Dual UV-Thermal Curing of Biobased Resorcinol Epoxy Resin-Diatomite Composites with Improved Acoustic Performance and Attractive Flame Retardancy Behavior


 ,
,  ,
,
Abstract
1. Introduction
2. Experimental
Materials
3. Sample preparation
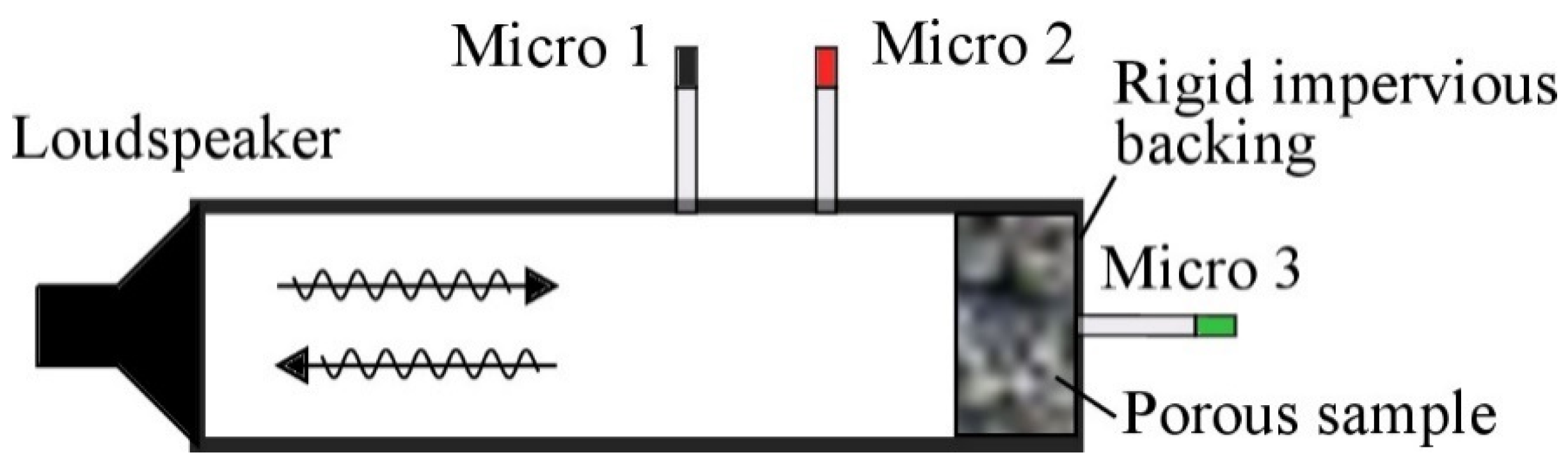
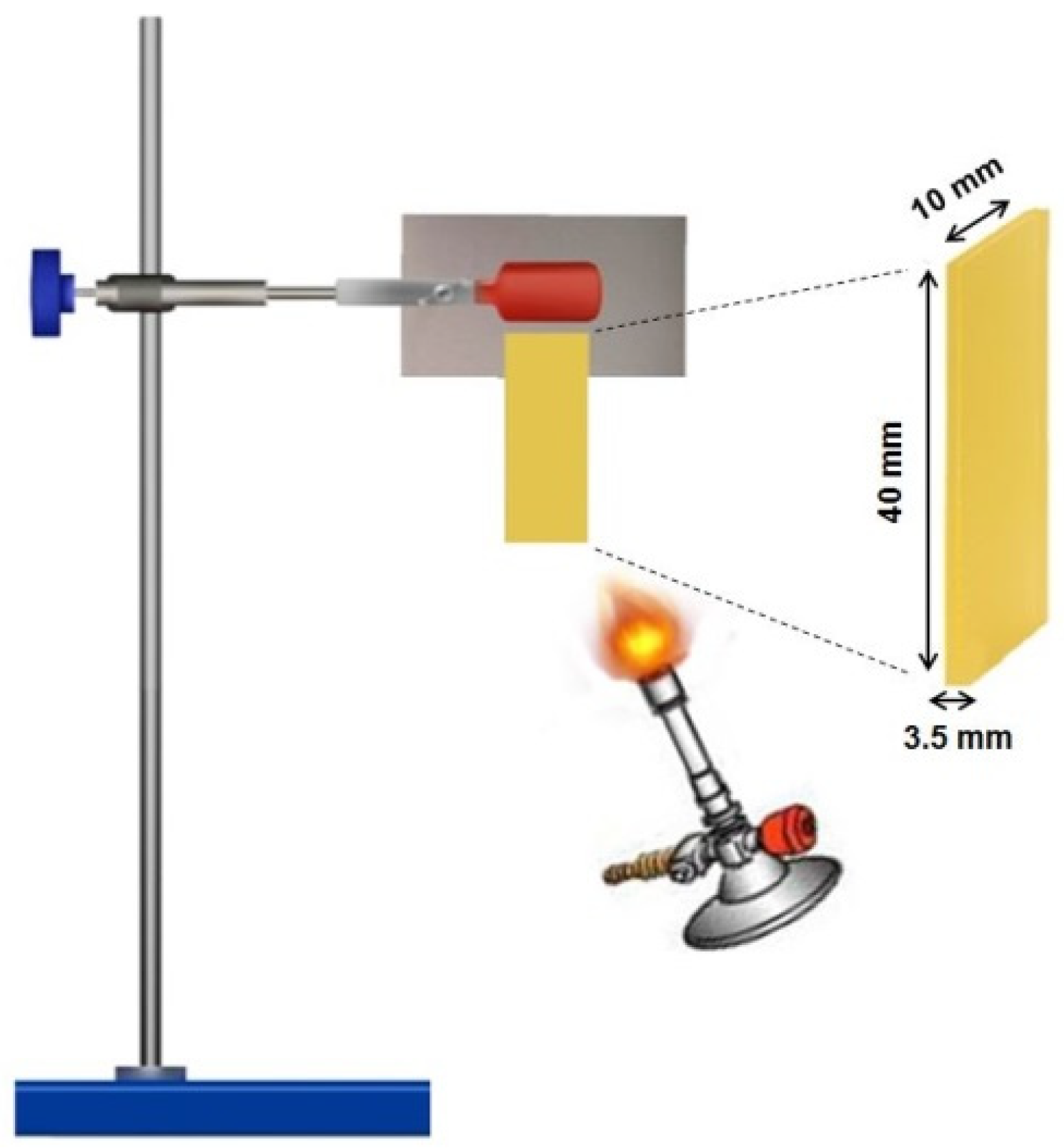
Experimental Techniques
4. Results and Discussion
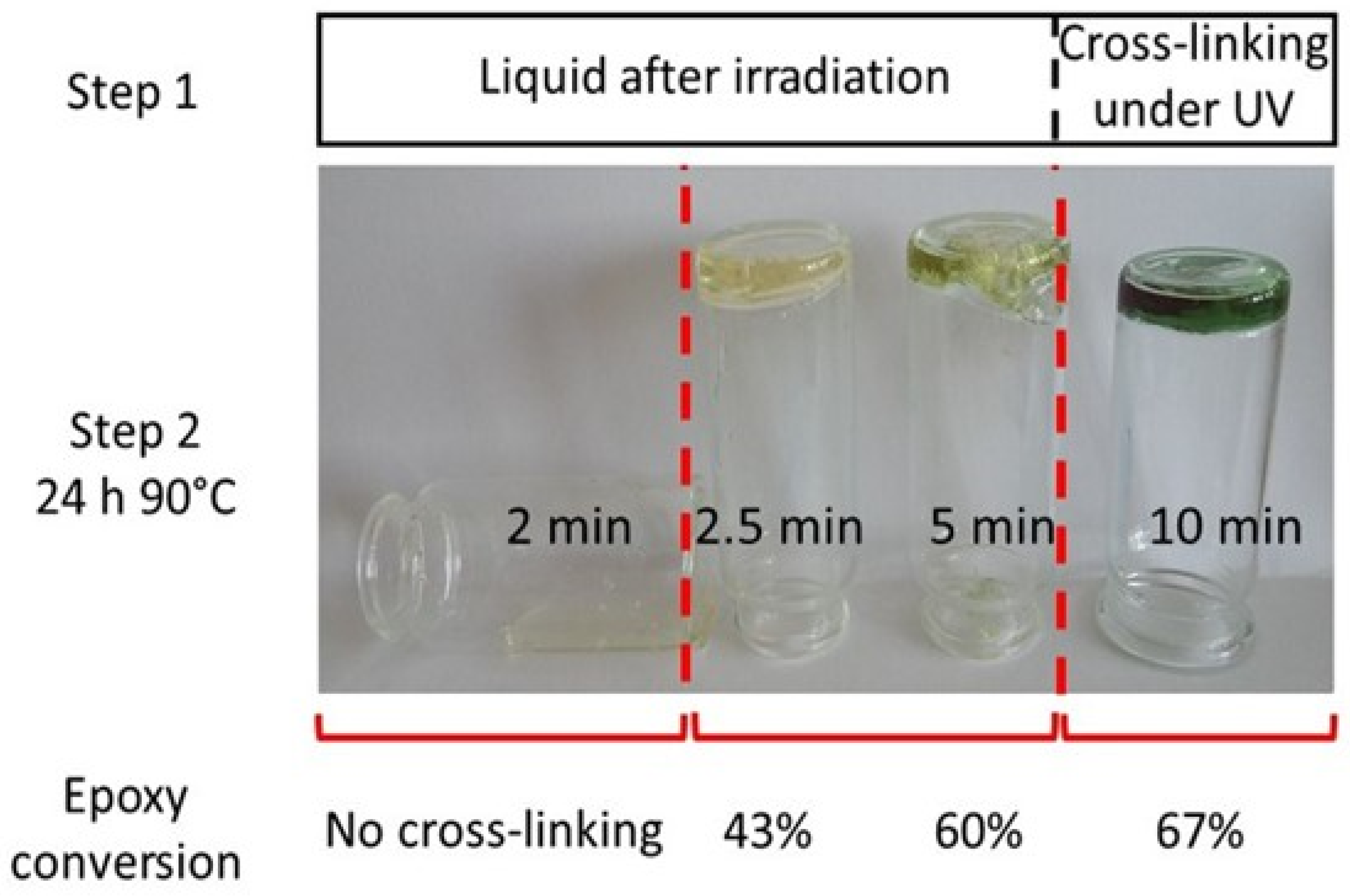
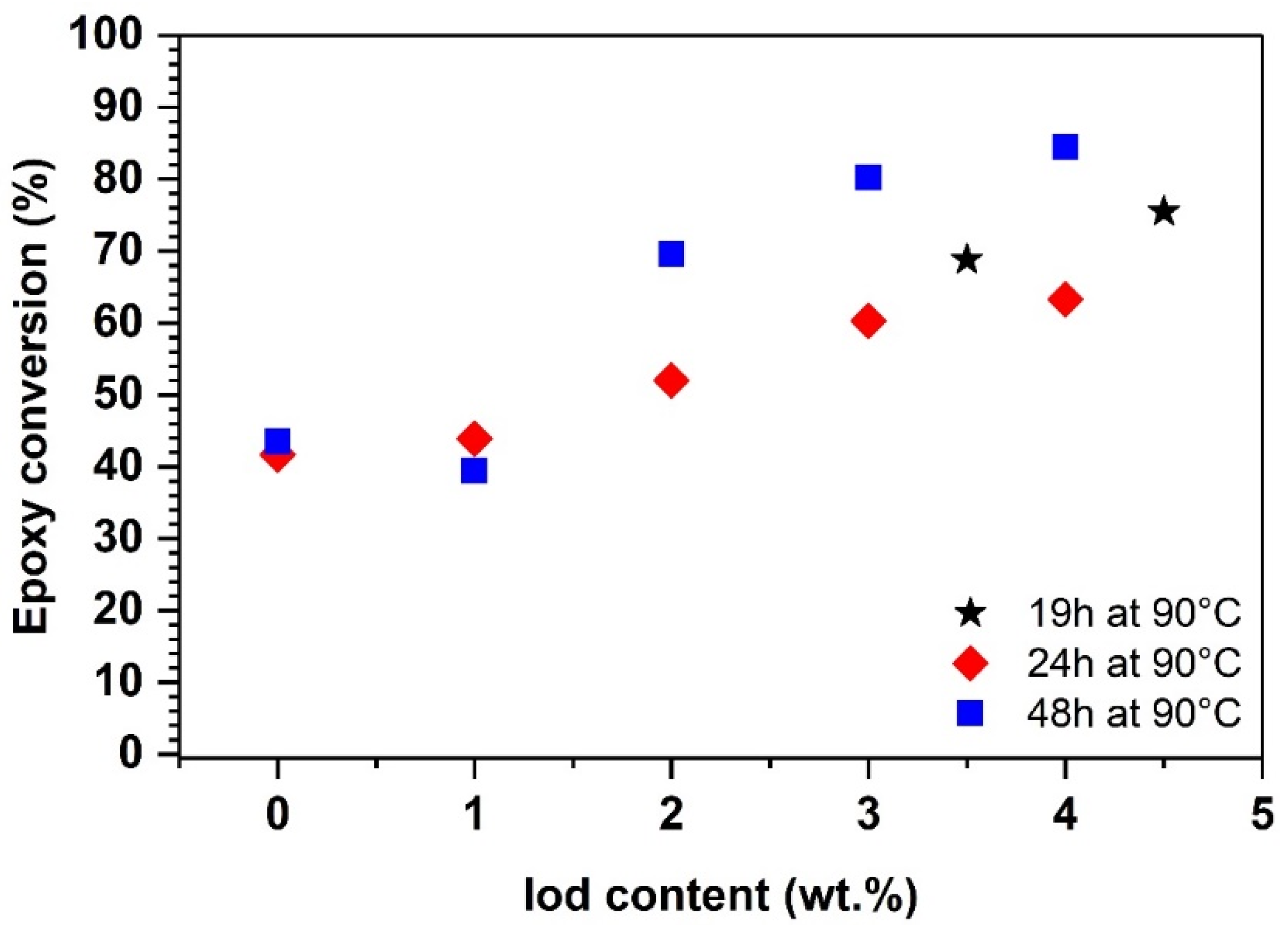
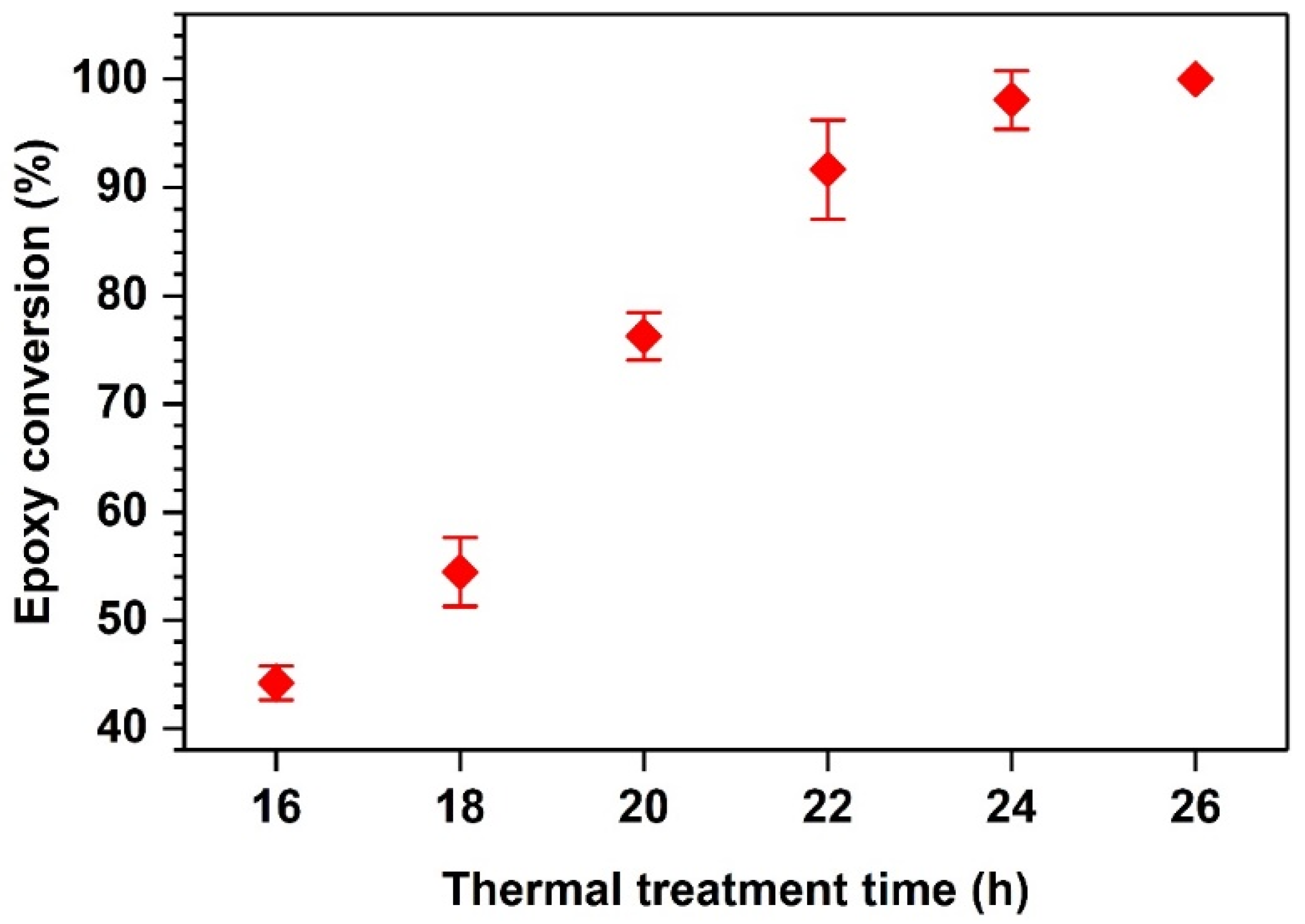
4.1. Photoinitiation and Thermal Dark Curing Process
4.2. UV-Induced Epoxy-Diatomite Composites with Thermal Post-Curing
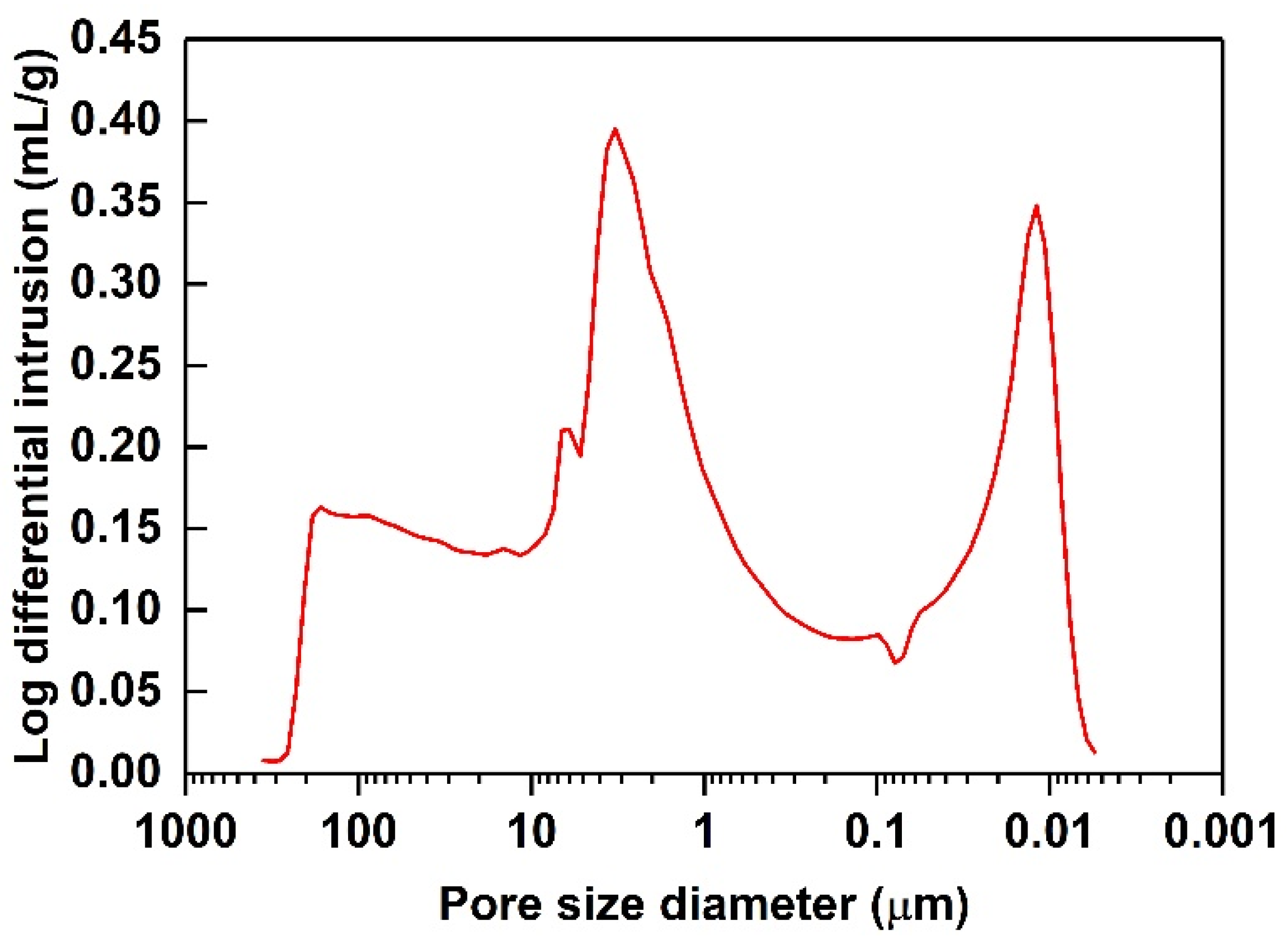
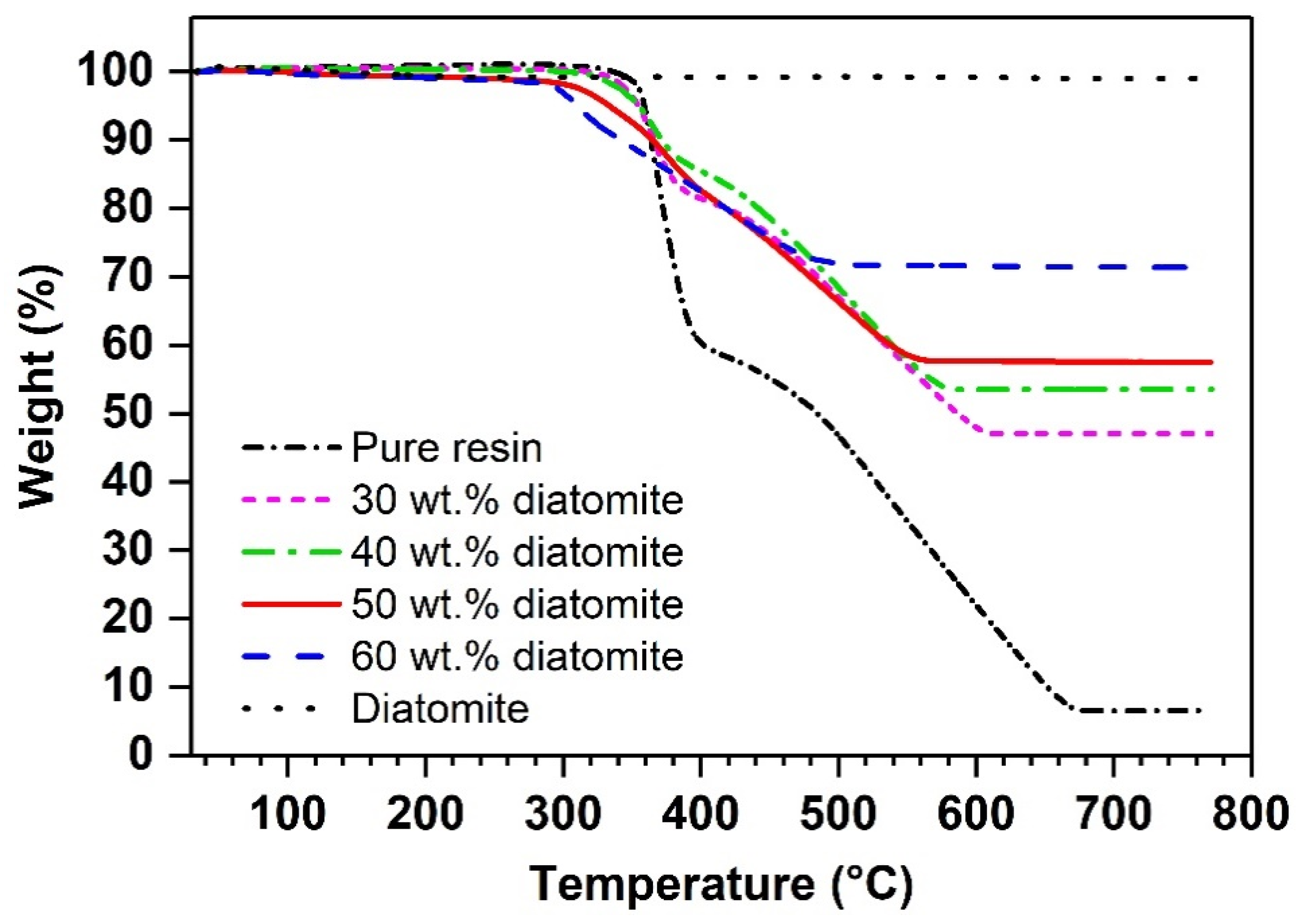
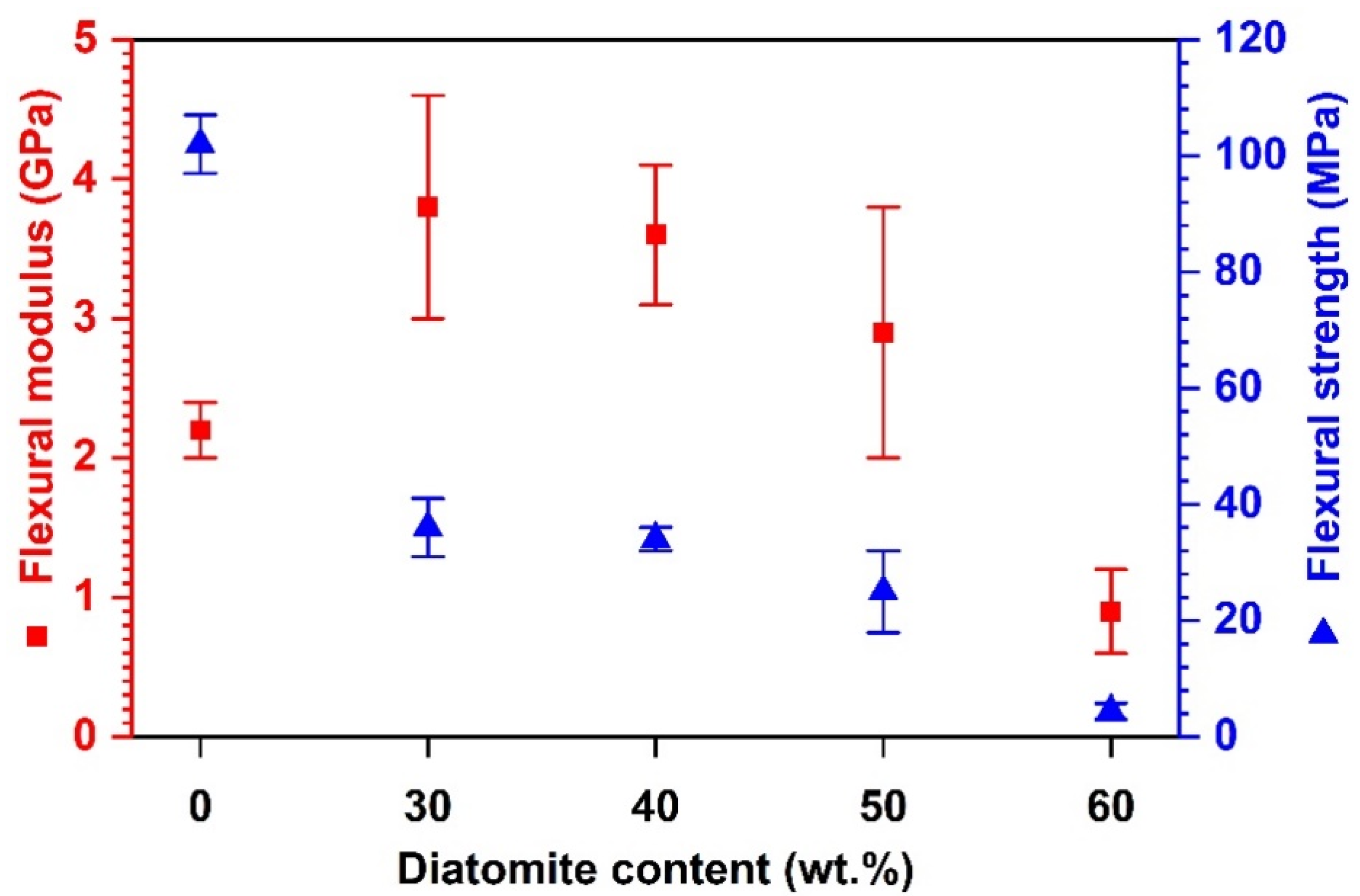
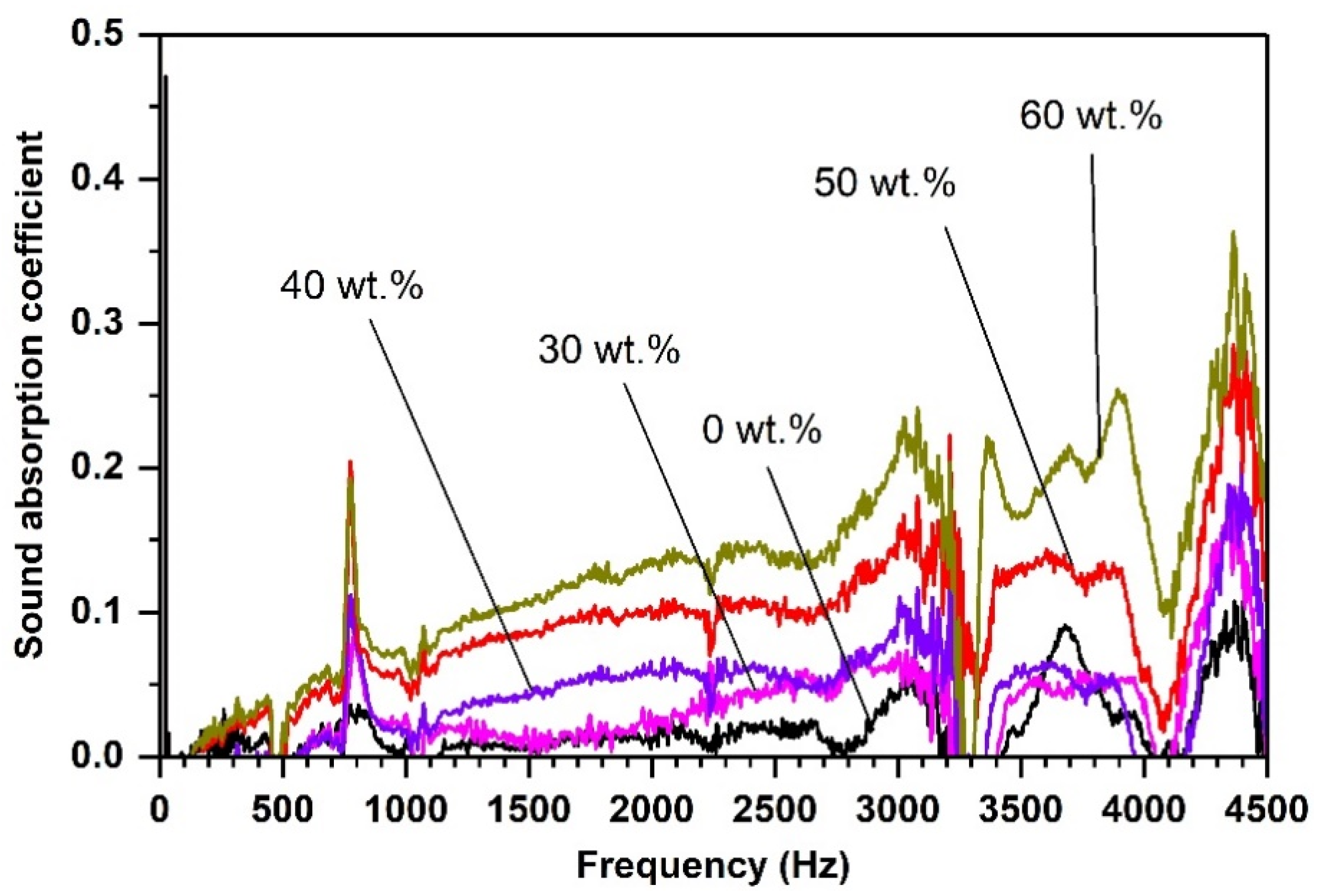
4.3. RDGE Composites with Diatomite Granules: Effects of Diatomite Content
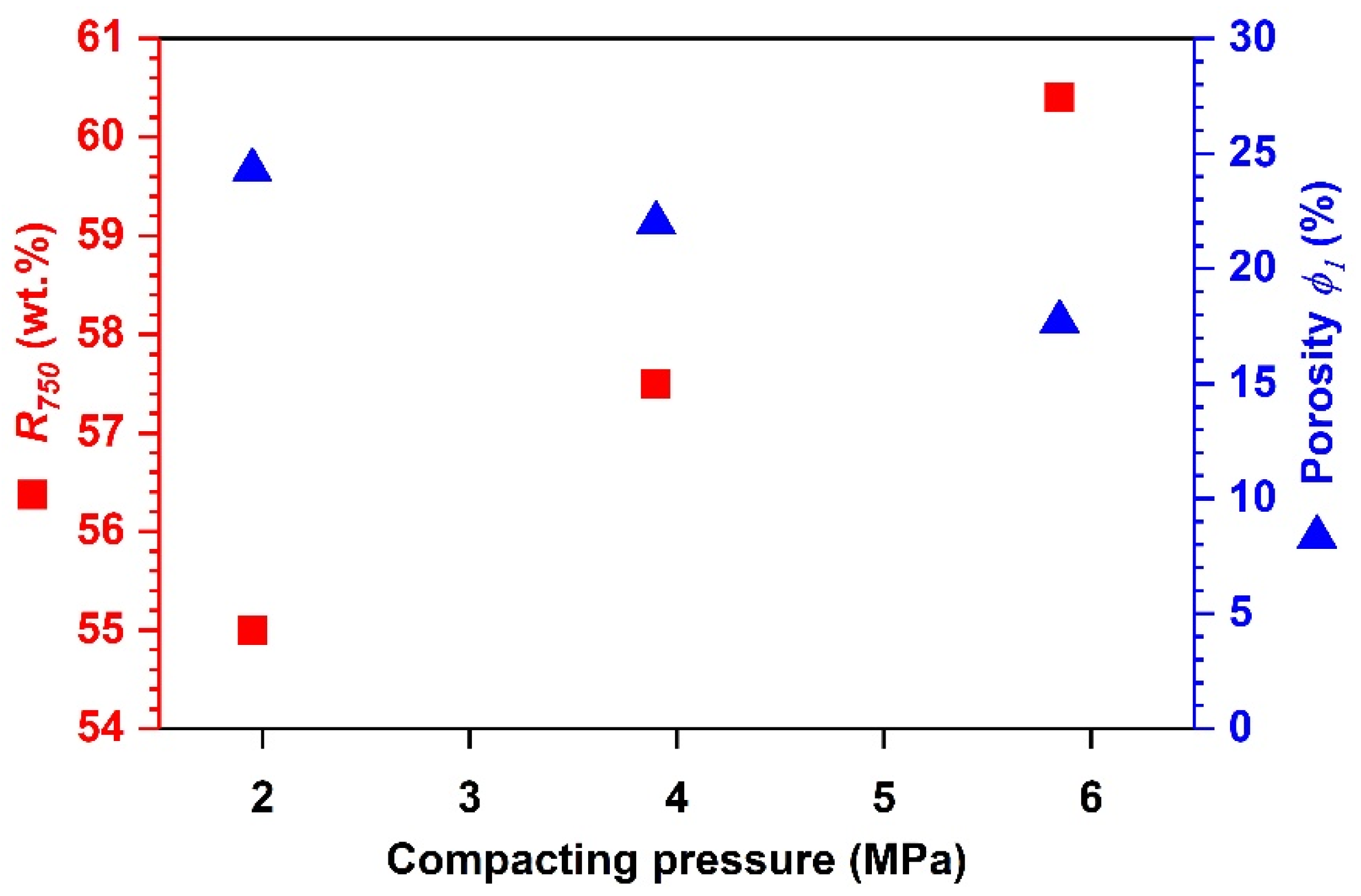
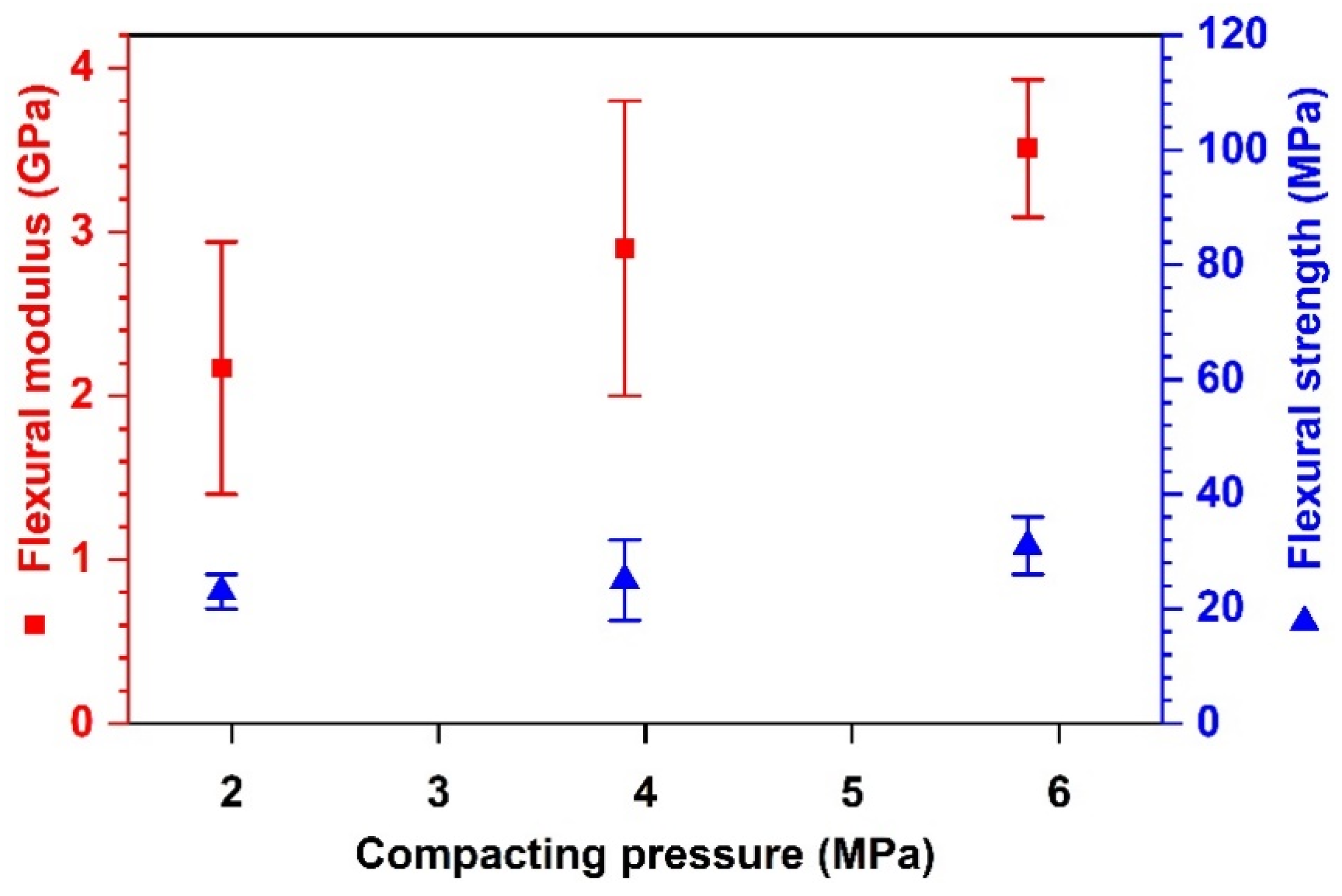
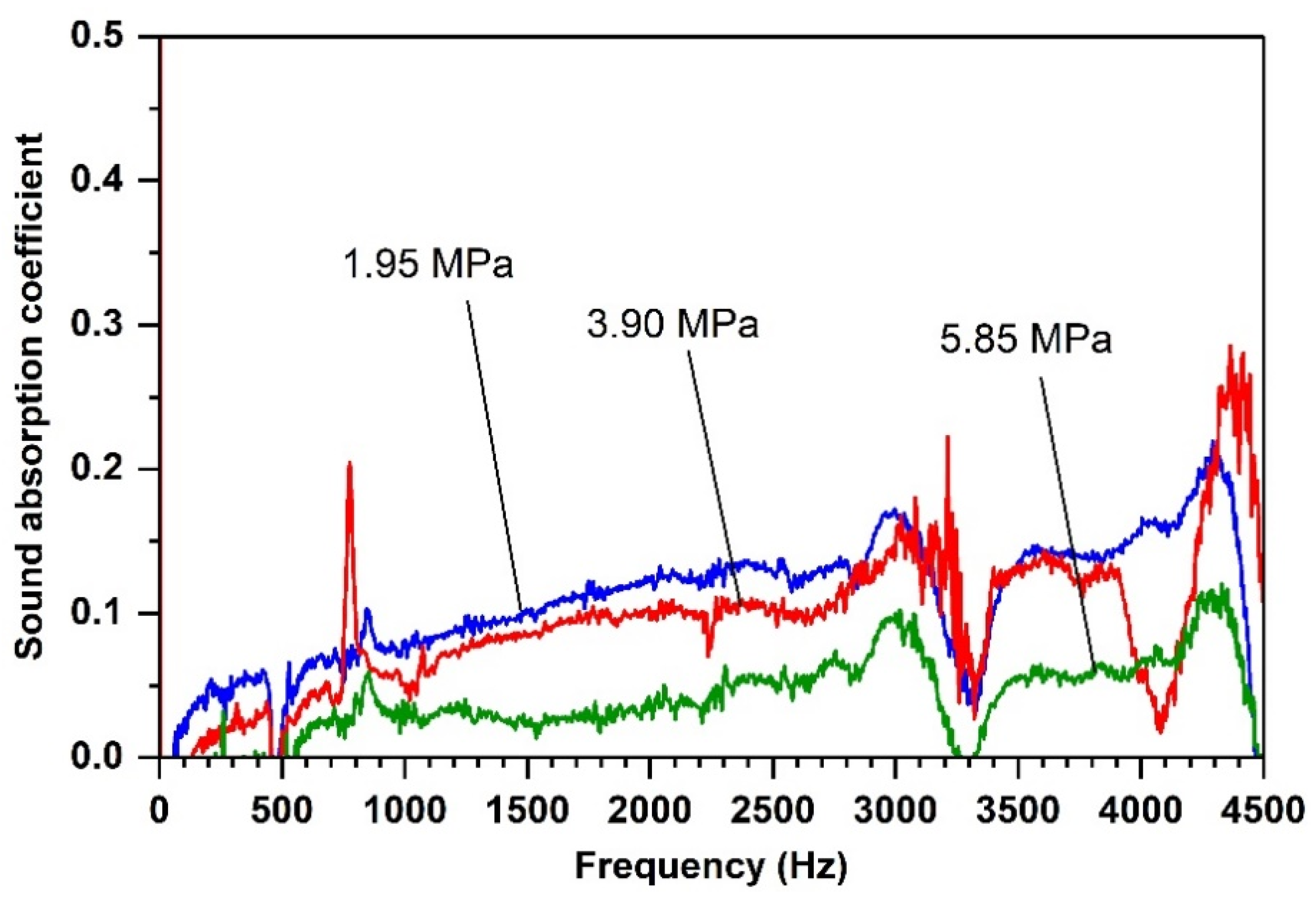
Effects of Compacting Pressure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Van Garderen, N.; Clemens, F.J.; Mezzomo, M.; Bergmann, C.P.; Graule, T. Investigation of Clay Content and Sintering Temperature on Attrition Resistance of Highly Porous Diatomite Based Material. Appl. Clay Sci. 2011, 52, 115–121. [Google Scholar] [CrossRef]
- Parkinson, J.; Gordon, R. Beyond Micromachining: The Potential of Diatoms. Trends Biotechnol. 1999, 17, 190–196. [Google Scholar] [CrossRef]
- Akin, S.; Schembre, J.M.; Bhat, S.K.; Kovscek, A.R. Spontaneous Imbibition Characteristics of Diatomite. J. Pet. Sci. Eng. 2000, 25, 149–165. [Google Scholar] [CrossRef]
- Lee, S.; Ha, J.-H.; Lee, J.; Song, I.-H.; Kwon, S.-H. Preparation and Characterization of a Low-Cost and Natural Material-Based Reticulated Porous Diatomite-Kaolin Composite. Appl. Sci. 2020, 10, 2125. [Google Scholar] [CrossRef]
- Mateo, S.; Cuevas, M.; La Rubia, M.D.; Eliche-Quesada, D. Preliminary Study of the Use of Spent Diatomaceous Earth from the Brewing Industry in Clay Matrix Bricks. Adv. Appl. Ceram. 2017, 116, 77–84. [Google Scholar] [CrossRef]
- Pimraksa, K.; Chindaprasirt, P. Lightweight Bricks Made of Diatomaceous Earth, Lime and Gypsum. Ceram. Int. 2009, 35, 471–478. [Google Scholar] [CrossRef]
- Escalera, E.; Garcia, G.; Terán, R.; Tegman, R.; Antti, M.-L.; Odén, M. The Production of Porous Brick Material from Diatomaceous Earth and Brazil Nut Shell Ash. Constr. Build. Mater. 2015, 98, 257–264. [Google Scholar] [CrossRef]
- Zheng, S.; Bai, C.; Gao, R. Preparation and Photocatalytic Property of TiO2 /Diatomite-Based Porous Ceramics Composite Materials. Int. J. Photoenergy 2012, 2012, 1–4. [Google Scholar] [CrossRef]
- Zeren, D.; Güden, M. The Increased Compression Strength of an Epoxy Resin with the Addition of Heat-Treated Natural Nano-Structured Diatom Frustules. J. Compos. Mater. 2017, 51, 1681–1691. [Google Scholar] [CrossRef]
- Leskovac, M.; Kovačević, V.; Lučić, S.; Perrott, H.R.; Šmit, I. Composites of Poly(Acrylate) Copolymer Filled with Diatomaceous Earth: Morphology and Mechanical Behaviour. Mater. Res. Innov. 2002, 6, 206–213. [Google Scholar] [CrossRef]
- Cacciotti, I.; Rinaldi, M.; Fabbrizi, J.; Nanni, F. Innovative Polyetherimide and Diatomite Based Composites: Influence of the Diatomite Kind and Treatment. J. Mater. Res. Technol. 2019, 8, 1737–1745. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, D.; Liu, Z.; Chen, H.; Zhou, Y.; Zhou, Y.; Zhu, B. Effects of Biomass Diatom Frustule on Structure and Properties of Polyurethane Elastomer. J. Appl. Polym. Sci. 2020, 137, 48452. [Google Scholar] [CrossRef]
- Dobrosielska, M.; Przekop, R.; Sztorch, B.; Brząkalski, D.; Zgłobicka, I.; Łępicka, M.; Dobosz, R.; Kurzydłowski, K. Biogenic Composite Filaments Based on Polylactide and Diatomaceous Earth for 3D Printing. Materials 2020, 13, 4632. [Google Scholar] [CrossRef]
- Fu, Y.; Xu, X.; Huang, Y.; Hu, J.; Chen, Q.; Wu, Y. Preparation of New Diatomite–Chitosan Composite Materials and Their Adsorption Properties and Mechanism of Hg(II). R. Soc. Open Sci. 2017, 4, 170829. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Cong, S. Modified Diatomite Forms in the Rubber Nanocomposites. J. Thermoplast. Compos. Mater. 2020, 33, 659–672. [Google Scholar] [CrossRef]
- Benayache, S.; Alleg, S.; Mebrek, A.; Suñol, J.J. Thermal and Microstructural Properties of Paraffin/Diatomite Composite. Vacuum 2018, 157, 136–144. [Google Scholar] [CrossRef]
- Xu, G.; Leng, G.; Yang, C.; Qin, Y.; Wu, Y.; Chen, H.; Cong, L.; Ding, Y. Sodium Nitrate – Diatomite Composite Materials for Thermal Energy Storage. Sol. Energy 2017, 146, 494–502. [Google Scholar] [CrossRef]
- Wang, R.-M.; Zheng, S.-R.; Zheng, Y.-P. Polymer Matrix Composites and Technology; Woodhead Publishing Limited: Cambridge, UK, 2011; ISBN 978-0-85709-221-2. [Google Scholar]
- Jin, F.-L.; Li, X.; Park, S.-J. Synthesis and Application of Epoxy Resins: A Review. J. Ind. Eng. Chem. 2015, 29, 1–11. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Hsu, C.-Y.; Wei, W.-L.; Jeng, R.-J. Preparation and Thermal Properties of Epoxy-Silica Nanocomposites from Nanoscale Colloidal Silica. Polymer 2003, 44, 5159–5167. [Google Scholar] [CrossRef]
- Kosbar, L.L.; Gelorme, J.D.; Japp, R.M.; Fotorny, W.T. Introducing Biobased Materials into the Electronics Industry. J. Ind. Ecol. 2000, 4, 93–105. [Google Scholar] [CrossRef]
- Pan, H. Synthesis of Polymers from Organic Solvent Liquefied Biomass: A Review. Renew. Sustain. Energy Rev. 2011, 15, 3454–3463. [Google Scholar] [CrossRef]
- Nikafshar, S.; Zabihi, O.; Hamidi, S.; Moradi, Y.; Barzegar, S.; Ahmadi, M.; Naebe, M. A Renewable Bio-Based Epoxy Resin with Improved Mechanical Performance That Can Compete with DGEBA. RSC Adv. 2017, 7, 8694–8701. [Google Scholar] [CrossRef]
- Nguyen, Q.; Nguyen, N.; Rios de Anda, A.; Nguyen, V.; Versace, D.; Langlois, V.; Naili, S.; Renard, E. Photocurable Bulk Epoxy Resins Based on Resorcinol Derivative through Cationic Polymerization. J. Appl. Polym. Sci. 2020, 137, 10. [Google Scholar] [CrossRef]
- Bourne, L.B.; Milner, F.J.M.; Alberman, K.B. Health Problems of Epoxy Resins and Amine-Curing Agents. Occup. Environ. Med. 1959, 16, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Xiao-Xue, W.; Qian-ying, W.; Ying, Z. Sound Absorption Performance of Diatom Mud Coating and Its Influence on Indoor Acoustic Environment. Ferroelectrics 2019, 549, 241–253. [Google Scholar] [CrossRef]
- Jin, H.-Y.; Yang, Y.-Q.; Xu, L.; Hou, S.-E. Effects of Spherical Silica on the Properties of an Epoxy Resin System. J. Appl. Polym. Sci. 2011, 121, 648–653. [Google Scholar] [CrossRef]
- Yang, P.; Ren, M.; Chen, K.; Liang, Y.; Lü, Q.-F.; Zhang, T. Synthesis of a Novel Silicon-Containing Epoxy Resin and Its Effect on Flame Retardancy, Thermal, and Mechanical Properties of Thermosetting Resins. Mater. Today Commun. 2019, 19, 186–195. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Wu, C.-S.; Chiu, Y.-S.; Ho, W.-H. Preparation, Thermal Properties, and Flame Retardance of Epoxy-Silica Hybrid Resins. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 2354–2367. [Google Scholar] [CrossRef]
- Gu, H.; Guo, J.; He, Q.; Tadakamalla, S.; Zhang, X.; Yan, X.; Huang, Y.; Colorado, H.A.; Wei, S.; Guo, Z. Flame-Retardant Epoxy Resin Nanocomposites Reinforced with Polyaniline-Stabilized Silica Nanoparticles. Ind. Eng. Chem. Res. 2013, 52, 7718–7728. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, J.; Song, S. Preparation of a Novel Type of Flame Retardant Diatomite and Its Application in Silicone Rubber Composites. Adv. Powder Technol. 2019, 30, 1567–1575. [Google Scholar] [CrossRef]
- Bulut, U.; Crivello, J.V. Investigation of the Reactivity of Epoxide Monomers in Photoinitiated Cationic Polymerization. Macromolecules 2005, 38, 3584–3595. [Google Scholar] [CrossRef]
- Goethals, E.; Duprez, F. Carbocationic Polymerizations. Prog. Polym. Sci. 2007, 32, 220–246. [Google Scholar] [CrossRef]
- Washburn, E.W. The Dynamics of Capillary Flow. Phys. Rev. 1921, 17, 273–283. [Google Scholar] [CrossRef]
- Lowell, S.; Shields, J.E.; Thomas, M.A.; Thommes, M. Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density; Particle Technology Series; Springer: Dordrecht, The Netherlands, 2004; Volume 16, ISBN 978-90-481-6633-6. [Google Scholar]
- De Anda, A.R.; Fillot, L.A.; Rossi, S.; Long, D.; Sotta, P. Influence of the Sorption of Polar and Non-Polar Solvents on the Glass Transition Temperature of Polyamide 6,6 Amorphous Phase. Polym. Eng. Sci. 2011, 51, 2129–2135. [Google Scholar] [CrossRef]
- Rios De Anda, A.; Fillot, L.A.; Preda, F.M.; Rossi, S.; Long, D.R.; Sotta, P. Sorption and Plasticization Effects of Ethanol–Toluene–Isooctane Ternary Mixtures in Polyamide 6,6 and Induced Plasticization Effects. Eur. Polym. J. 2014, 55, 199–209. [Google Scholar] [CrossRef]
- Rios de Anda, A.; Fillot, L.-A.; Long, D.R.; Sotta, P. Influence of the Amorphous Phase Molecular Mobility on Impact and Tensile Properties of Polyamide 6,6. J. Appl. Polym. Sci. 2016, 133, 9. [Google Scholar] [CrossRef]
- ASTM D790-03: Standard Test Methods for Flexural Properties of Unreinforced and Reinforced Plastics and Electrical Insulating Materials; ASTM International: West Conshohocken, PA, USA, 2003.
- ISO 10534-2: Acoustics-Determination of Sound Absorption Coefficient and Impedance in Impedance Tubes. Part 2: Transfer-Function Method; International Organization for Standardization: Geneva, Switzerland, 1998.
- Salissou, Y.; Panneton, R. Wideband Characterization of the Complex Wave Number and Characteristic Impedance of Sound Absorbers. J. Acoust. Soc. Am. 2010, 128, 2868–2876. [Google Scholar] [CrossRef]
- ASTM C423: Standard Test Method for Sound Absorption and Sound Absorption Coefficients by the Reverberation Room Method; ASTM International: West Conshohocken, PA, USA, 2002.
- Huggett, C. Estimation of Rate of Heat Release by Means of Oxygen Consumption Measurements. Fire Mater. 1980, 4, 61–65. [Google Scholar] [CrossRef]
- Chaisena, A.; Rangsriwatananon, K. Effects of Thermal and Acid Treatments on Some Physico-Chemical Properties of Lampang Diatomite. Suranaree J. Sci. Technol. 2004, 11, 289–299. [Google Scholar]
- Kulpe, J.A.; Lee, C.-Y.; Leamy, M.J. Computation of Acoustic Absorption in Media Composed of Packed Microtubes Exhibiting Surface Irregularity. J. Acoust. Soc. Am. 2011, 130, 826–834. [Google Scholar] [CrossRef]
- Swift, M.J.; Bris, P.; Horoshenkov, K.V. Acoustic Absorption in Re-Cycled Rubber Granulate. Appl. Acoust. 1999, 57, 203–212. [Google Scholar] [CrossRef]
- Bifulco, A.; Parida, D.; Salmeia, K.A.; Nazir, R.; Lehner, S.; Stämpfli, R.; Markus, H.; Malucelli, G.; Branda, F.; Gaan, S. Fire and Mechanical Properties of DGEBA-Based Epoxy Resin Cured with a Cycloaliphatic Hardener: Combined Action of Silica, Melamine and DOPO-Derivative. Mater. Des. 2020, 193, 108862. [Google Scholar] [CrossRef]
- Butler, S.; Fotsing, E.R.; Ross, A. Acoustic Thermoset Open-Cell Foams Produced by Particulate Leaching Process. J. Mater. Sci. 2019, 54, 12553–12572. [Google Scholar] [CrossRef]
- Ali, M.S.; Mohamed Ariff, A.H.; Jaafar, C.N.A.; Tahir, S.M.; Mazlan, N.; Maori, K.A.; Naser, H. Factors Affecting the Porosity and Mechanical Properties of Porous Ceramic Composite Materials. In Reference Module in Materials Science and Materials Engineering; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 978-0-12-803581-8. [Google Scholar]
- Patel, P.S.; Shepherd, D.E.; Hukins, D.W. Compressive Properties of Commercially Available Polyurethane Foams as Mechanical Models for Osteoporotic Human Cancellous Bone. BMC Musculoskelet. Disord. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-Y. Advanced Polyimide Materials: Synthesis, Characterization, and Applications; Elsevier: Amsterdam, The Netherlands, 2018; ISBN 978-0-12-812641-7. [Google Scholar]
- Sonnier, R.; Vahabi, H.; Ferry, L.; Lopez-Cuesta, J.-M. Pyrolysis-Combustion Flow Calorimetry: A Powerful Tool To Evaluate the Flame Retardancy of Polymers. In Fire and Polymers VI: New Advances in Flame Retardant Chemistry and Science; Morgan, A.B., Wilkie, C.A., Nelson, G.L., Eds.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2012; Volume 1118, pp. 361–390. ISBN 978-0-8412-2780-4. [Google Scholar]
- ASTM D7309-20: Standard Test Method for Determining Flammability Characteristics of Plastics and Other Solid Materials Using Microscale Combustion Calorimetry; ASTM International: West Conshohocken, PA, USA, 2020.
- Lyon, R.E.; Walters, R.N. A Microscale Combustion Calorimeter; Federal Aviation Administration, Office of Aviation Research: Washington, DC, USA, 2002. [Google Scholar]
- Wu, H.; Sulkis, M.; Driver, J.; Saade-Castillo, A.; Thompson, A.; Koo, J.H. Multi-Functional ULTEMTM1010 Composite Filaments for Additive Manufacturing Using Fused Filament Fabrication (FFF). Addit. Manuf. 2018, 24, 298–306. [Google Scholar] [CrossRef]
- Butnaru, I.; Bruma, M.; Gaan, S. Phosphine Oxide Based Polyimides: Structure–Property Relationships. RSC Adv. 2017, 7, 50508–50518. [Google Scholar] [CrossRef]
- Schartel, B.; Wilkie, C.A.; Camino, G. Recommendations on the Scientific Approach to Polymer Flame Retardancy: Part 1—Scientific Terms and Methods. J. Fire Sci. 2016, 34, 447–467. [Google Scholar] [CrossRef]
- Nguyen, Q.-B.; Nguyen, V.-H.; Perrot, C.; Rios de Anda, A.; Renard, E.; Naili, S. Multiscale Approach to Characterize Effective Mechanical, Hydraulic and Acoustic Properties of a New Bio-Based Porous Material. Mater. Today Commun. 2021, 26, 101938. [Google Scholar] [CrossRef]
- Yorov, K.E.; Kottsov, S.Y.; Baranchikov, А.Е.; Boytsova, O.V.; Kiskin, M.A.; Varaksina, E.A.; Kopitsa, G.P.; Lermontov, S.A.; Sidorov, A.A.; Pipich, V.; et al. Photoluminescent Porous Aerogel Monoliths Containing ZnEu-Complex: The First Example of Aerogel Modified with a Heteronuclear Metal Complex. J. Sol Gel Sci. Technol. 2019. [Google Scholar] [CrossRef]
- Brinker, C.J.; Scherer, G.W. Sol-Gel Science-The Physics and Chemistry of Sol-Gel Processing; Academic Press Inc.: Cambridge, MA, USA, 1990. [Google Scholar]
- Al-Oweini, R.; El-Rassy, H. Synthesis and Characterization by FTIR Spectroscopy of Silica Aerogels Prepared Using Several Si(OR)4 and R′′Si(OR′)3 Precursors. J. Mol. Struct. 2009, 919, 140–145. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diatomite Content (wt.%) | 0 | 30 | 40 | 50 | 60 |
|---|---|---|---|---|---|
| (°C) | 153 ± 3 | 105 ± 1 | 100 ± 1 | 99 ± 1 | 96 ± 1 |
| (°C) | 359 | 355 | 355 | 334 | 310 |
| (wt.%) | 6.5 | 47.0 | 53.5 | 57.5 | 71.4 |
| Calculated diatomite cntent (wt.%) * | - | 43.8 | 50.8 | 55.1 | 70.2 |
| (%) * | - | 17.5 | 19.3 | 24.2 | 41.9 |
| (%) * | - | 19.6 | 25.6 | 31.5 | 37.2 |
| Density (g/cm3) | 1.318 | 1.437 | 1.427 | 1.380 | 1.181 |
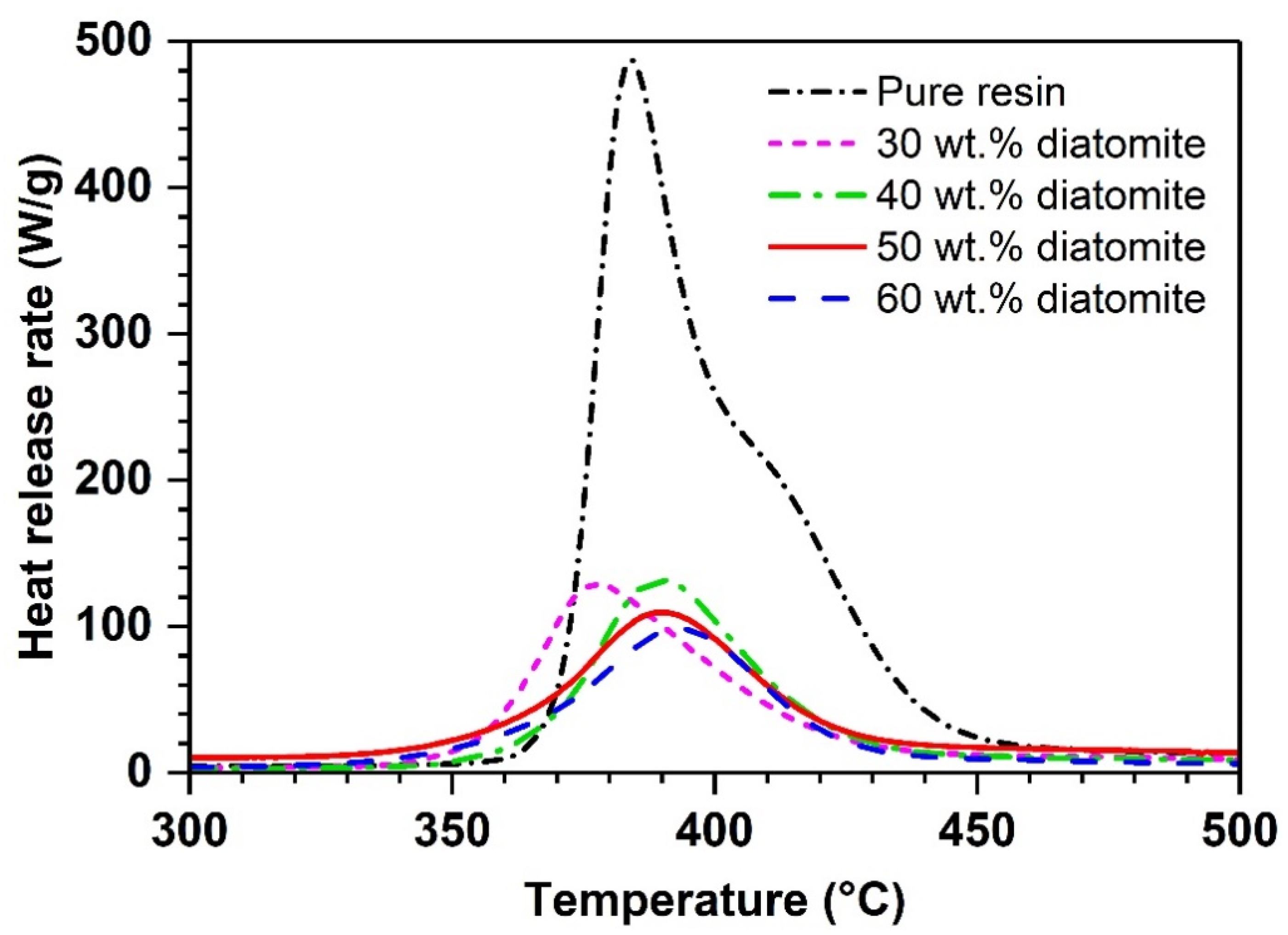
| Diatomite Content (wt.%) | pHRR (W/g) | TpHRR (°C) | THR (kJ/g) |
|---|---|---|---|
| 0 | 487 | 384 | 16.2 |
| 30 | 129 | 377 | 5.7 |
| 40 | 132 | 390 | 6 |
| 50 | 109 | 390 | 5 |
| 60 | 97 | 394 | 4.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, Q.-B.; Vahabi, H.; Rios de Anda, A.; Versace, D.-L.; Langlois, V.; Perrot, C.; Nguyen, V.-H.; Naili, S.; Renard, E. Dual UV-Thermal Curing of Biobased Resorcinol Epoxy Resin-Diatomite Composites with Improved Acoustic Performance and Attractive Flame Retardancy Behavior. Sustain. Chem. 2021, 2, 24-48. https://doi.org/10.3390/suschem2010003
Nguyen Q-B, Vahabi H, Rios de Anda A, Versace D-L, Langlois V, Perrot C, Nguyen V-H, Naili S, Renard E. Dual UV-Thermal Curing of Biobased Resorcinol Epoxy Resin-Diatomite Composites with Improved Acoustic Performance and Attractive Flame Retardancy Behavior. Sustainable Chemistry. 2021; 2(1):24-48. https://doi.org/10.3390/suschem2010003
Chicago/Turabian StyleNguyen, Quoc-Bao, Henri Vahabi, Agustín Rios de Anda, Davy-Louis Versace, Valérie Langlois, Camille Perrot, Vu-Hieu Nguyen, Salah Naili, and Estelle Renard. 2021. "Dual UV-Thermal Curing of Biobased Resorcinol Epoxy Resin-Diatomite Composites with Improved Acoustic Performance and Attractive Flame Retardancy Behavior" Sustainable Chemistry 2, no. 1: 24-48. https://doi.org/10.3390/suschem2010003
APA StyleNguyen, Q.-B., Vahabi, H., Rios de Anda, A., Versace, D.-L., Langlois, V., Perrot, C., Nguyen, V.-H., Naili, S., & Renard, E. (2021). Dual UV-Thermal Curing of Biobased Resorcinol Epoxy Resin-Diatomite Composites with Improved Acoustic Performance and Attractive Flame Retardancy Behavior. Sustainable Chemistry, 2(1), 24-48. https://doi.org/10.3390/suschem2010003