J. Am. Chem. Soc. 2016, 138, 16639–16644. [Google Scholar] [CrossRef]
- Hammouche, M.; Lexa, D.; Savéant, J.M. Catalysis of the Electrochemical Reduction of Carbon Dioxide by Iron(“0”) Porphyrins. J. Electroanal. Chem. 1988, 249, 347–351. [Google Scholar] [CrossRef]
- Mondal, B.; Rana, A.; Sen, P.; Dey, A. Intermediates Involved in the 2e−/2H+ Reduction of CO2 to CO by Iron(0) Porphyrin. J. Am. Chem. Soc. 2015, 137, 11214–11217. [Google Scholar] [CrossRef]
- Nichols, E.M.; Derrick, J.S.; Nistanaki, S.K.; Smith, P.T.; Chang, C.J. Positional Effects of Second-Sphere Amide Pendants on Electrochemical CO2 Reduction Catalyzed by Iron Porphyrins. Chem. Sci. 2018, 9, 2952–2960. [Google Scholar] [CrossRef]
- Margarit, C.G.; Schnedermann, C.; Asimow, N.G.; Nocera, D.G. Carbon Dioxide Reduction by Iron Hangman Porphyrins. Organometallics 2019, 38, 1219–1223. [Google Scholar] [CrossRef]
- Guo, K.; Li, X.; Lei, H.; Zhang, W.; Cao, R. Unexpected Effect of Intramolecular Phenolic Group on Electrocatalytic CO2 Reduction. ChemCatChem 2020, 12, 1591–1595. [Google Scholar] [CrossRef]
- Guo, H.; Liang, Z.; Guo, K.; Lei, H.; Wang, Y.; Zhang, W.; Cao, R. Iron Porphyrin with Appended Guanidyl Group for Significantly Improved Electrocatalytic Carbon Dioxide Reduction Activity and Selectivity in Aqueous Solutions. Chin. J. Catal. 2022, 43, 3089–3094. [Google Scholar] [CrossRef]
- Sen, P.; Mondal, B.; Saha, D.; Rana, A.; Dey, A. Role of 2nd Sphere H-bonding Residues in Tuning the Kinetics of CO2 Reduction to CO by Iron Porphyrin Complexes. Dalton Trans. 2019, 48, 5965–5977. [Google Scholar] [CrossRef] [PubMed]
- Gotico, P.; Boitrel, B.; Guillot, R.; Sircoglou, M.; Quaranta, A.; Halime, Z.; Leibl, W.; Aukauloo, A. Second-Sphere Biomimetic Multipoint Hydrogen-Bonding Patterns to Boost CO2 Reduction of Iron Porphyrins. Angew. Chem. Int. Ed. 2019, 58, 4504–4509. [Google Scholar] [CrossRef]
- Gotico, P.; Roupnel, L.; Guillot, R.; Sircoglou, M.; Leibl, W.; Halime, Z.; Aukauloo, A. Atropisomeric Hydrogen Bonding Control for CO2 Binding and Enhancement of Electrocatalytic Reduction at Iron Porphyrins. Angew. Chem. Int. Ed. 2020, 59, 22451–22455. [Google Scholar] [CrossRef] [PubMed]
- Derrick, J.S.; Loipersberger, M.; Nistanaki, S.K.; Rothweiler, A.V.; Head-Gordon, M.; Nichols, E.M.; Chang, C.J. Templating Bicarbonate in the Second Coordination Sphere Enhances Electrochemical CO2 Reduction Catalyzed by Iron Porphyrins. J. Am. Chem. Soc. 2022, 144, 11656–11663. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Fan, Y.-J.; Zhang, J.-L. Construction of Secondary Coordination Sphere Boosts Electrochemical CO2 Reduction of Iron Porphyrins. J. Porphyr. Phthaloc. 2020, 24, 465–472. [Google Scholar] [CrossRef]
- Amanullah, S.; Saha, P.; Dey, A. Activating the Fe(I) State of Iron Porphyrinoid with Second-Sphere Proton Transfer Residues for Selective Reduction of CO2 to HCOOH via Fe(III/II)−COOH Intermediate(s). J. Am. Chem. Soc. 2021, 143, 13579–13592. [Google Scholar] [CrossRef]
- Guo, K.; Li, X.; Lei, H.; Guo, H.; Jin, X.; Zhang, X.-P.; Zhang, W.; Apfel, U.-P.; Cao, R. Role-Specialized Division of Labor in CO2 Reduction with Doubly-Functionalized Iron Porphyrin Atropisomers. Angew. Chem. Int. Ed. 2022, 61, e202209602. [Google Scholar] [CrossRef]
- Ramuglia, A.R.; Budhija, V.; Ly, K.H.; Marquardt, M.; Schwalbe, M.; Weidinger, I.M. An Iron Porphyrin Complex with Pendant Pyridine Substituents Facilitates Electrocatalytic CO2 Reduction via Second Coordination Sphere Effects. ChemCatChem 2021, 13, 3934–3944. [Google Scholar] [CrossRef]
- Narouz, M.R.; De La Torre, P.; An, L.; Chang, C.J. Multifunctional Charge and Hydrogen-Bond Effects of Second- Sphere Imidazolium Pendants Promote Capture and Electrochemical Reduction of CO2 in Water Catalyzed by Iron Porphyrins. Angew. Chem. Int. Ed. 2022, 61, e202207666. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J.-M. Catalysis of the Electrochemical Reduction of Carbon Dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Gotico, P.; Halime, Z.; Aukauloo, A. Recent Advances in Metalloporphyrin-Based Catalyst Design Towards Carbon Dioxide Reduction: From Bio-Inspired Second Coordination Sphere Modifications to Hierarchical Architectures. Dalton Trans. 2020, 49, 2381–2396. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
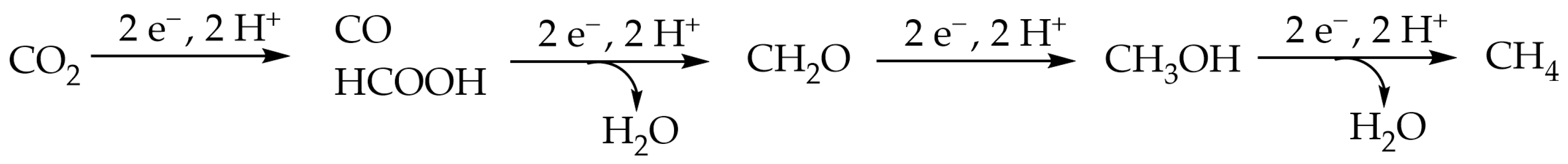{kind=link}
| Porphyrin | Conditions | η′/V | log TOFmax/s−1 | FE/% | Ref. | |
|---|---|---|---|---|---|---|
| FeTPP | DMF + 3 M PhOH | 0.74 | 4.5 | 87 | [16] | |
| 1 | DMF + 2 M H2O | 0.64 | 6.0 | 94 | [12,14] | |
| 2 | DMF + 3 M PhOH | 0.22 | 6.0 | ~100 | [16] | |
| 3 | DMF + 0.1 M PhOH | −2.122 | 4.4 | 83 | [19] | |
| 4 | DMF + 0.1 M PhOH | −2.182 | 6.7 | 92 | [19] | |
| 5 | DMF + 0.1 M PhOH | −2.152 | 2.2 | 74 | [19] | |
| 6 | DMF + 0.1 M PhOH | −2.162 | 3.8 | 79 | [19] | |
| 7 | DMF + 0.04 M PhOH | 0.76 | 2.7 | 94 | [20] | |
| 8 | DMF + 0.04 M PhOH | 0.77 | 2.5 | 93 | [20,33] | |
| 11 | ACN + 0.25 M H2O | 5.6 | 98 | [22] | ||
| 12 | ACN + 0.25 M H2O | 4.2 | 96 | [22] | ||
| 13 | ACN + 3 M PhOH | −1.273 | 5.7 | 87 | [23] | |
| 14 | ACN + 3 M PhOH | −1.193 | 3.0 | [23] | ||
| 15 | ACN + 3 M PhOH | −1.153 | 2.4 | [23] | ||
| 16 | DMF + 5 M H2O | 0.63 | 3.85 | [24] | ||
| 17 | DMF + 5 M H2O | 0.43 | 3.83 | 91 | [24] | |
| 18 | DMF + 2.22 M H2O | 0.56 | 4.08 | 91 | [25] | |
| 19 | DMF + 2.22 M H2O | 0.61 | 4.71 | 95 | [25] | |
| 20 | DMF + 0.1 M TEAHCO3 | −2.062 | 93 | [26] | ||
| 21 | DMF + 0.1 M TEAHCO3 | −2.182 | 54 | [26] | ||
| 22 | DMF + 1 M PhOH | 0.76 | 4.0 | 88 | [27] | |
| 23 | DMF + 1 M PhOH | 0.82 | 3.4 | 87 | [27] | |
| 24 | DMF + 1 M H2O | −1.554 | 2.2 | 974 | [28] | |
| 25 | ACN + 0.25 M PhOH | −1.825 | 5.6 | 95 | [29] | |
| 26 | ACN + 0.25 M PhOH | −1.885 | 5.4 | 93 | [29] | |
| 27 | ACN + 0.25 M PhOH | −1.985 | 5.0 | 93 | [29] | |
| 28 | ACN + 0.25 M PhOH | −2.055 | 4.3 | 91 | [29] | |
| 29 | ACN + 0.128 M PhOH | 0.95 | 8.3 | [30] | ||
| 30 | ACN + 0.128 M PhOH | 0.86 | 6.9 | [30] | ||
| 31 | ACN + 3 M TFE | −1.782 | 9.1 | 100 | [31] | |
| 32 | ACN + 3 M TFE | −1.792 | 4.9 | 28 | [31] | |
| 33 | ACN + 3 M TFE | −1.792 | 7.5 | 100 | [31] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Briš, A.; Margetić, D. Hydrogen-Bonding Secondary Coordination Sphere Effect on CO2 Reduction. Organics 2023, 4, 277-288. https://doi.org/10.3390/org4020022
Briš A, Margetić D. Hydrogen-Bonding Secondary Coordination Sphere Effect on CO2 Reduction. Organics. 2023; 4(2):277-288. https://doi.org/10.3390/org4020022
Chicago/Turabian StyleBriš, Anamarija, and Davor Margetić. 2023. "Hydrogen-Bonding Secondary Coordination Sphere Effect on CO2 Reduction" Organics 4, no. 2: 277-288. https://doi.org/10.3390/org4020022
APA StyleBriš, A., & Margetić, D. (2023). Hydrogen-Bonding Secondary Coordination Sphere Effect on CO2 Reduction. Organics, 4(2), 277-288. https://doi.org/10.3390/org4020022