1. Introduction
The use of plastic waste to develop high value-added materials, known also as chemical-upcycling, is seen nowadays as an interesting strategy to promote more sustainable materials [
1]. Indeed, the world plastic consumption has increased to 359 million metric tons in 2018, of which almost 50% of these plastics are used for short-term applications [
2]. The constant increase of the plastic consumption of our society and their deficient waste managements, with only 32.5% of the collected plastic from municipal solid waste (MSW) being recycled in Europe (2019) and only 8.7% in the United States (2018), is causing major environmental problems [
2,
3,
4,
5]. One of the plastics contributing to this problem is poly(ethylene terephthalate) (PET), the fourth most produced plastic in the world, currently exceeding 30 million tons per year. In spite of the robust recycling programs devoted to PET, no country in the world reclaims more than 60% of PET bottles for recycling, relying on mechanical methods that usually reduce the value of virgin PET [
2,
5].
Alternatively, chemical recycling, unlike mechanical recycling, transforms PET into a terephthalate unit containing precursors such as terephthalic acid, dimethyl terephthalate or bis-hydroxy (2-ethyl) terephthalate (BHET) in high yield and purity [
2]. The latter quality is of great importance in electrochemical storage application, as impurities can have a significant impact on the cycling stability of organic electrode materials. As illustrated by Tarascon et al. [
6], small molecules with similar chemistries to the ones produced by chemical recycling (i.e., terephthalate lithium salts), have already been shown to be redox-active and have shown great promise as anode material in lithium-ion batteries [
6]. In particular, the di-lithium terephthalate small molecule exhibited one of the lowest redox potentials reported for an organic anode material (i.e., 0.8 V vs. Li
+ /Li, respectively) [
6]. However, as commonly reported for organic-based electrodes, fast capacity fading was also observed for the the di-lithium terephthalate small molecule, where only 70–80% capacity retention was observed after 50 cycles, due to dissolution of the redox active material into the electrolyte [
6]. Similar observations were made by Ghosh et al., who reported the use of disodium terephthalate, obtained from the chemical recycling of PET waste, as organic anode materials for application in Li-ion and Na-ion batteries [
7].
A common approach to improve the cycling stability of the redox-active small molecules is to covalently bond them to a polymer backbone in order to reduce their solubility into the electrolyte. Consequently, redox polymers have attracted a lot of attention as electrode materials for energy storage application, due to their inherent features such as enhanced cycling stability, high rate performance, processability (i.e., extrusion, roll-to-roll and 2D/3D-printing processes), flexibility, recyclability and their potential to be derived from biomass resources [
8,
9,
10,
11,
12]. In addition, redox polymers are considered as a promising alternative to the scarce inorganic electrode materials that are currently used in Li-ion battery technology [
13,
14], as they are essentially composed of naturally abundant elements (i.e., carbon, hydrogen, oxygen, nitrogen and sulphur) [
10]. The redox-active site in redox polymers, which can undergo reversible oxidization (loss of electrons) and reduction (gain of electrons) reactions, can be either incorporated in the main polymer backbone (e.g., conducting polymers) or as a side chain (e.g., a polymer bearing a quinone pendant group). The vast majority of redox chemistries developed to date, which are based on conjugated carbonyls, conjugated amines, and nitroxide radicals, was generally employed as a cathode electrode material due to their relatively high redox potential (i.e., >1.5 V vs. Li/Li
+) [
8,
9,
10]. The design of redox polymers with lower redox potential, which is appealing in term of the energy density output of the electrochemical devices, is a more challenging task, with so far less diversity available in terms of redox chemistry. Most organic anode materials reported to date are based on conjugated dicarboxylates, Schiff bases, conjugated azo groups and viologens moieties [
12]. Finally, redox polymers can be designed using a wide range of polymerization methods such as free-radical polymerization, controlled radical polymerization or emulsion polymerization, allowing high tuneability of their macromolecular architecture to suit application requirements. Interestingly, the use of redox polymer nanoparticles has been recently shown to have a beneficial impact on the rate capability of redox polymer electrodes, where higher capacity retention was observed at fast (dis-) charge rate with smaller nanoparticles (i.e., <200 nm) [
15,
16,
17,
18,
19].
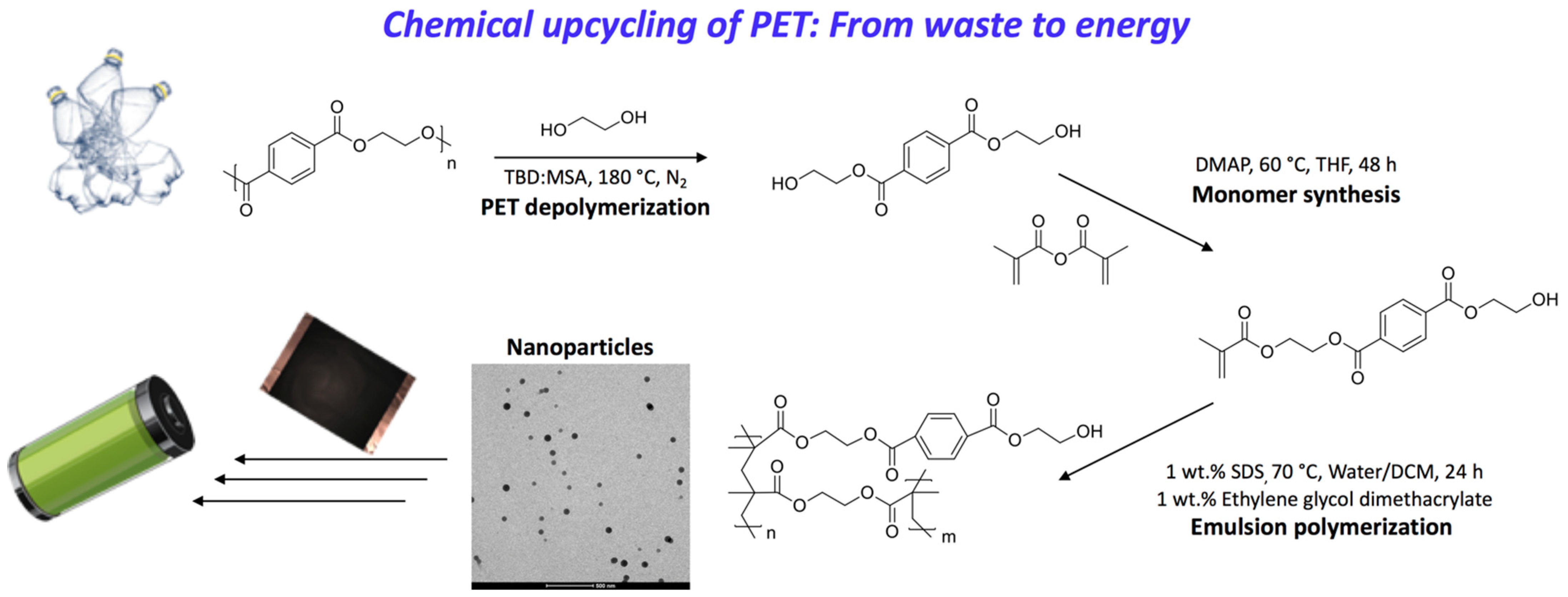
Herein, a novel application for PET plastic waste as a sustainable anode material precursor is proposed via a chemical-upcycling approach (
Figure 1). The goal of this article is to highlight the potential of using PET waste to produce redox-active polymer nanoparticles as anode material for energy storage application. First, bis-hydroxy(2-ethyl) terephthalate (BHET) obtained from PET plastic waste via organocatalyzed chemical recycling was functionalized into a monosubstituted methacrylic monomer. Then, methacrylic terephthalate polymer nanoparticles were synthesized by emulsion polymerization. The electrochemical characterization of the terephthalate precursor (BHET) in solution was investigated by cyclic voltammetry using either a 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF
6) or a 0.1 M lithium bis(trifluoromethane) sulfonimide (LiTFSI) electrolyte systems. Finally, the cycling performance of the redox polymer nanoparticle anode was assessed in a three-electrode cell using a 0.1 M TBAPF
6 electrolyte system for application as anode material in all-organic batteries.
2. Materials and Methods
2.1. Materials
Dimethylaminopyridine (DMAP, 99%, Acros), Methacrylic Anhydride (MetAn, 94%, Sigma, St. Louis, MO, USA), Tetrahydrofuran (extra dry, Fisher, 99.8%), Bis(hydroxyethyl) terephtalate (BHET, Sigma), Triethylamine (TEA, Sigma) were all used as received. Acetone, dichloromethane (DCM), ethyl acetate (EtOAc) and methanol (MeOH), all used for purification of products, were technical grade furnished by Scharlab.
2.2. Monomer Synthesis
Bis-hydroxy (2-ethyl) terephthalate (BHET, 14 g, 55.07 mmol) and 4-(methylamino) pyridine (DMAP, 0.07 g, 0.55 mmol) were dissolved in 200 mL anhydrous THF under nitrogen atmosphere and heated until completely dissolved. Once at room temperature, a solution of methacrylic anhydride (4.1 mL, 27.53 mmol) in 100 mL of anhydrous THF was added dropwise over 15 min and the reaction mixture was heated up to 50 °C for 48 h. Afterwards, the solvent was removed under reduced pressure and the crude product extracted from DCM with water (3 × 250 mL). Then, the organic phase was dried over MgSO4 and purified by column chromatography starting with (95:5 DCM:EtOAc) until the undesired bismethacrylic monomer was eluted. Then (70:30 DCM:EtOAc) was used to afford the desired product as a white solid (6.16 g, 19.1 mmol, 34.7%). It is noteworthy that the bismethacrylic monomer was also isolated in 9% yield using the reported portocol. 1H NMR (300 MHz, DMSO-d6, δ): 8.11 (m, 4H, Ar), 6.03 (dd, 1H, C = C-Ha), 5.68 (dd, 1H, C = C-Hb), 4.95 (t, 1H,OH), 4.58 (m, 2H, Ar-COO-CH2), 4.46 (m, 2H Ar-COO-CH2-CH2), 4.32 (t, 2H, CH2-CH2-OH), 3.72 (q, 2H, CH2-OH), 1,86 (dd, 3H CH3). 13C NMR (300 MHz, DMSO-d6, δ):166.43, d 165.12, 135.63, 133.95, 133.34, 129.62, 129.46, 126.13, 67.05, 63.10, 62.30, 59.01, 17.92.
2.3. Terephthalate-Based Nanoparticles: Emulsion Polymerization
Sodium dodecyl sulfate (SDS) (20 mg, 1 wt. % with respect to monomer weight, 0.07 mmol) and potassium persulfate (KPS) (12 mg, 0.6 wt.% with respect to monomer weight, 0.04 mmol) were dissolved in 24 mL of deionized water in a round bottom flask and deoxygenated for 10 min by nitrogen flow. A solution of hydroxyethyl-methacrylate monomer (HETMA) (2 g, 6.21 mmol) in dichloromethane (6 mL) was then added to previous aqueous solution and stirred vigorously. Then, the cross-linking agent, ethyleneglycol dimethacrylate, (EGDMA) (20 mg, 1 wt.% with respect to monomer weight, 0.10 mmol) was added to the mixture via a syringe. The mixture was then heated to 70 °C overnight under nitrogen atmosphere. The terephthalate-based nanoparticles were then purified via dialysis for 3 days at 25 °C (Spectra/Por®4 standard regenerated cellulose membrane, MWCO 12–14 KD) and finally freeze-dried using a Telstar LyoQest−85 Lyophilizer at −80 °C and 0.089 mbar during 3 days (yield 64 w/w%). NMR analysis was not performed due limited solubility in common organic solvent of the crosslinked terephthalate-based nanoparticles.
2.4. Preparation of Terephthalate-based Electrode
The terephthalate-based electrodes were prepared via a doctor blade coating method onto a carbon coated copper foil current collector (Gelon energy Co. Limited, Linyi, China, 11 µm thick). For this purpose, a slurry of terephthalate nanoparticles (50 wt.%), conductive carbon (Super C65, Timical, 40 wt.%) and binder (PVDF, Solef® 5130, 10 wt.%) in N-methylpyrrolidone (NMP) was prepared, resulting in an active material loading around 0.5 mg.cm−2.
2.5. Characterization Methods
The hydrodynamic diameter of the terephthalate-based nanoparticles was measured by dynamic light scattering (DLS) using a Malvern Zetasizer Nano ZS spectrophotometer. TEM measurements were conducted on a TECNAI G2 20 TWIN transmission electron microscope, operating at an accelerating voltage of 200 kV. Fourier transform infrared (FTIR) spectra were recorded using a Bruker Alpha II spectrophotometer in the wavenumber range of 4000 cm−1 to 360 cm−1 with 124 scans at a resolution of 4 cm−1. Cyclic voltammetry (CV) measurements in a three-electrode cell were recorded using a multi-channel Potentiostat (Biologic, VMP3). A platinum wire and an Ag/AgNO3 electrode were used as counter and reference electrodes, respectively. CV of the model molecules in solution were performed in either 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) in dimethyl sulfoxide or 0.1 M lithium bis(trifluoromethane)sulfonimide (LiTFSI) in dimethyl sulfoxide. Galvanostatic cycling measurements in a three-electrode cell were recorded using an VMP3 potentiostat (Biologic). A platinum wire and an Ag/AgNO3 electrode were used as counter and reference electrodes, respectively.
3. Results and Discussion
The chemical upcycling approach, proposed herein, consists of the conversion of PET plastic waste into high performance anode material for application in energy storage (
Figure 1). As first step, the depolymerization of PET is performed via a glycolysis reaction using a recyclable organocatalyst in high yields, via a recently developed method by our research group, resulting in the present case in bis(2-hydroxyethyl) terephthalate, BHET, as the depolymerization product [
20]. Organocatalyst is considered as a promising “green” alternative compared to traditional organometallic catalysts due to their lower environmental footprint, high selectivity and recyclability [
2,
21], complementing the sustainable aspect of the synthesis route proposed herein. In the next step, the depolymerization product (i.e., bis(2-hydroxyethyl) terephthalate, BHET) is converted into a methacrylate-based terephthalate monomer using methacrylic anhydride, which is considered as a greener alternative to more traditional chlorinated reagent (i.e., methacryloyl chloride). Finally, the incorporation of methacrylic functionality facilitates the polymerization of the terephthalate-based monomer by radical polymerization, allowing the preparation of well-defined nanoparticles by emulsion polymerization, which is considered as a more sustainable polymerization technique due to the use of water as dispersing media [
22].
To initially assess the viability of this chemical upcycling approach, the electrochemical characterization of both bis(2-hydroxyethyl) terephthalate (BHET) and bis(2-methoxyethyl) terephthalate (BMEET), as model molecules, was performed using cyclic voltammetry in order to evaluate the impact of the esterification of the terephthalate acid as well as the presence of a hydroxyl terminated group (i.e., BHET) on their redox properties. The latter will also allow to assess if a mono or di-methacrylate functionalization of the BHET is required, knowing that both compounds can be easily synthesized with the reported method, herein, with only a slight modification of the protocol (i.e., by varying the number of equivalents of methacrylic anhydride used, currently 0.5 eq.).
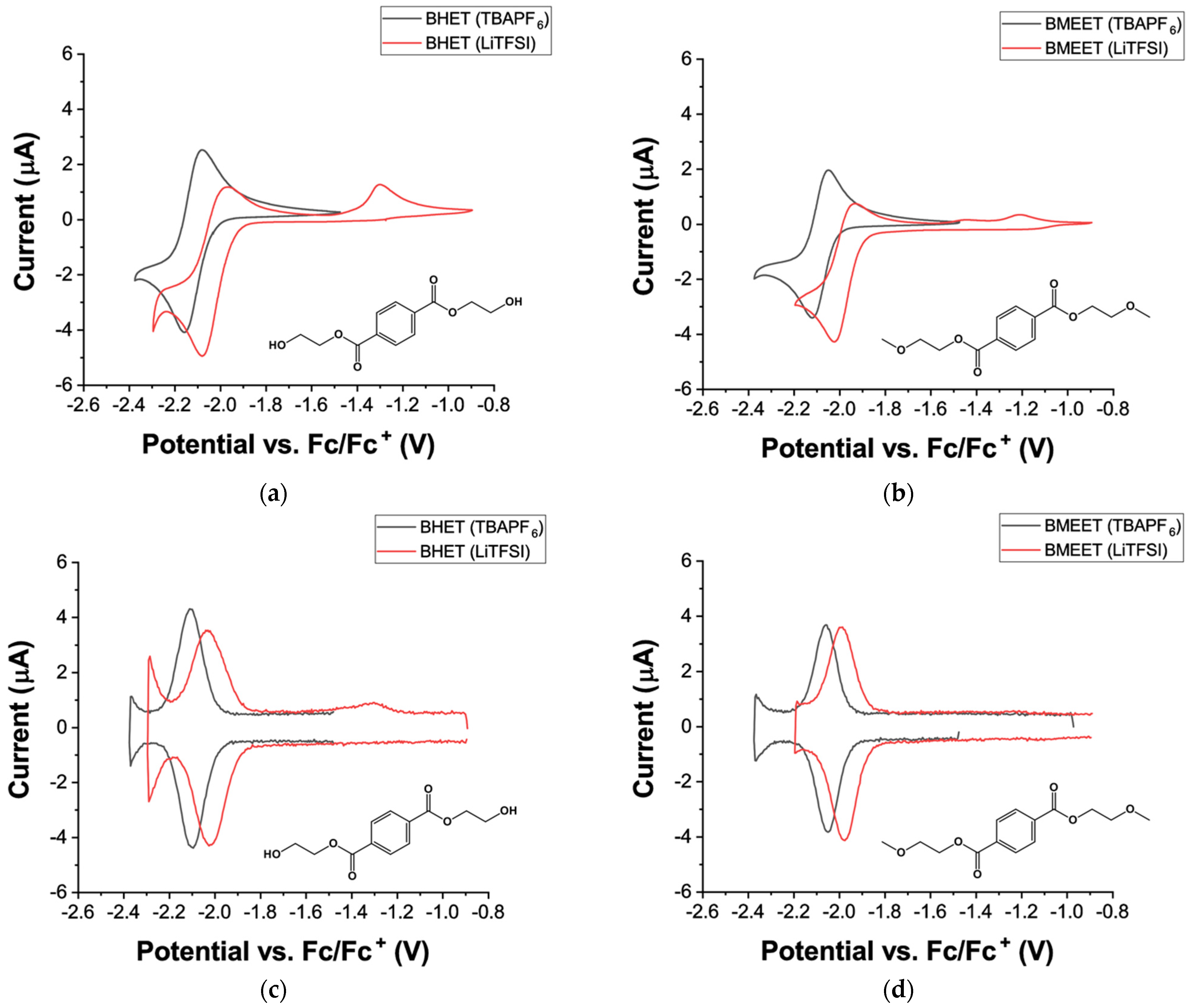
Figure 2 shows the cyclic and differential pulse voltammograms of BHET and BMEET in solution with either tetrabutylammonium hexafluorophosphate (TBAPF
6) or lithium bis(trifluoromethane)sulfonimide (LiTFSI), as electrolyte salts. As can be seen in
Figure 2a, BHET exhibits a reversible redox process, when using a TBAPF
6/DMSO electrolyte system, with a potential of –2.16 V and –2.08 V vs. Fc/Fc
+ for the reduction and oxidation processes, respectively. Such results are in agreement with previous studied on dimethyl 2-vinyl terephthalate or diethyl terephthalate molecules [
23,
24]. Interestingly, the use of a lithium-based electrolyte (i.e., LiTFSI/DMSO) results in an anodic shift of the redox potential associated to the BHET (i.e., ~80 mV), as well as a strong irresistibility between the reduction and oxidation processes, which are located at a potential of −2.08 V and −1.97 V vs. Fc/Fc
+, respectively. This phenomenon is more apparent in the corresponding differential pulse voltammograms (
Figure 2c), from which a coulomb efficiency (CE) of 98.4 % and 88.5 % can be extracted, for the TBAPF
6/DMSO and the LiTFSI/DMSO electrolyte systems, respectively. Additionally, a second oxidation process is observed at a potential of –1.3 V vs. Fc/Fc
+. When cycling BHET in LiTFSI/DMSO in a voltage range between –2.3 and –1.4 V vs. Fc/Fc
+, a fast fading of the reduction peak is observed. (
Figure S2a in Supplementary Materials). On the other hand, a stable peak current is observed for the reduction process, when anodic voltage limit is increased to –0.9 V vs. Fc/Fc
+ (
Figure S2b in Supplementary Materials), which suggests that the second oxidation process is likely also related to the oxidation of the reduced BHET to its initial neutral state. The cycling voltammetry of the BMEET was also performed in order to investigate if the irreversibility of the BHET in lithium-containing electrolyte was due to the presence of the hydroxyl terminated group, and the results are shown in
Figure 2b for both electrolyte systems (i.e., TBAPF
6/DMSO and LiTFSI/DMSO). Similarly to BHET, BMEET in TBAPF
6/DMSO exhibits a pair of well-reversible oxidation and reduction peaks at a potential of –1.93 V and –2.02 V vs. Fc/Fc
+, respectively. Again, strong irreversibility between the reduction and oxidation processes is observed when employing a lithium-containing electrolyte, as highlighted in
Figure 2b. CE of 97.7% and 83.8% are obtained for the BMEET in either TBAPF
6/DMSO or LiTFSI/DMSO electrolyte, respectively. Overall, both BHET and BMEET show good redox reversibility in solution, when using a TBAPF
6/DMSO electrolyte system, suggesting that the presence of hydroxyl terminated group is not detrimental for redox processes. Surprisingly, both BHET and BMEET show strong irreversibility of their redox process, in particular the oxidation one, when employing lithium-containing electrolytes. Indeed, Armand et. al previously showed that the terephthalate lithium salt exhibited good reversibility in lithium-based electrolyte (i.e., 1 M LiPF
6 in ethylene carbonate-dimethyl carbonate (1:1
w/w)), suggesting the esterification of the terephthalate acid by ethyl glycol has a detrimental impact on redox process of terephthalate moiety, in lithium containing electrolyte [
6]. Overall, the initial electrochemical characterization indicates that the molecules produced by chemical depolymerization of PET could be used as precursor to develop redox-active polymers for application as anode materials in all-organic batteries, where no lithium-based electrolyte is required.
Based on the initial electrochemical results, mono-methacrylate functionalization of the depolymerization product (i.e., bis(2-hydroxyethyl) terephthalate, BHET) was selected for the further development of redox-active polymer nanoparticles. The etherification of BHET was performed using methacrylic anhydride and 4-(methylamino) pyridine (DMAP) as catalyst, resulting in 2-hydroxyethyl (2-(methacryloyloxy)ethyl) terephthalate (HETMA). Terephthalate polymer nanoparticles (TP NPs) were synthesized using an optimized emulsion polymerization method, as described in
Figure 1. The terephthalate methacrylic monomer was polymerized in a water/dichloromethane mixture at 70 °C, in the presence of sodium dodecyl sulfate (SDS) and potassium persulfate (KPS) as surfactant and radical initiator, respectively, leading to terephthalate polymer nanoparticles in medium-to-high yield (i.e., 64%). As commonly reported for organic-based electrode, the dissolution of redox active materials is a recognized issue, resulting in fast capacity fading. Such phenomena was also reported by Tarascon et al. [
6]. For the di-lithium terephthalate small molecule, where only 70–80% capacity retention was observed after 50 cycles. For this reason, ethylene glycol dimethacrylate (i.e., 1 wt.%) was employed as crosslinking agent in order to produce non-soluble polymeric redox-active nanoparticles. The hydrodynamic and dried-state diameters of the terephthalate polymer nanoparticles were determined by Dynamic Light Scattering (DLS) and Transmission Electron Microscopy (TEM), resulting in average sizes of 115 nm and 71 nm, respectively (
Figure 3a,b). As previously mentioned, redox-active nanoparticles with a diameter below 200 nm have demonstrated enhanced rate capability due to the reduced diffusion pathway to access the redox active moieties of the nanoparticles inner core, suggesting fast redox-kinetics can be obtained for the size range of the crosslinked terephthalate nanoparticles [
15,
16,
18,
25].
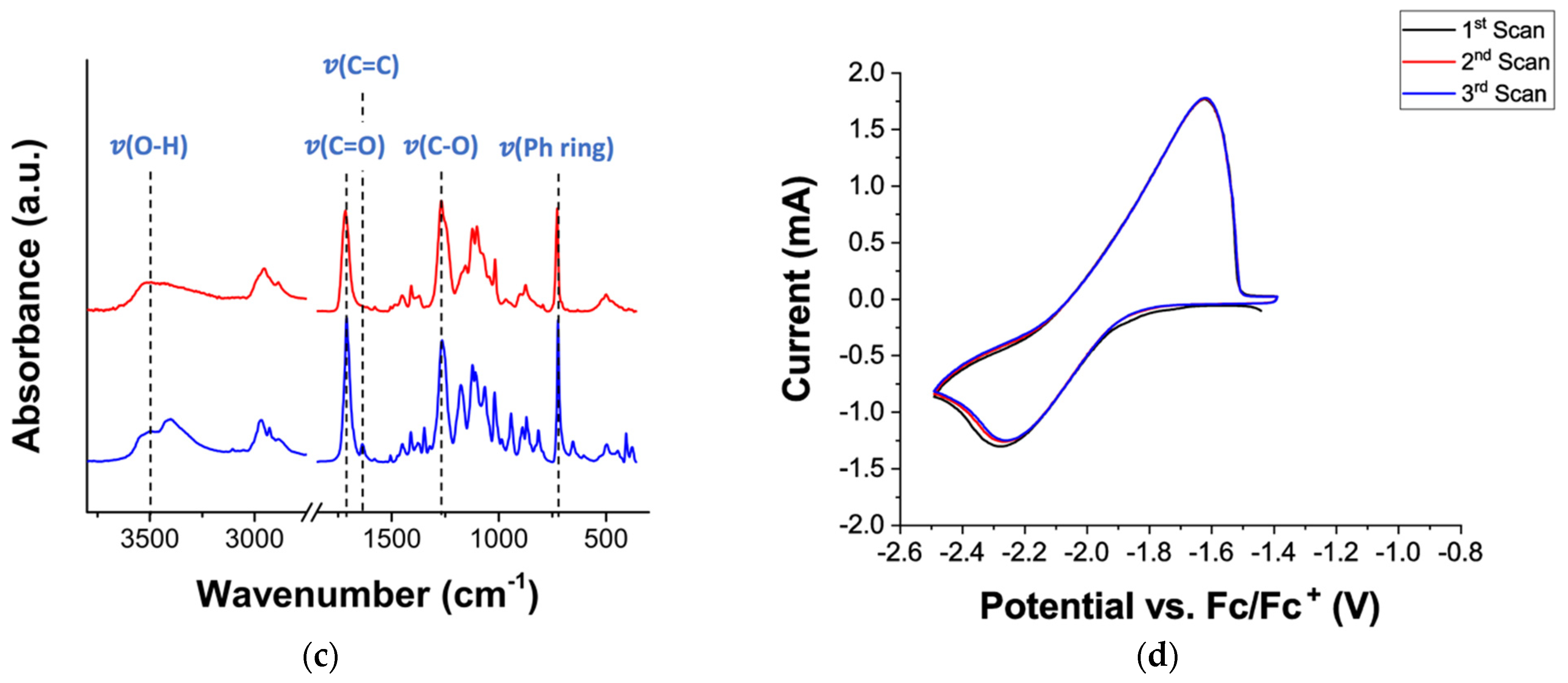
The chemical composition of the terephthalate polymer nanoparticles was analysed by FT-IR and the spectra are shown in
Figure 3c. The absence of the vibration bands of the methacrylate group at 1637 cm
−1 (ν (C=C)) as well as the decrease in intensity of the (β (C=C)) vibration bands at 943 cm
−1 and 815 cm
−1 in polymeric nanoparticles confirms full monomer conversion [
26]. Additionally, characteristic vibration bands associated with aromatic ester groups are observed in the terephthalate-based nanoparticles at 1716 cm
−1 (ν (C=O)) and 1270/1250 cm
−1 (ν (C-O)) [
27,
28]. Vibration bands of the phenyl ring are observed at 1696 cm
−1 (ν (C=C)) and 730 cm
−1 (ν
s (C-C)), as well as aliphatic alcohol groups at 3500 cm
−1 (ν (O-H)) and 2950/2882 cm
−1 (ν (C-H)) [
27,
28]. Overall, the FT-IR analysis confirms the chemical nature of the methacrylate terephthalate monomer and the successful polymerization of the terephthalate NPs. The electrochemical characterization of the terephthalate nanoparticles was carried out by cyclic voltammetry, using a 0.1 M TBAPF
6 electrolyte solution in acetonitrile, and the resulted voltammogram is shown in
Figure 3c,d. For this purpose, a slurry of terephthalate nanoparticles was prepared by mixing terephthalate nanoparticles (50 wt.%), Super carbon C65 (40 wt.%) and poly(vinylidene) (10 wt.%), as redox-active material, carbon additive and binder, respectively. Reversible oxidation and reduction peaks are observed at −1.62 V and −2.26 V vs. Fc/Fc
+, respectively, confirming their potential use as an ultra-low potential anode material for all-organic batteries.
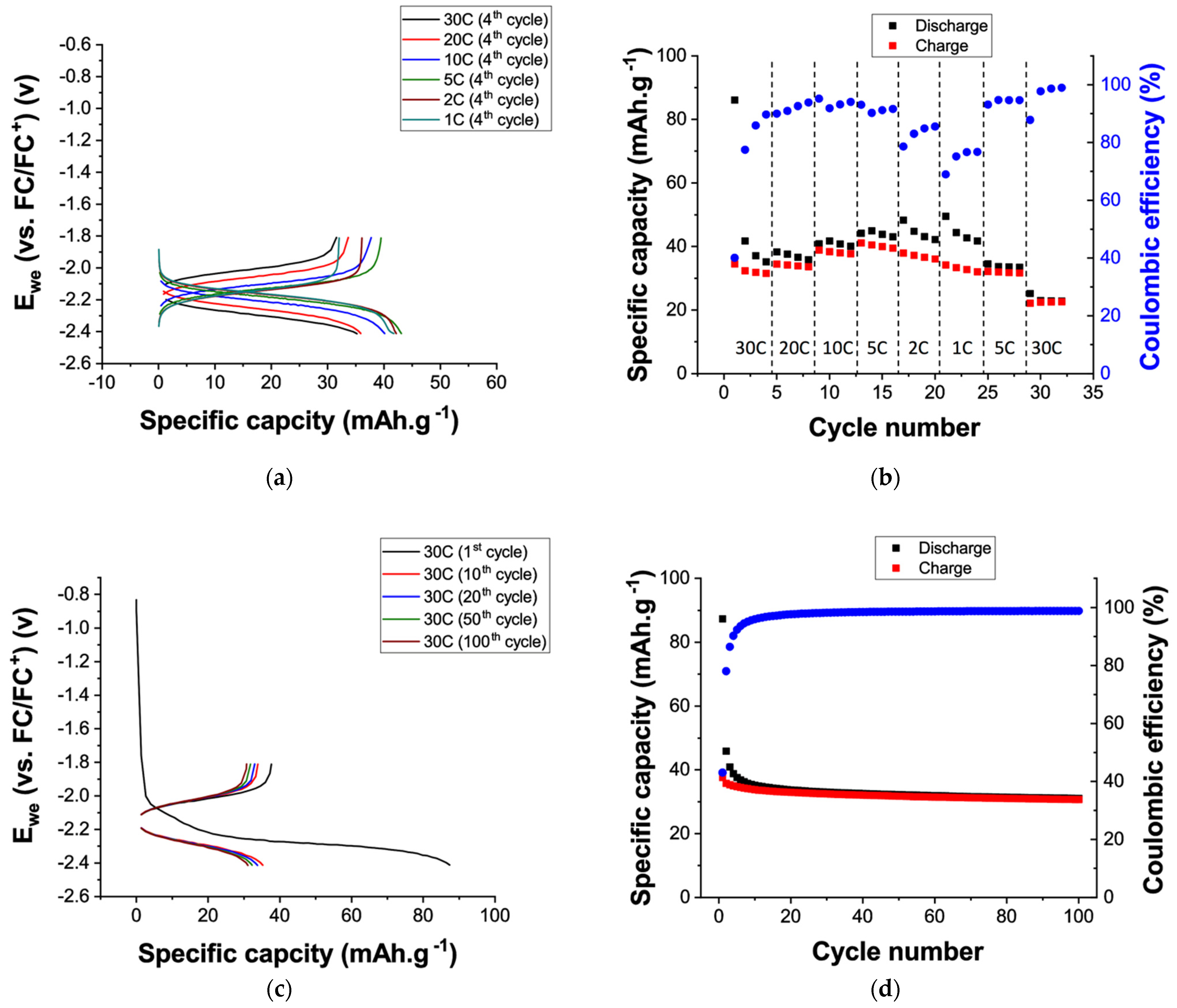
To further assess the electrochemical performance of the terephthalate nanoparticles, galvanostatic cycling experiments were performed using a 0.1 M TBAPF
6 solution in acetonitrile as electrolyte in three-electrode cell and the results are shown in
Figure 4. Based on the cyclic voltammetry data (
Figure 3d) a voltage range between −2.4 V and −1.8 V vs. Fc/Fc
+, was used for the galvanostatic cycling experiments. Discharge and charge plateaus are observed at a potential of −2.18 V and −2.12 V vs. Fc/Fc
+, respectively, for C-rate below 5 C (
Figure 4a). Increasing the C-rate from 5 C to 30 C only results in a small polarization, going from 60 to 300 mV, which is in agreement with the cyclic voltammetry data of terephthalate nanoparticles performed at a scan rate of 50 mV.s
−1. As can be seen in
Figure 4a and
Figure S3a in the Supplementary Materials, poor coulombic efficiency (CE) is obtained during the first cycle, with a specific discharge and charge capacities of 86 and 35 mAh.g
−1, respectively. This is likely due to solid electrolyte interphase formation and initial irreversible side reactions at such low potential, as suggested by the increase of CE upon cycling, going 40 % to 89 % in 4 cycles. As expected, a small increase of the specific charge capacity, going from 31.5 to 39.5 mAh.g
−1, is observed when the C-rate decreases from 30 C to 5 C. Interestingly, further decrease of the C-rate below 5 C results in a more pronounced hysteresis between the delivered specific discharge and charge capacities. Indeed, an increase of the specific discharge capacities, going from 43.1 to 48.3 mAh.g
−1, is observed when decreasing C-rate from 5 C to 2 C. While the opposite trend is observed for the specific charge capacities, going from 39.5 to 38.0 mAh.g
−1. This also coincides with the observation of a second discharge plateau at a potential of −2.40 V vs. Fc/Fc
+ at low C-rate (i.e., 2 C and 1 C). Additionally, lower capacities are obtained when cycling back the terephthalate electrode at a C-rate of 5 C, suggesting that irreversible degradation of the terephthalate electrode have occurred during the cycling at low C-rate (i.e., 2 C and 1 C). To determine whether this second discharge plateau is related to some degradation reactions associated to the terephthalate nanoparticles and results in the specific charge capacity fading observed, galvanostatic cycling experiments were performed using a more conservative cathodic voltage limits of –2.30 V vs. Fc/Fc
+ and the result are shown in
Figure S4 in the Supplementary Materials. As expected with more conservative cathodic voltage limits, lower specific discharge and charge capacities are reached at high C-rates (i.e., ≥5 C). At lower C-rate, the second discharge plateau is not observed even with a C-rate as low as 1 C. Under these cycling conditions, similar trends are observed with the formation of a larger hysteresis between the delivered specific discharge and charge capacities and fast fading of the specific charge capacity, when cycling at a C-rate of 2 C or 1 C (
Figure S4 in the Supplementary Materials). This result suggests that the capacity fading at low C-rate is not caused by the second discharge plateau observed previously, but rather by some irreversibility of redox reactions. It is not clear if this irreversibility is due to some instability/degradation of the terephthalate nanoparticles (e.g., methacrylate linker) during the discharge process or is due to difficulties in oxidizing the reduced terephthalate moiety back to its neutral state, similarly to cyclic voltammetry results of the BHET in lithium containing electrolytes, despite the fact that in this case a TBAPF
6-based electrolyte was used.
Long-term cycling stability of terephthalate nanoparticles was then investigated at high C-rate, due to the instability of the terephthalate nanoparticles at low C-rate, and the results are shown in
Figure 4c,d. For these experiment, the initial voltage range limits (i.e., –2.4 V and –1.8 V vs. Fc/Fc
+) was used in order to maximize capacities delivered, since no second degradation plateau was observed during discharge. As can be seen in
Figure 4c, a stable specific discharge capacity of 32.8 mAh.g
−1 is reached after some activation cycles (i.e., 35 cycles), with a 98.8 % coulombic efficiency. After 100 cycles, terephthalate nanoparticles still delivered a specific discharge capacity of 30.8 mAh.g
−1, corresponding to a capacity retention of 94 %. Decreasing the cathodic voltage limit to an even lower potential (i.e., –2.4 V and –2.6 V vs. Fc/Fc
+) results in fast capacity fading of 49 % after only 100 cycles (
Figure S5a in the Supplementary Materials). This coincides again with the observation of a second discharge plateau at a potential of −2.5 vs. Fc/Fc
+ (
Figure S5b in the Supplementary Materials). The observation of this second discharge plateau at a much lower potential compared to the one observed at a C-rate of 1 C is likely due to faster (dis)charging rate, resulting in cell polarization. To confirm the capacity obtained from the terephthalate nanoparticles electrode is not due to carbon contribution, present in the electrode (i.e., 40 wt.%), galvanostatic cycling of electrodes containing only carbon were performed using the same voltage range limits at various C-rate (
Figure S6 in Supplementary Materials). Specific discharge capacities of 0.17, 0.30, 0.45, 0.60, 0.79 and 0.92 mAh.g
−1 of carbon were obtained at a C-rate of 30 C, 20 C, 10 C, 5 C, 2 C and 1 C, respectively, suggesting that the carbon contribution present in the terephthalate nanoparticles electrode is not significant.
Overall, the chemical upcycling approach of PET proposed herein is quite unique in the sense that most of the upcycling approaches of plastic wastes, including PET, employed combustion methods to produce carbonaceous materials for application in energy storage devices [
29,
30,
31,
32,
33]. An exception to this was a 2020 study by Ghosh et al. who proposed a chemical upcycling approach of PET waste to produce disodium terephthalate for application in sodium-ion and lithium-ion batteries [
7]. The approach proposed here is the first example of chemical upcycling of PET to produce a new terephthalate-based monomer and its corresponding polymer nanoparticles. Additionally, the synthesis route of the methacrylic-terephthalate polymer, proposed herein, is relatively step efficient (i.e., 3 steps) when compared to the vinylic dilithium terephthalate-based polymer reported by Nauroozi et al. [
23], which involved a multiple-step synthesis route (i.e., 7 steps). In term of cycling performance, the terephthalate NPs, at high C-rate, compares favorably to the vinylic dilithium terephthalate-based polymer reported by Nauroozi et al. [
23], which is delivered a stable specific discharge capacity of 48 mAh.g
−1 after 200 cycles. It should be noted that direct comparison between the two systems is difficult as the electrolyte systems are different (i.e., 1 M LiPF
6 in ethylene carbonate/dimethyl carbonate vs. 0.1 M TBAPF
6 in acetonitrile) as well as the nature of the redox moieties (i.e., conjugated dicarboxylate vs. conjugated diester).


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




