
Electrochemical Bromofunctionalization of Alkenes and Alkynes—To Sustainability and Beyond



Abstract

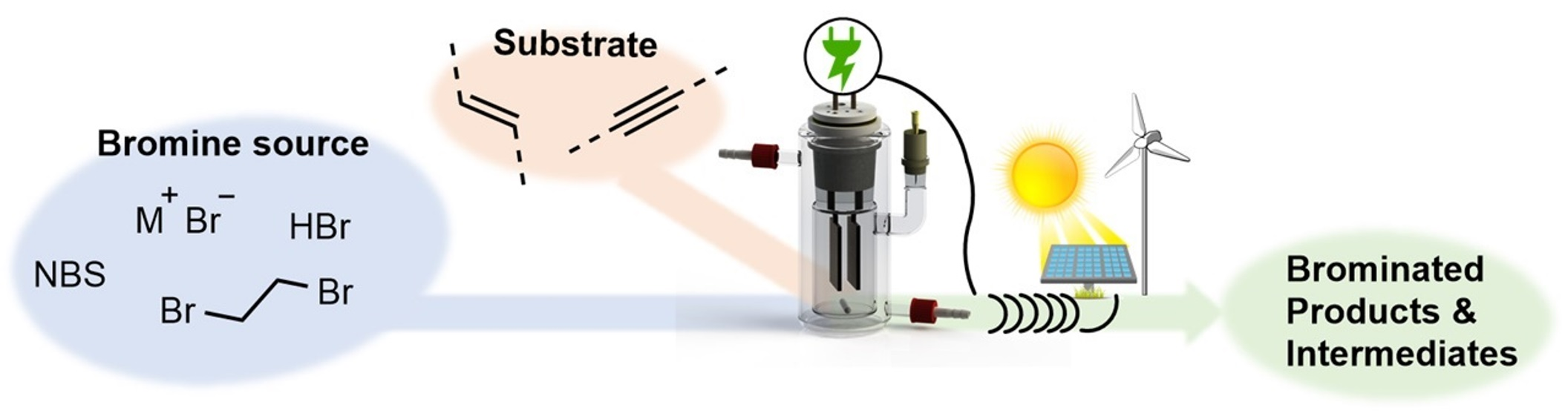
1. Introduction
2. Bromine and Bromination Agents
3. Electrochemical Bromofunctionalization of Alkenes
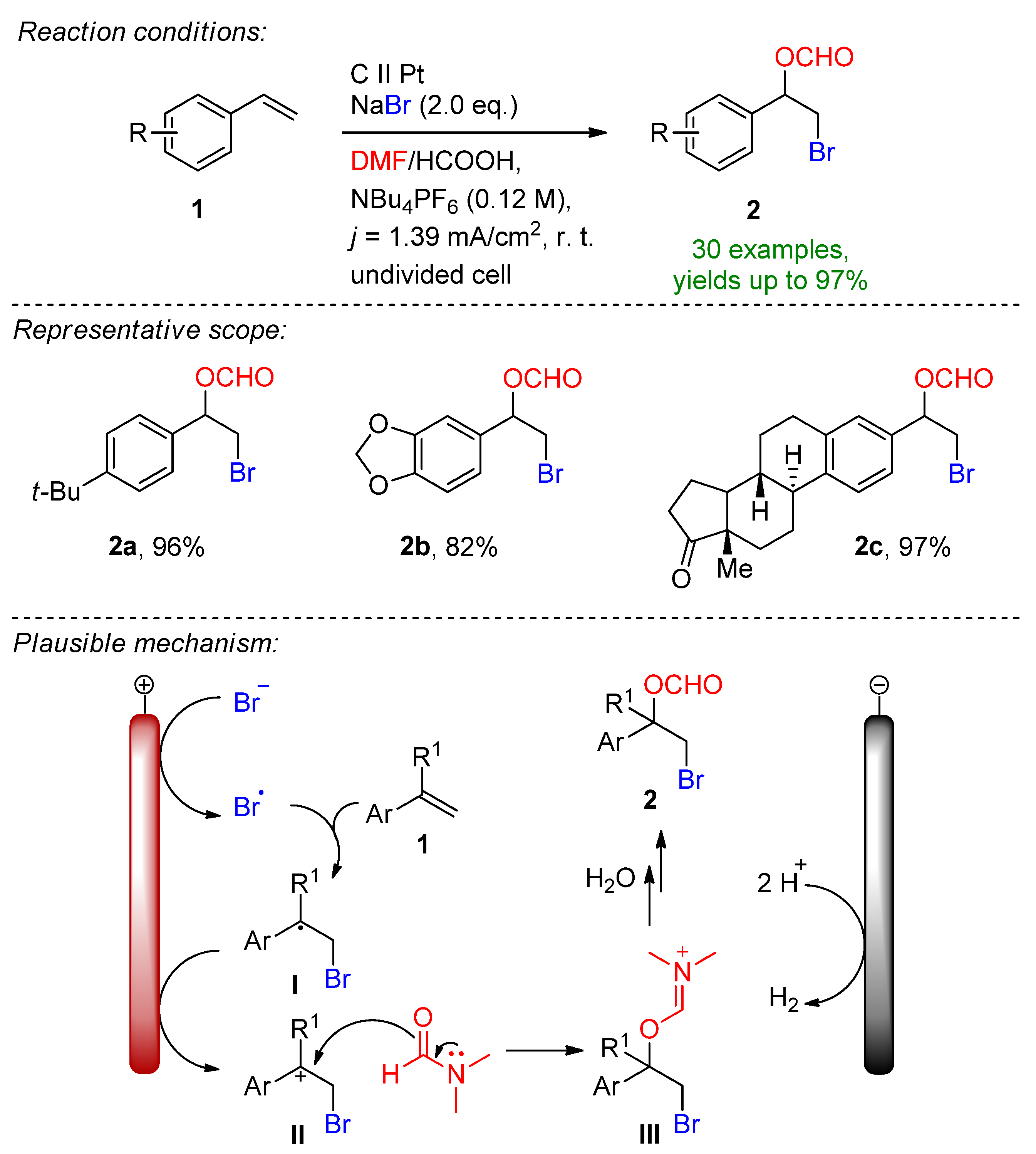
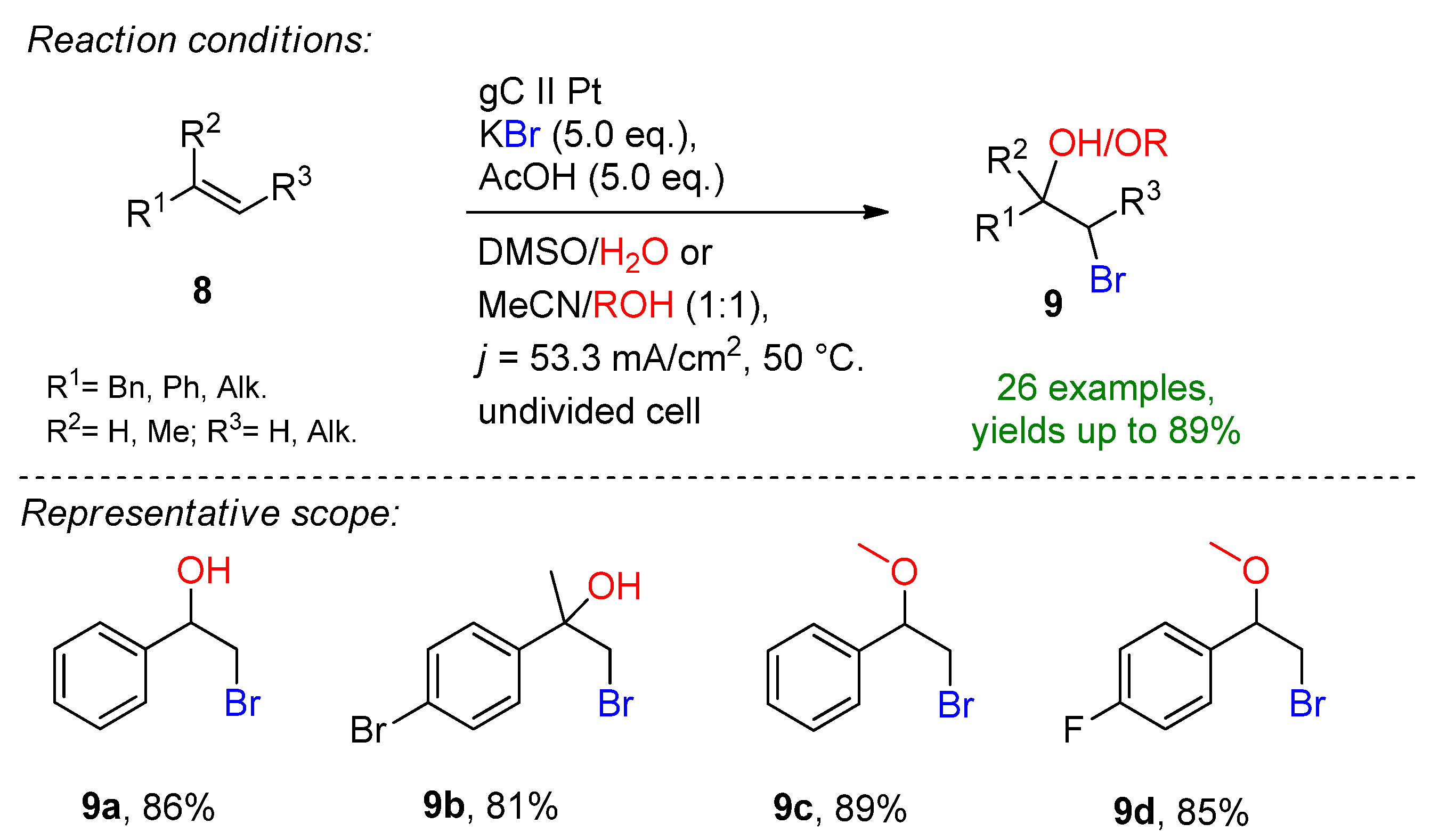
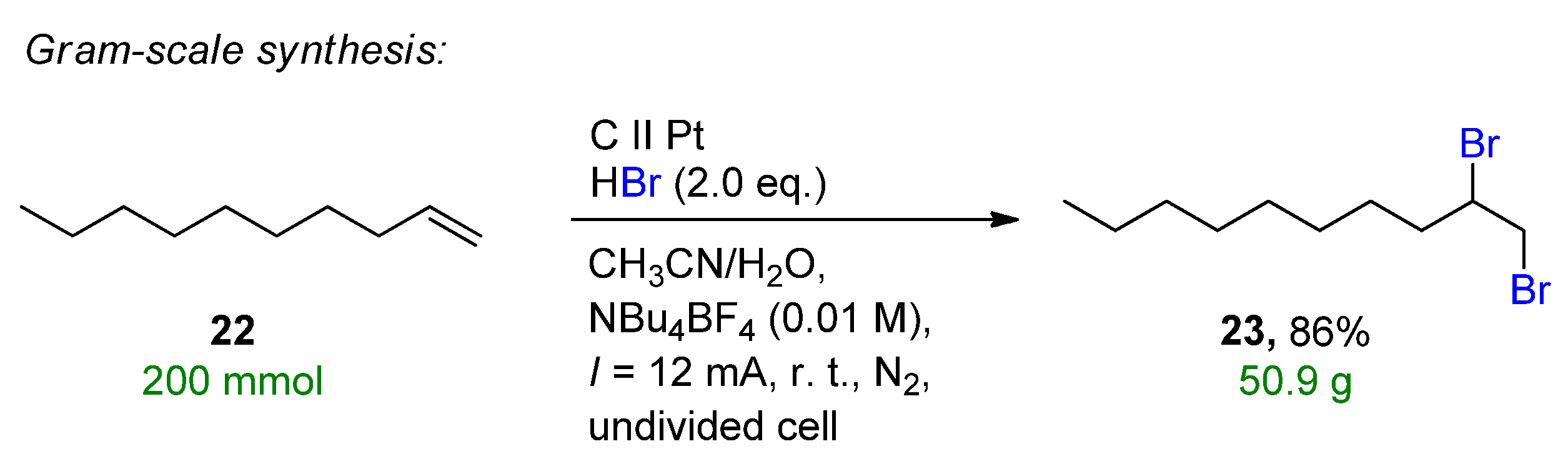
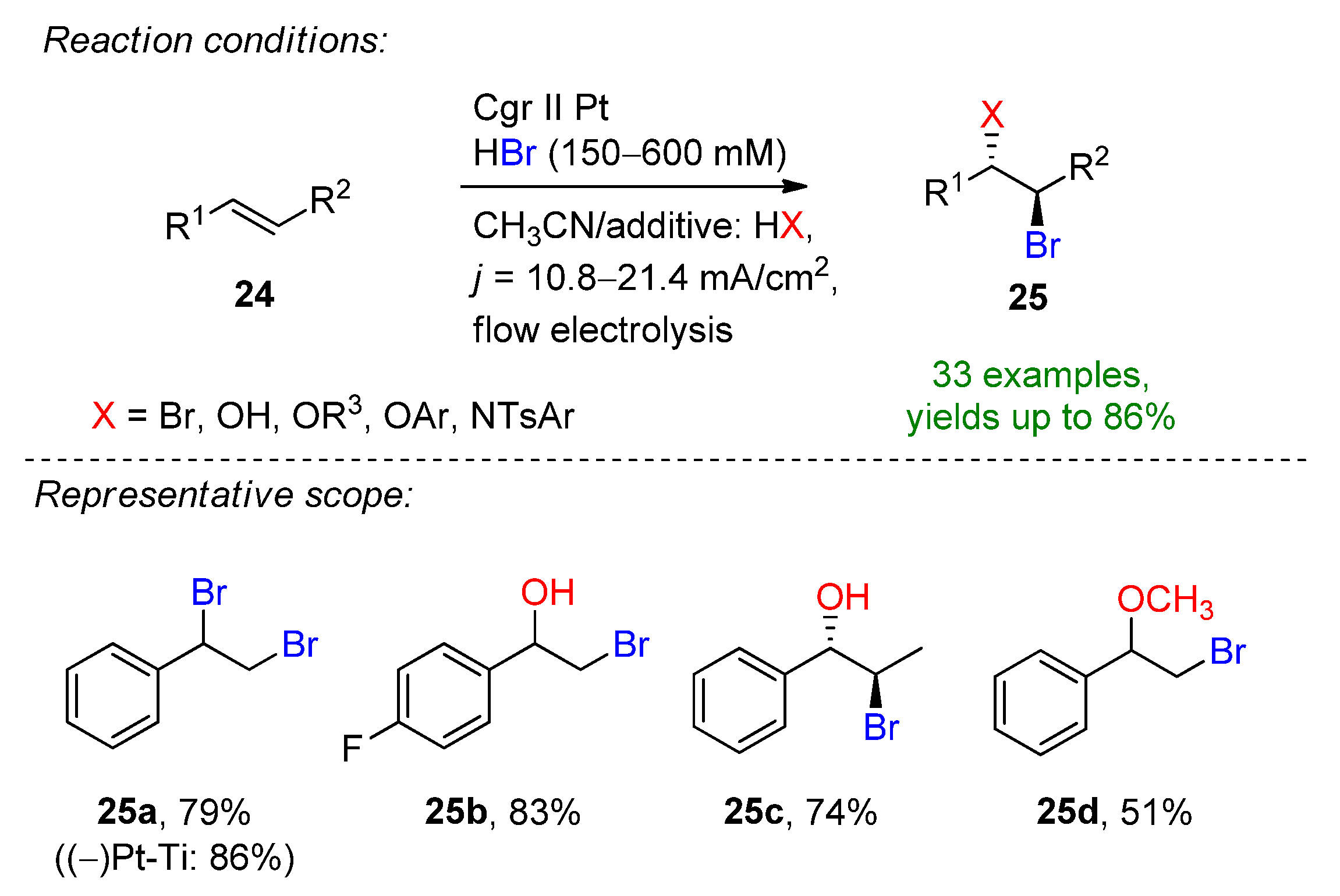
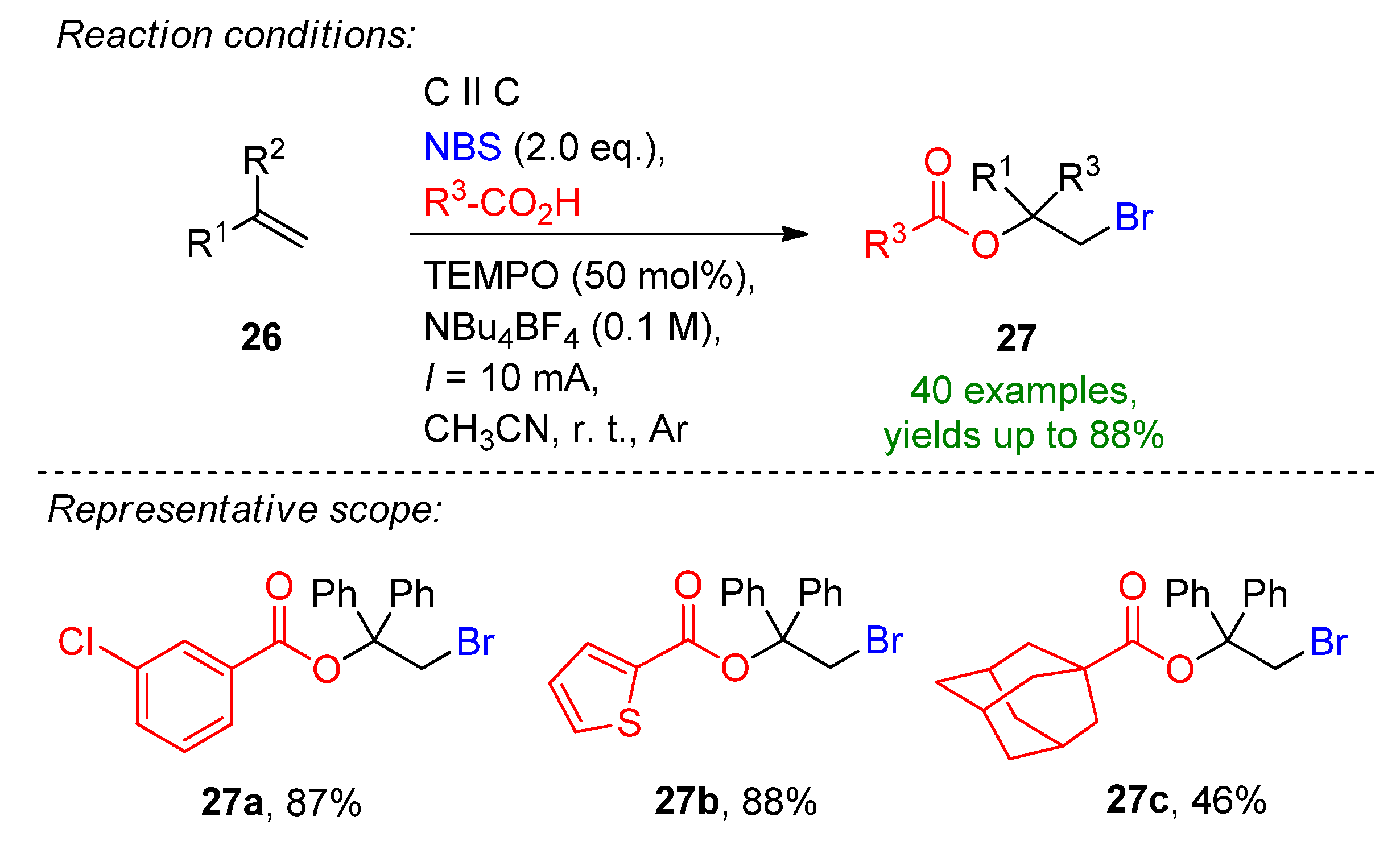
3.1. HBr, MBr and Alkylammonium Bromides as Bromine Sources
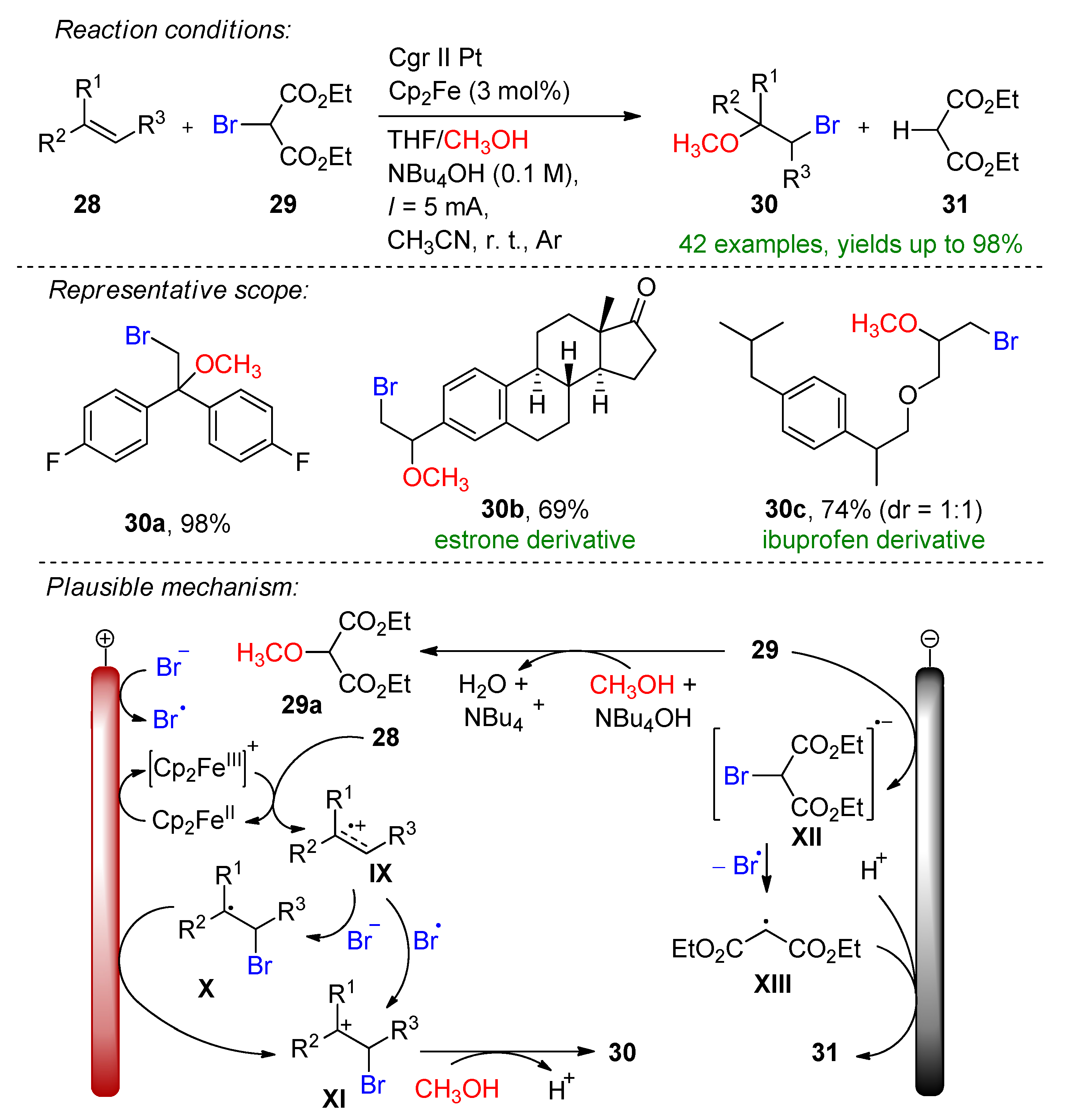
3.2. Organohalides as Bromine Sources
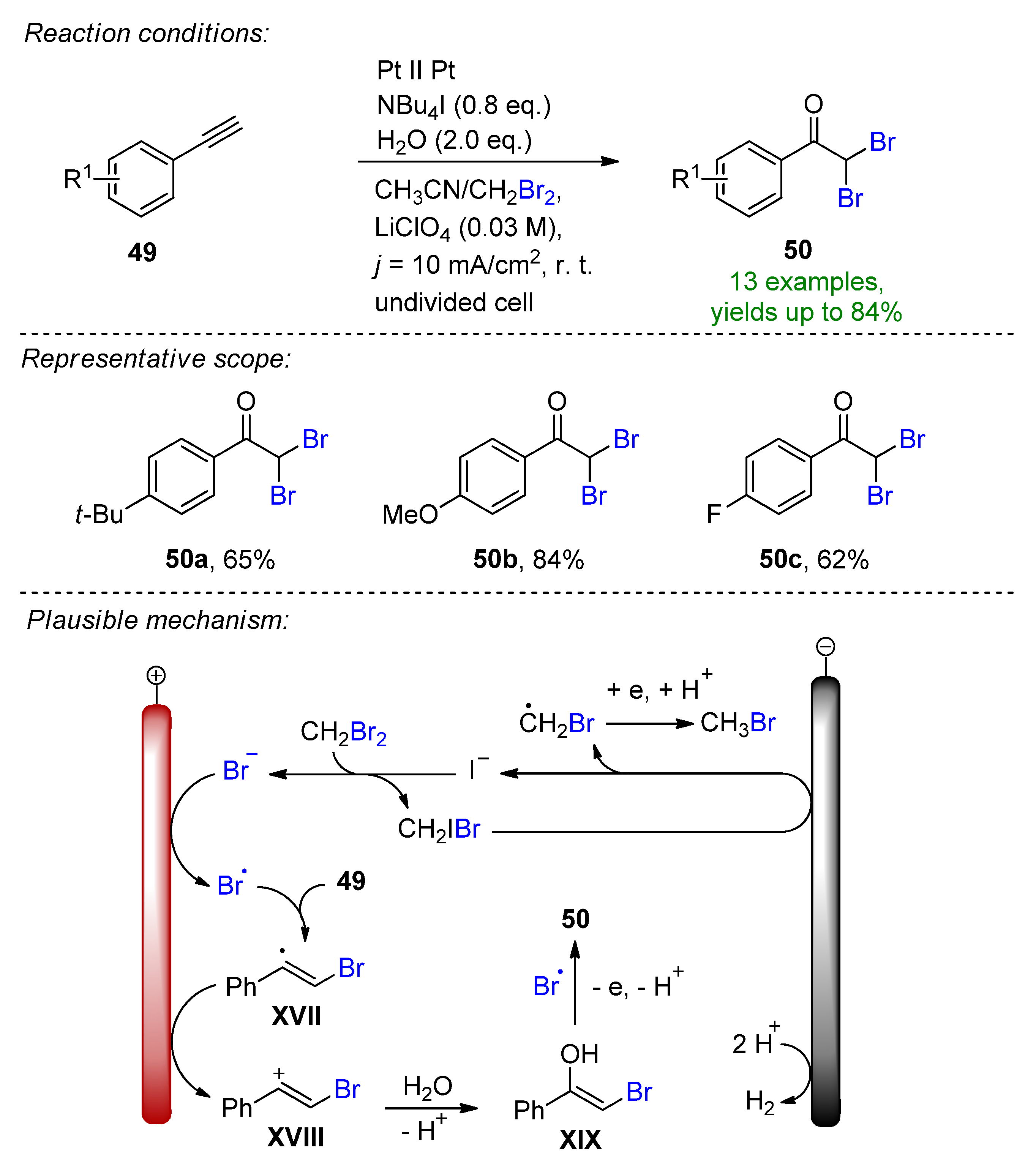
4. Electrochemical Bromofunctionalization of Alkynes
4.1. HBr, MBr and Alkylammonium Bromides as Bromine Sources
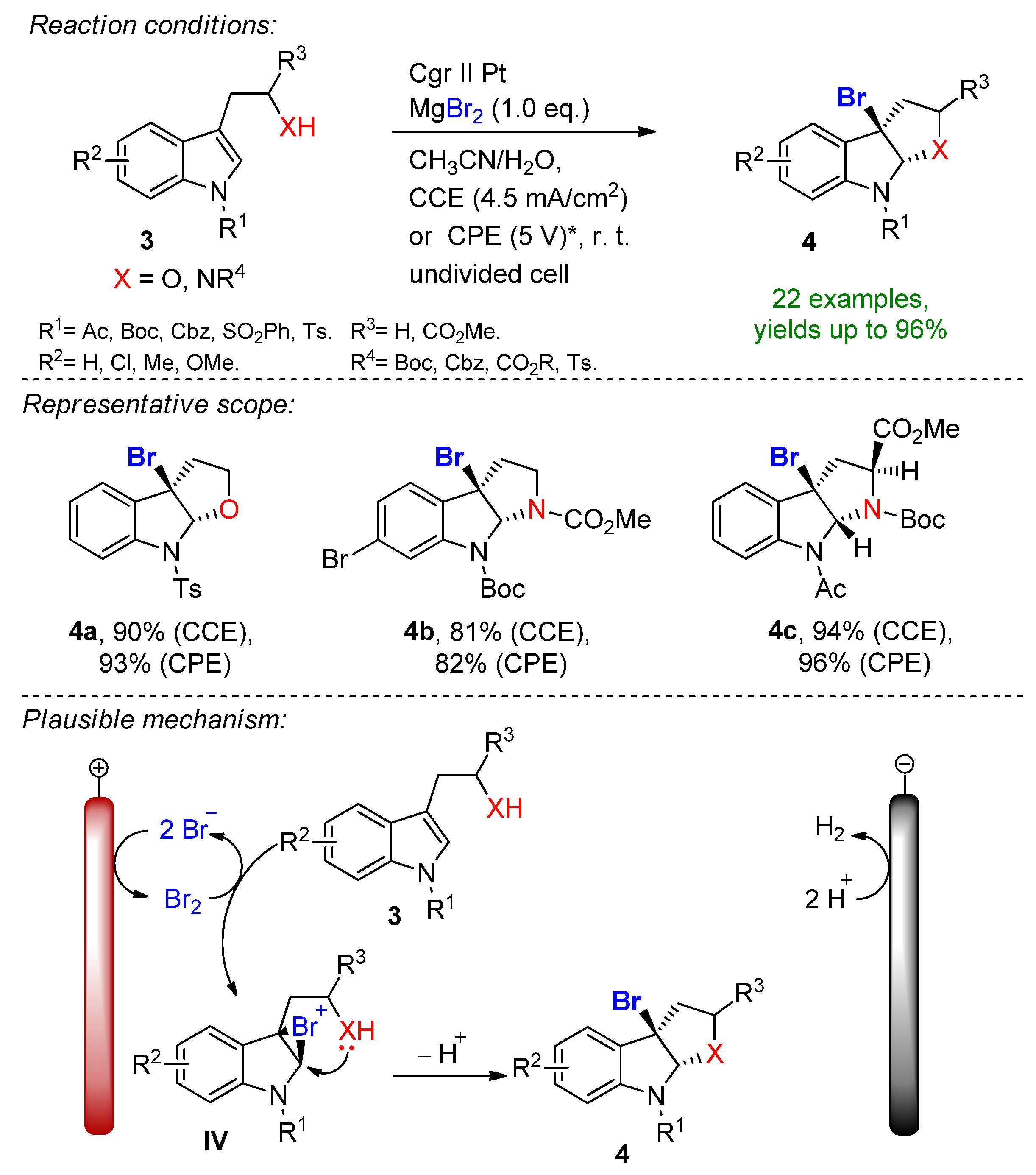
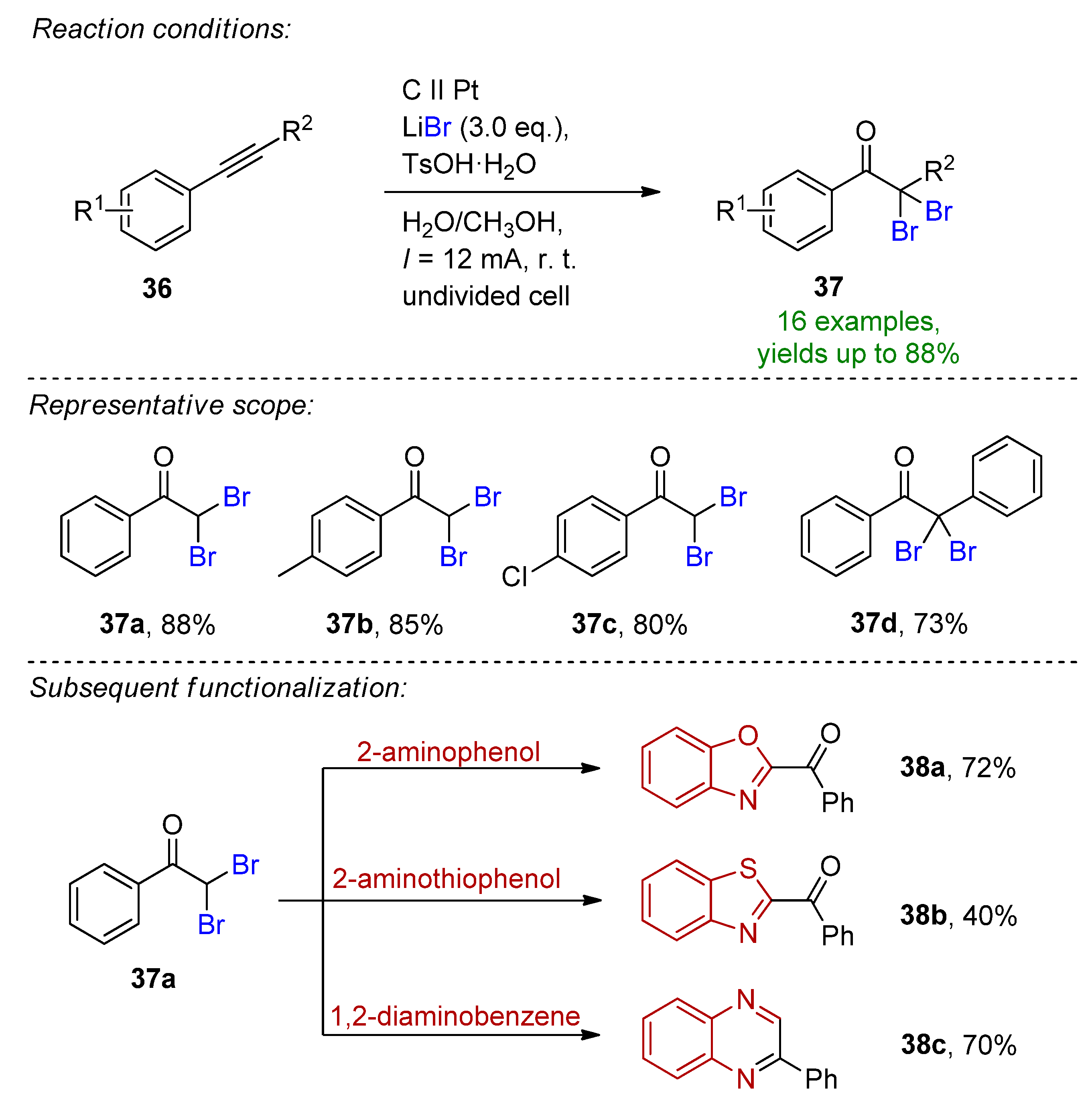
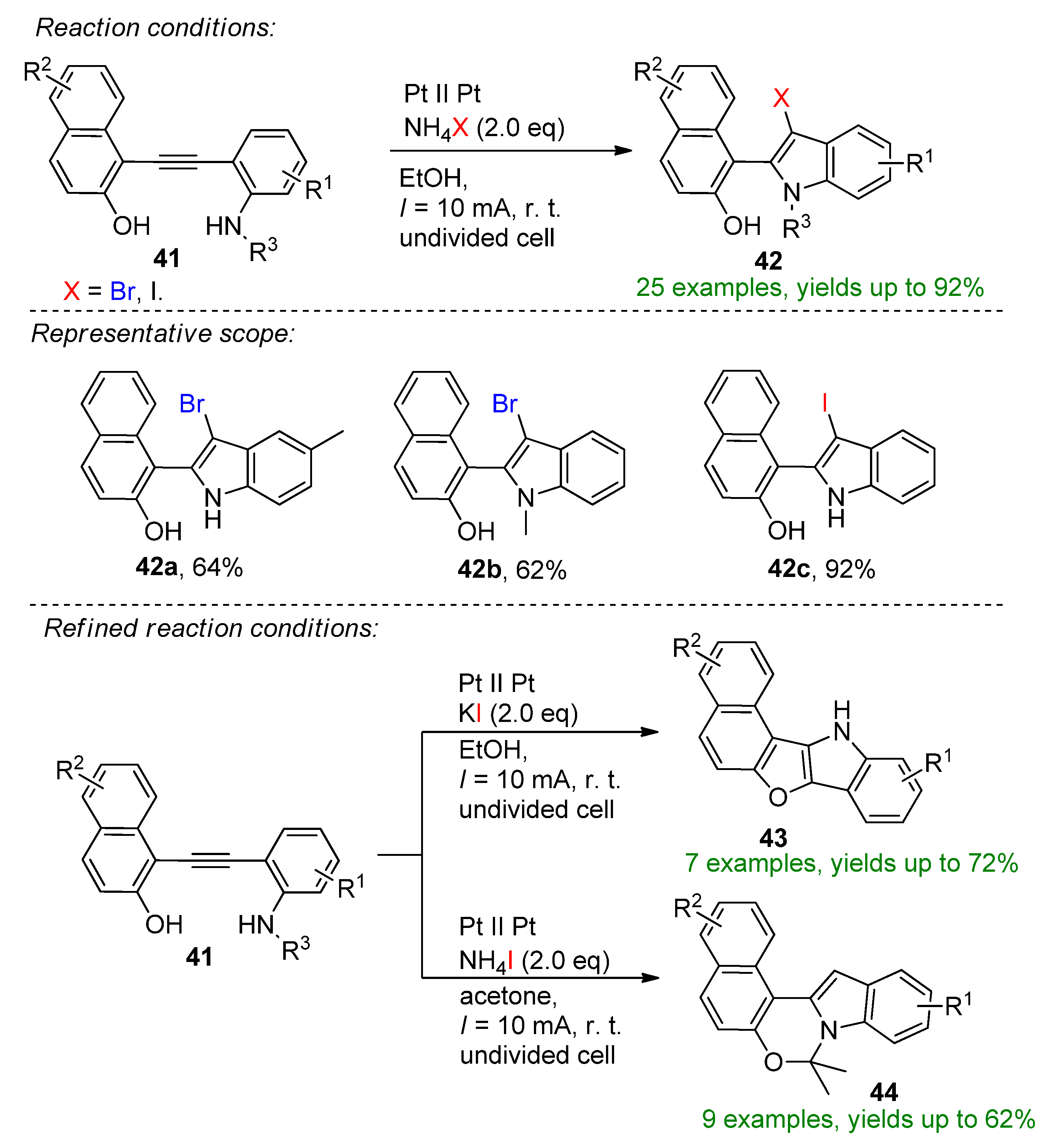
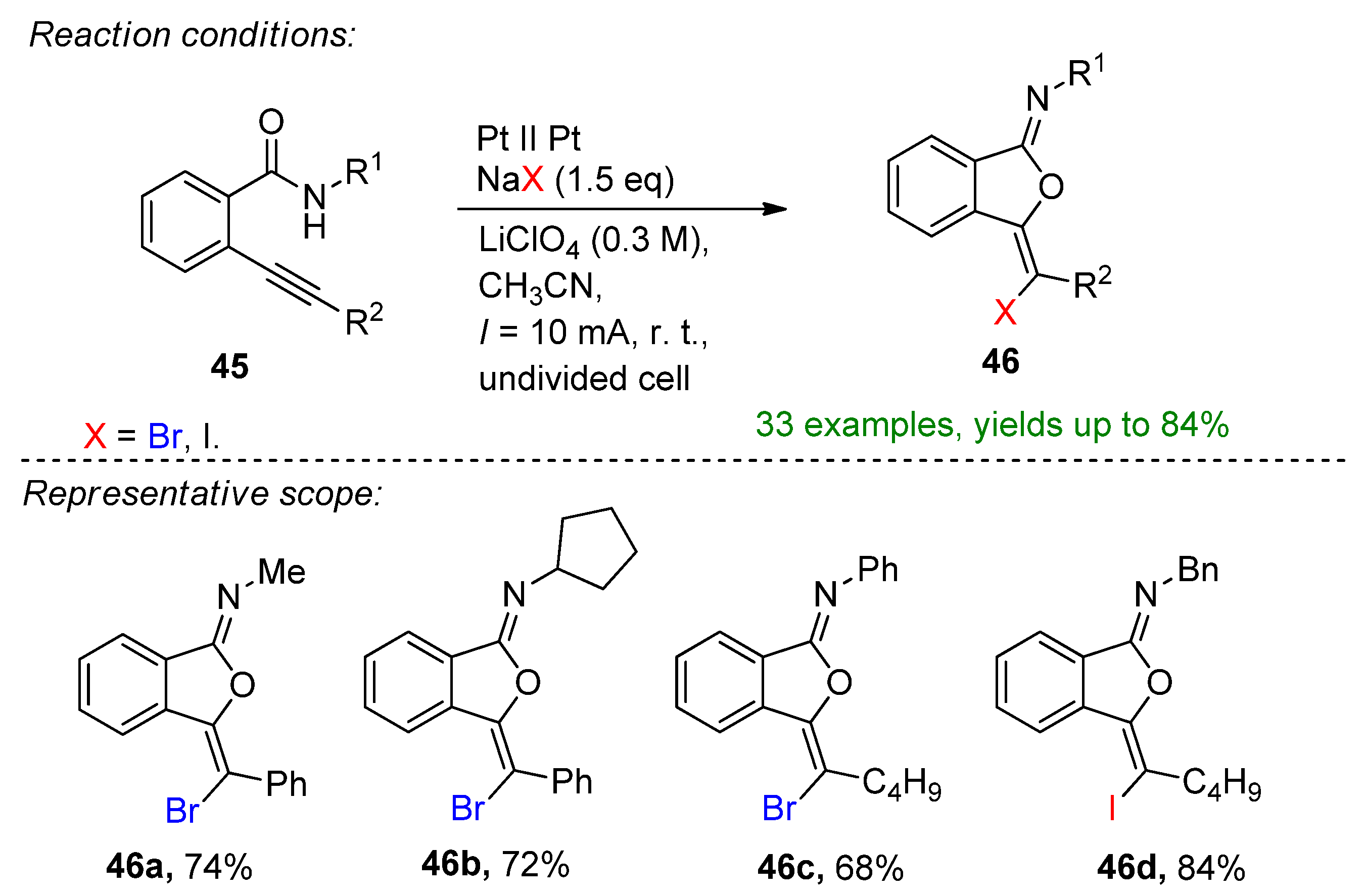
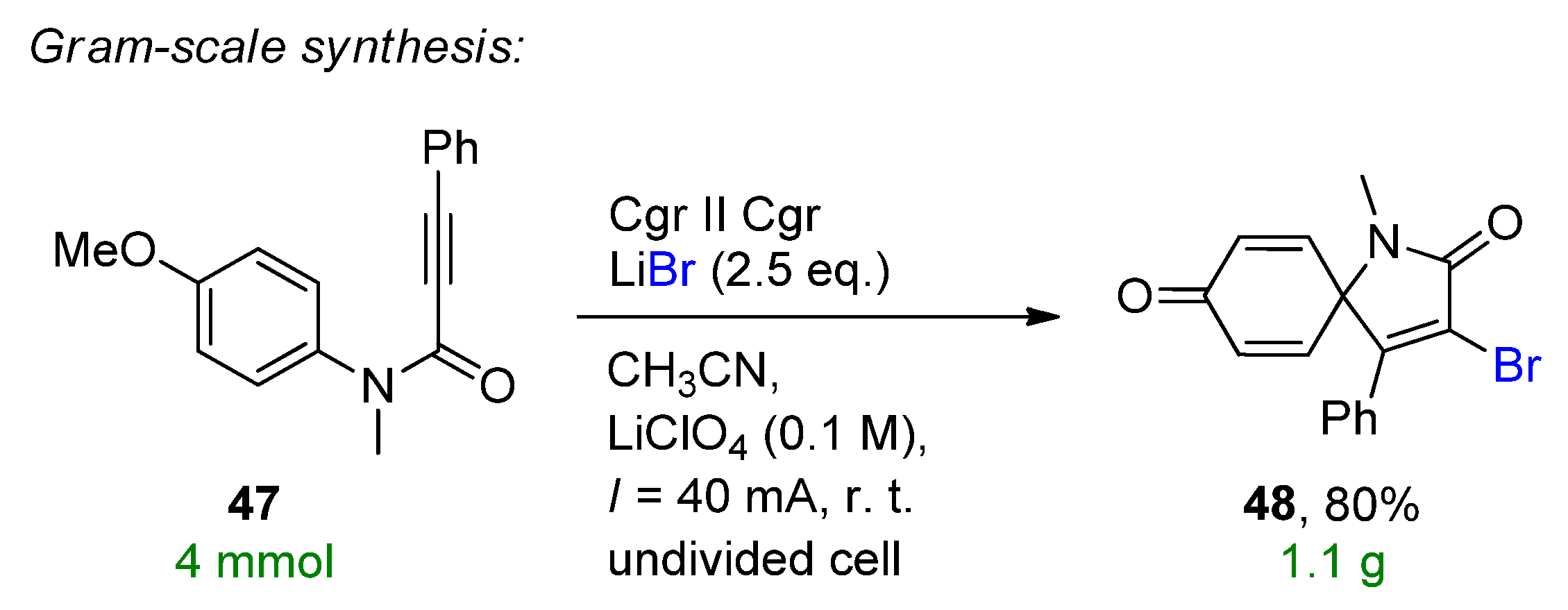
4.2. Organohalides as Bromine Sources
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoegh-Guldberg, O.; Jacob, D.; Taylor, M. Global Warming of 1.5 °C. Available online: https://www.ipcc.ch/sr15/ (accessed on 25 August 2022).
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev. 2017, 117, 13230–13319. [Google Scholar] [CrossRef]
- Shatskiy, A.; Lundberg, H.; Kärkäs, M.D. Organic electrosynthesis: Applications in complex molecule synthesis. ChemElectroChem 2019, 6, 4067–4092. [Google Scholar] [CrossRef]
- Hilt, G. Basic strategies and types of applications in organic electrochemistry. ChemElectroChem 2020, 7, 395–405. [Google Scholar] [CrossRef]
- Pollok, D.; Waldvogel, S.R. Electro-organic synthesis—A 21st century technique. Chem. Sci. 2020, 11, 12375–12592. [Google Scholar] [CrossRef]
- Novaes, L.F.T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J.M.; Lin, S. Electrocatalysis as an enabling technology for organic synthesis. Chem. Soc. Rev. 2021, 50, 7941–8002. [Google Scholar] [CrossRef]
- Beil, S.B.; Pollok, D.; Waldvogel, S.R. Reproducibility in electroorganic synthesis—Myths and misunderstandings. Angew. Chem. Int. Ed. 2021, 60, 14750–14759. [Google Scholar] [CrossRef]
- Yoshida, J.; Shimizu, A.; Hayashi, R. Electrogenerated cationic reactive intermediates: The pool method and further advances. Chem. Rev. 2017, 118, 4702–4730. [Google Scholar] [CrossRef]
- Röckl, J.L.; Pollok, D.; Franke, R.; Waldvogel, S.R. A decade of electrochemical dehydrogenative C,C-coupling of aryls. Acc. Chem. Res. 2020, 53, 45–61. [Google Scholar] [CrossRef]
- Waldvogel, S.R.; Lips, S.; Selt, M.; Riehl, B.; Kampf, C.J. Electrochemical arylation reactions. Chem. Rev. 2018, 118, 6706–6765. [Google Scholar] [CrossRef]
- Seidler, J.; Strugatchi, J.; Gärtner, T.; Waldvogel, S.R. Does electrifying organic synthesis pay off—The energy efficiency of electro-organic conversions. MRS Energy Sustain. 2020, 7, E42. [Google Scholar] [CrossRef]
- Margarita, C.; Lundberg, H. Recent advances in asymmetric catalytic electrosynthesis. Catalysts 2020, 10, 982. [Google Scholar] [CrossRef]
- Chicas-Baños, D.F.; Frontana-Uribe, B.A. Electrochemical generation and use in organic synthesis of C-,O-, and N-centered radicals. Chem. Rec. 2021, 21, 2538–2573. [Google Scholar] [CrossRef]
- Klüh, D.; Waldmüller, W.; Gaderer, M. Kolbe electrolysis for the conversion of carboxylic acids to valuable products—A process design study. Clean Technol. 2021, 3, 1–18. [Google Scholar] [CrossRef]
- Gleede, B.; Selt, M.; Franke, R.; Waldvogel, S.R. Developments in the dehydrogenative electrochemical synthesis of 3,3´,5,5´-tetramethyl-2,2´-biphenol. Chem. Eur. J. 2021, 27, 8252–8263. [Google Scholar] [CrossRef]
- Zheng, Y.; Shao, X.; Ramadoss, V.; Tian, L.; Wang, Y. Recent developments in photochemical and electrochemical decarboxylative C(sp3)-N bond formation. Synthesis 2020, 52, 1357–1368. [Google Scholar] [CrossRef]
- Listratova, A.V.; Sbei, N.; Voskressensky, L.G. Catalytic electrosynthesis of N,O-heterocycles—Recent advances. Eur. J. Org. Chem. 2020, 2020, 2012–2027. [Google Scholar] [CrossRef]
- Wirtanen, T.; Rodrigo, E.; Waldvogel, S.R. Recent advances in the electrochemical reduction of substrates involving N-O bonds. Adv. Synth. Catal. 2020, 362, 2088–2101. [Google Scholar] [CrossRef]
- Mei, H.; Pajkert, R.; Wang, L.; Li, Z.; Röschenthaler, G.; Han, J. Chemistry of electrochemical oxidative reactions of sulfinate salts. Green Chem. 2020, 22, 3028–3059. [Google Scholar] [CrossRef]
- Blum, S.P.; Hofman, K.; Manolikakes, G.; Waldvogel, S.R. Advances in photochemical and electrochemical incorporation of sulfur dioxide for the synthesis of value-added compounds. Chem. Commun. 2021, 57, 8236–8249. [Google Scholar] [CrossRef]
- Sbei, N.; Martins, G.M.; Shirinfar, B.; Ahmed, N. Electrochemical phosphorylation of organic molecules. Chem. Rec. 2020, 20, 1530–1552. [Google Scholar] [CrossRef]
- Thang, S.; Liu, Y.; Lei, A. Electrochemical oxidative cross-coupling with hydrogen evolution: A green and sustainable way for bond formation. Chem 2018, 4, 27–45. [Google Scholar] [CrossRef]
- Yang, Q.; Fang, P.; Mei, T. Recent Advances in organic electrochemical C-H functionalization. Chin. J. Chem. 2018, 36, 338–352. [Google Scholar] [CrossRef]
- Scheide, M.R.; Nicoleti, C.R.; Martins, G.M.; Braga, A.L. Electrohalogenation of organic compounds. Org. Biomol. Chem. 2021, 19, 2578–2602. [Google Scholar] [CrossRef]
- Kisukuri, C.M.; Fernandes, V.A.; Delgado, J.A.C.; Häring, A.P.; Paixão, M.W.; Waldvogel, S.R. Electrochemical installation of CFH2−, CF2H−, CF3−, and perfluoroalkyl groups into small organic molecules. Chem. Rec. 2021, 21, 2502–2525. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Tiz, D.B.; Bagnoli, L.; Rosati, O.; Marini, F.; Sancineto, L.; Santi, C. New halogen-containing drugs approved by FDA in 2021: An overview on their synthesis and pharmaceutical use. Molecules 2022, 27, 1643. [Google Scholar] [CrossRef]
- Bidleman, T.F.; Andersson, A.; Jantunen, L.M.; Kucklick, J.R.; Kylin, H.; Letcher, R.J.; Tysklind, M.; Wong, F. A review of halogenated natural products in Arctic, Subarctic and Nordic ecosystems. Emerg. Contam. 2019, 5, 89–115. [Google Scholar] [CrossRef]
- Häggblom, M.M.; Bossert, I.D. Halogenated organic compounds: A global perspective. In Dehalogenation: Microbial Processes and Environmental Applications; Springer: Boston, MA, USA, 2003; pp. 3–29. [Google Scholar] [CrossRef]
- Yuan, Y.; Lei, A. Is electrosynthesis always green and advantageous compared to traditional methods? Nat. Commun. 2020, 11, 802. [Google Scholar] [CrossRef]
- Möhle, S.; Zirbes, M.; Rodrigo, E.; Gieshoff, T.; Wiebe, A.; Waldvogel, S.R. Modern electrochemical aspects for the synthesis of value-added organic products. Angew. Chem. Int. Ed. 2018, 57, 6018–6041. [Google Scholar] [CrossRef]
- Wiebe, A.; Gieshoff, T.; Möhle, S.; Rodrigo, E.; Zirbes, M.; Waldvogel, S.R. Electrifying organic synthesis. Angew. Chem. Int. Ed. 2018, 57, 5594–5619. [Google Scholar] [CrossRef]
- Tang, H.; Jia, J.; Pan, Y. Halogen-mediated electrochemical organic synthesis. Org. Biomol. Chem. 2020, 18, 5315–5333. [Google Scholar] [CrossRef]
- Lian, F.; Xu, K.; Zeng, C. Indirect electrosynthesis with halogen ions as mediators. Chem. Rec. 2021, 21, 2290–2305. [Google Scholar] [CrossRef]
- Inokuchi, T.; Matsumoto, S.; Torii, S. Indirect electrooxidation of alcohols by a double mediatory system with two redox couples of [R2N+=O]/R2NO· and [Br· or Br+]/Br− in an organic-aqueous two-phase solution. J. Org. Chem. 1991, 56, 2416–2421. [Google Scholar] [CrossRef]
- Limborg, F.; Clauson-Kaas, N. A small cell for electrolytic alkoxylation of furans. Acta Chem. Scand. 1953, 7, 234–235. [Google Scholar] [CrossRef][Green Version]
- Nieto-Mendoza, E.; Guevara-Salazar, J.A.; Ramírez-Apan, M.T.; Frontana-Ulribe, B.A.; Cogordan, B.A.; Cárnedas, J. Electro-oxidation of hispanolone and anti-inflammatory properties of the obtained derivates. J. Org. Chem. 2005, 70, 4538–4541. [Google Scholar] [CrossRef]
- Salameh, E.; Tarawneh, A.; Al-Raggad, M. Origin of high bromide concentration in the water sources in Jordan and in the Dead Sea water. Arab. J. Geosci. 2016, 9, 414. [Google Scholar] [CrossRef]
- Fernández, L. Market Volume of Bromine Worldwide from 2015 to 2021, with a Forecast for 2022 to 2029. Available online: https://www.statista.com/statistics/1245240/bromine-market-volume-worldwide/ (accessed on 25 August 2022).
- Wu, N.; Herrmann, T.; Paepke, O.; Tickner, J.; Hale, R.; Harvey, E.; La Guardia, M.; McClean, M.D.; Webster, T.F. Human exposure to PBDEs: Association of PBDE body burdens with food consumption and house dust concentrations. Environ. Sci. Technol. 2007, 41, 1584–1589. [Google Scholar] [CrossRef]
- Hales, B.F.; Robaire, B. Effects of brominated and organophospate ester flame retardants on male reproduction. Andrology 2020, 8, 915–923. [Google Scholar] [CrossRef]
- Addis, D.R.; Molyvdas, A.; Ambalavanan, N.; Matalon, S.; Jilling, T. Halogen exposure injury in the developing lung. Ann. N. Y. Acad. Sci. 2020, 1480, 30–43. [Google Scholar] [CrossRef]
- Addis, D.R.; Aggarwal, S.; Lazrak, A.; Jilling, T.; Matalon, S. Halogen-induced chemical injury to the mammalian cardiopulmonary systems. Physiology 2021, 36, 272–291. [Google Scholar] [CrossRef]
- Saikira, I.; Borah, A.J.; Phukan, P. Use of bromine and bromo-organic compounds in organic synthesis. Chem. Rev. 2016, 116, 6837–7042. [Google Scholar] [CrossRef]
- Eissen, M.; Lenoir, D. Electrophilic bromination of alkenes: Environmental, health and safety aspects of new alternative methods. Chem. Eur. J. 2008, 14, 9830–9841. [Google Scholar] [CrossRef]
- Van Kerrebroeck, R.; Horsten, T.; Stevens, C.V. Bromide oxidation: A safe strategy for electrophilic brominations. Eur. J. Org. Chem. 2022, in press. [CrossRef]
- Tariq, Z.M. Electrochemistry of Br−/Br2 Redox Couple in Acetonitrile, Methanol and Mix Media of Acetonitrile–Methanol: An Insight into Redox Behavior of Bromide on Platinum (Pt) and Gold (Au) Electrode. Phys. Chem. 2019, 234, 295–312. [Google Scholar] [CrossRef]
- Halász, D.; Visy, C.; Szűcs, A.; Novák, M. Bromide ion oxidation of various Pt surfaces. React. Kinet. Catal. Lett. 1992, 48, 177–188. [Google Scholar] [CrossRef]
- Vojinovic, V.; Mentus, S.; Komnenic, V.J. Bromide oxidation and bromine reduction in propylene carbonate. Electroanal. Chem. 2003, 547, 109–113. [Google Scholar] [CrossRef]
- Haynes, W.M. Abundance of elements in the earth’s crust and in the sea. In CRC Handbook of Chemistry and Physics; CRC Press: Boulder, CO, USA, 2016; pp. 14–17. ISBN 978-1-4822-0868-9. [Google Scholar]
- Sigma Aldrich. Available online: https://www.sigmaaldrich.com/DE/en (accessed on 14 September 2022).
- Kingston, C.; Palkowitz, M.D.; Takahira, Y.; Vantourout, J.C.; Peters, B.K.; Kawamata, Y.; Baran, P.S. A survival guide for the “Electro-curious”. Acc. Chem. Rev. 2020, 53, 72–83. [Google Scholar] [CrossRef]
- Schotten, C.; Nicholls, T.P.; Bourne, R.A.; Kapur, N.; Nguen, B.N.; Williams, C.E. Making electrochemistry easily accessible to the synthetic chemist. Green Chem. 2020, 22, 3358–3375. [Google Scholar] [CrossRef]
- Wu, T.; Nguyen, B.H.; Daugherty, M.C.; Moeller, K.D. Paired electrochemical reactions and the on-site generation of a chemical reagent. Angew. Chem. Int. Ed. 2019, 58, 3562–3565. [Google Scholar] [CrossRef]
- Wu, T.; Moeller, K.D. Organic electrochemistry: Expanding the scope of paired reactions. Angew. Chem. Int. Ed. 2021, 60, 12883–12890. [Google Scholar] [CrossRef]
- Marken, F.; Cresswell, A.J.; Bull, S.T. Recent advances in paired electrosynthesis. Chem. Rec. 2021, 21, 2585–2600. [Google Scholar] [CrossRef]
- Klein, M.; Waldvogel, S.R. Counter electrode reactions—Important stumbling blocks on the way to a working electro-organic synthesis. Angew. Chem. Int. Ed. 2022. accepted. [Google Scholar] [CrossRef]
- Clark, J.H. Green Chemistry: Challenges and opportunities. Green Chem. 1998, 1, 1–8. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Frontana-Ulribe, B.A.; Little, R.D.; Ibanez, J.G.; Palma, A.; Vasquez-Medrano, R. Organic electrosynthesis: A promising green methodology in organic synthesis. Green Chem. 2010, 12, 2099–2119. [Google Scholar] [CrossRef]
- Prat, D.; Wells, A.; Hayler, J.; Sneddon, H.; McElroy, R.; Abou-Shehada, S.; Dunn, P.J. CHEM21 selection guide of classical- and less classical solvents. Green Chem. 2016, 18, 288–296. [Google Scholar] [CrossRef]
- Dong, X.; Roeckl, J.L.; Waldvogel, S.R.; Morandi, B. Merging shuttle reactions and paired electrolysis for reversible vicinal dihalogenations. Science 2021, 371, 507–514. [Google Scholar] [CrossRef]
- Salom-Rolg, X.; Bauder, C. Recent applications in the use of sulfoxides as chiral auxiliaries for the asymetric synhesis of natural and biologically active products. Synthesis 2020, 52, 964–978. [Google Scholar] [CrossRef]
- Torii, S.; Uneyama, K.; Ueda, K. Electrochemical procedure for a practical preparation of piperonal from isosafrole. J. Org. Chem. 1984, 49, 1830–1832. [Google Scholar] [CrossRef]
- Uneyama, K.; Masatsugu, Y.; Torii, S. Electrochemical epoxidation and carbon-carbon bond cleavage for the preparation of 3-methyl-4-oxo-2-phenyl-4H-1-benzopyran-8-carboxylic acid from 3-methyl-2-phenyl-8-(1-prophenyl)-4H-1-benzopyran-4-one. Bull. Chem. Soc. Jpn. 1985, 58, 2361–2365. [Google Scholar] [CrossRef]
- Inokuchi, T.; Matsumoto, S.; Tsuji, M.; Torii, S. Electrohalogenation of propargyl acetates and amides to form the 1,1-dibromo-2-oxo functionality and a facile synthesis of furaneol. J. Org. Chem. 1992, 57, 5023–5027. [Google Scholar] [CrossRef]
- Inês, M.; Mendonça, A.J.; Esteves, A.P.; Mendonça, D.I.; Medeiros, M.J. Electroepoxidation of natural and synthetic alkenes mediated by sodium bromide. Comptes Rendus Chim. 2009, 12, 841–849. [Google Scholar] [CrossRef]
- Kulangiappar, K.; Ramaprakash, M.; Vasudevan, D.; Raju, T. Electrochemical bromination of cyclic and acyclic enes using biphasic electrolysis. Synth. Commun. 2016, 46, 145–153. [Google Scholar] [CrossRef]
- Yang, C. An impending platinum crisis and its implications for the future of the automobile. Energy Policy 2009, 37, 1805–1808. [Google Scholar] [CrossRef]
- Das, K.K.; Reddy, R.C.; Bagoji, I.B.; Das, S.; Bagali, S.; Mullur, L.; Khodnapur, J.P.; Biradar, M.S. Primary concept of nickel toxicity—An overview. J. Basic Clin. Physiol. Pharmacol. 2019, 30, 141–152. [Google Scholar] [CrossRef]
- Savignan, L.; Faucher, S.; Chéry, P.; Lespes, G. Platinum group elements contamination in soils: Review of the current state. Chemosphere 2021, 271, 129517. [Google Scholar] [CrossRef]
- Sun, X.; Ma, H.; Mei, T.; Fang, P.; Hu, Y. Electrochemical radical formyloxylation-bromination, -chlorination, and -trifluoromethylation of alkenes. Org. Lett. 2019, 21, 3167–3171. [Google Scholar] [CrossRef]
- Bormann, S.; Van Schie, M.M.C.H.; De Almeida, T.P.; Zhang, W.; Stöckl, M.; Ulber, R.; Hollman, F.; Holtmann, D. H2O2 production at low overpotential for electroenzymatic halogenation reactions. ChemSusChem 2019, 12, 4759–4763. [Google Scholar] [CrossRef]
- Wever, R.; Renirie, R.; Hollmann, F. Vanadium chloroperoxidases as versatile biocatalysts. In Vanadium Catalysis; Sudradhar, M., Pombeiro, A.J.L., da Silva, J.A.L., Eds.; Royal Society of Chemistry: London, UK, 2021; Chapter 24; ISBN 978-1-78801-857-9. [Google Scholar]
- Wu, J.; Abou-Hamdan, H.; Guillot, R.; Kouklovsky, C.; Vincent, G. Electrochemical synthesis of 3a-bromofuranoindolines and 3a-bromopyrroloindolines mediated by MgBr2. Chem. Commun. 2020, 56, 1713–1716. [Google Scholar] [CrossRef]
- Hakamata, H.; Sato, S.; Ueda, H.; Tokuyama, H. AgNTf2-Mediated Allylation with Allylsilanes at C3a-Position of Hexahydropyrroloindoles: Application to Total Syntheses of Amauromine Alkaloids. Org. Lett. 2017, 19, 5308–5311. [Google Scholar] [CrossRef]
- Ramos-Villaseñor, J.M.; Rodríguez-Cárdenas, E.; Díaz, C.E.B.; Frontana-Uribe, B.A. Review—Use of 1,1,1,3,3,3–hexafluoro–2–propanol (HFIP) Co-Solvent Mixtures in Organic Electrosynthesis. J. Electrochem. Soc. 2020, 167, 155509. [Google Scholar] [CrossRef]
- Eberson, L.; Hartshorn, M.P.; Persson, O. 1,1,1,3,3,3-Hexafluoropropan-2-ol as a solvent for the generation of highly persistent radical cations. J. Chem. Soc. Perkin Trans. 2 1995, 1735–1744. [Google Scholar] [CrossRef]
- Eberson, L.; Hartshorn, M.P.; Persson, O. Generation of solutions of highly persistent radical cations by 4-tolylthallium(III) bis(trifluoroacetate) in 1,1,1,3,3,3-hexafluoropropan-2-ol. J. Chem. Soc. Chem. Commun. 1995, 1131–1132. [Google Scholar] [CrossRef]
- Eberson, L.; Persson, O.; Hartshorn, M.P. Detection and Reactions of Radical Cations Generated by Photolysis of Aromatic Compounds with Tetranitromethane in 1,1,1,3,3,3-Hexafluoro-2-propanol at Room Temperature. Angew. Chem. Int. Ed. 1995, 34, 2268–2269. [Google Scholar] [CrossRef]
- Schulz, L.; Waldvogel, S.R. Solvent control in electro-organic synthesis. Synlett 2018, 30, 275–286. [Google Scholar] [CrossRef]
- Röckl, J.L.; Dörr, M.; Waldvogel, S.R. Electrosynthesis 2.0 in 1,1,1,3,3,3-Hexafluoroisopropanol/Amine Mixtures. ChemElectroChem 2020, 7, 3686–3694. [Google Scholar] [CrossRef]
- Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. Halogen and Chalcogen Cation Pools Stabilized by DMSO. Versatile Reagents for Alkene Difunctionalization. J. Am. Chem. Soc. 2013, 135, 16070–16073. [Google Scholar] [CrossRef]
- Shimizu, A.; Hayashi, R.; Ashikari, Y.; Nokami, T.; Yoshida, J. Switching the reaction pathways of electrochemically generated β-haloalkoxysulfonium ions—Synthesis of halohydrins and epoxides. Beilstein J. Org. Chem. 2015, 11, 242–248. [Google Scholar] [CrossRef]
- Bityukov, O.V.; Vil’, V.A.; Nikishin, G.I.; Terent’ev, A.O. Alkene, Bromide, and ROH—How To Achieve Selectivity? Electrochemical Synthesis of Bromohydrins and Their Ethers. Adv. Synth. Catal. 2021, 363, 3070–3078. [Google Scholar] [CrossRef]
- Shang, X.; Liu, X.; Sun, Y. Flexible on-site halogenation paired with hydrogenation using halide electrolysis. Green Chem. 2021, 23, 2037–2043. [Google Scholar] [CrossRef]
- Strehl, J.; Abraham, M.L.; Hilt, G. Linear Paired Electrolysis—Realising 200 % Current Efficiency for Stoichiometric Transformations—The Electrochemical Bromination of Alkenes. Angew. Chem. Int. Ed. 2021, 60, 9996–10000. [Google Scholar] [CrossRef] [PubMed]
- Pitzer, L.; Schäfers, F.; Glorius, F. Rapid Assessment of the Reaction-Condition-Based Sensitivity of Chemical Transformations. Angew. Chem. Int. Ed. 2019, 58, 8572–8576. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.H.; Quach, L.; Paulisch, T.; Glorius, F. Visible-light-induced, metal-free carbene insertion into B-H bonds between acylsilanes and pinacolborane. J. Am. Chem. Soc. 2019, 141, 16227–16231. [Google Scholar] [CrossRef]
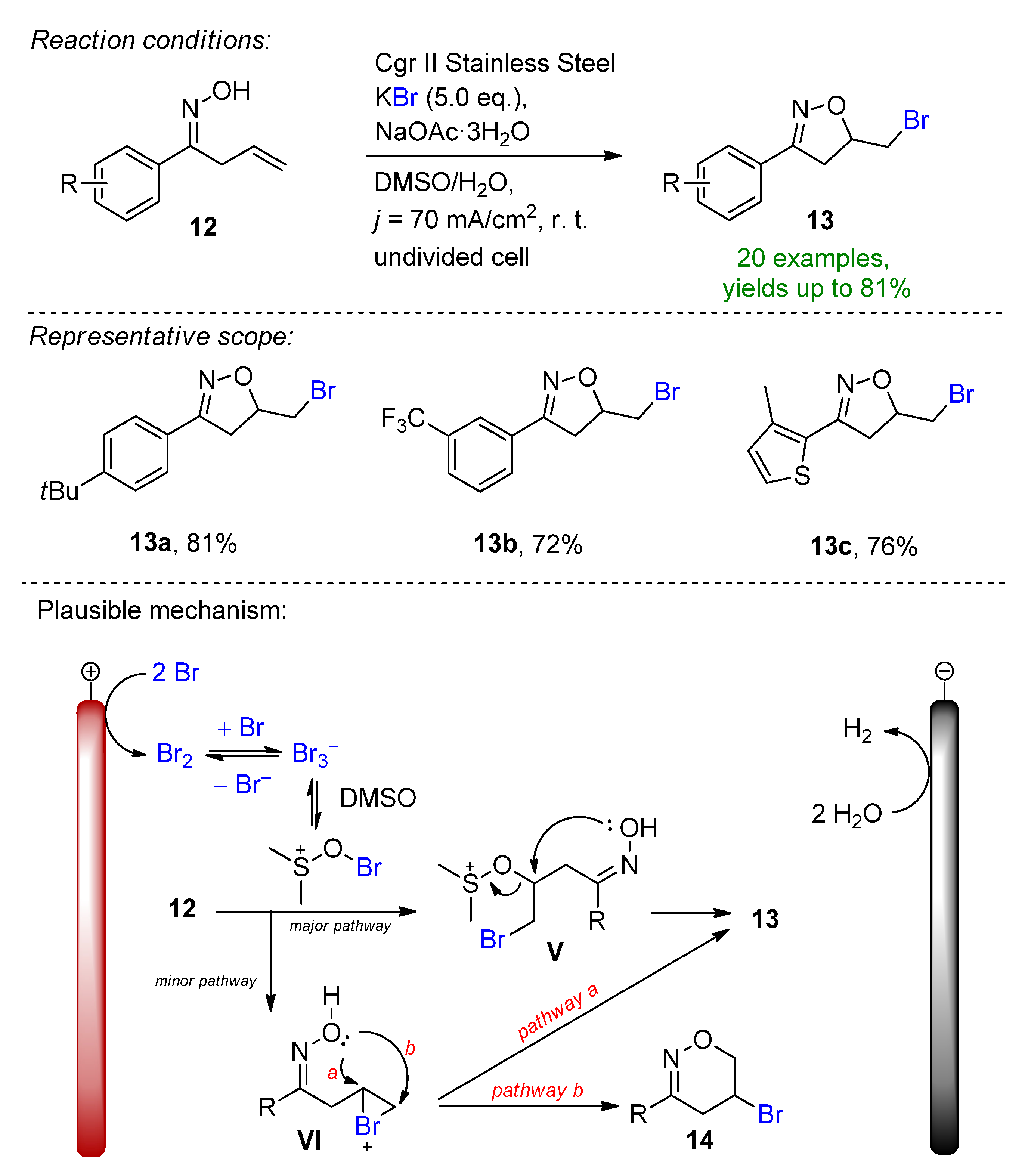
- Kale, A.P.; Nikolaienko, P.; Smirnova, K.; Rueping, M. Intramolecular Electrochemical Oxybromination of Olefins for the Synthesis of Isoxazolines in Batch and Continuous Flow. Eur. J. Org. Chem. 2021, 2021, 3496–3500. [Google Scholar] [CrossRef]
- He, Y.; Qin, X.; He, X.; Wu, X.; Yin, Z. Practical Synthesis of Halogenated N-Heterocycles via Electrochemical Anodic Oxidation of Unactivated Alkenes. Eur. J. Org. Chem. 2021, 2021, 5831–5834. [Google Scholar] [CrossRef]
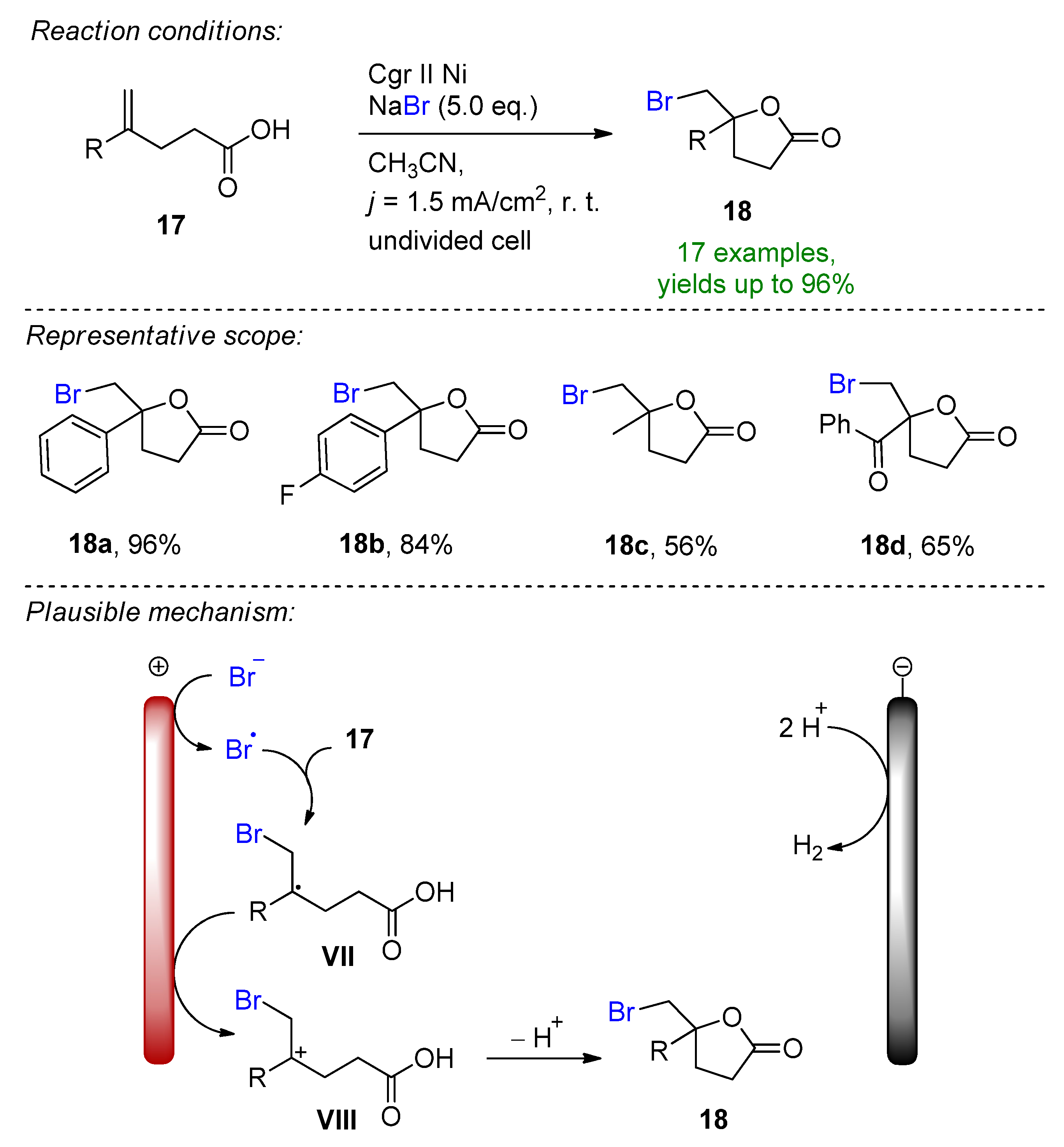
- Kim, R.; Ha, J.; Woo, J.; Kim, D.Y. Electrochemical oxidative bromolactonization of unsaturated carboxylic acids with sodium bromide: Synthesis of bromomethylated γ-lactones. Tetrahedron Lett. 2022, 88, 153567. [Google Scholar] [CrossRef]
- Yu, D.; Ji, R.; Sun, Z.; Li, W.; Liu, Z. Electrochemical chlorination and bromination of electron-deficient C-H bonds in quinones, coumarins, quinoxalines and 1,3-diketones. Tetrahedron Lett. 2021, 86, 153514. [Google Scholar] [CrossRef]
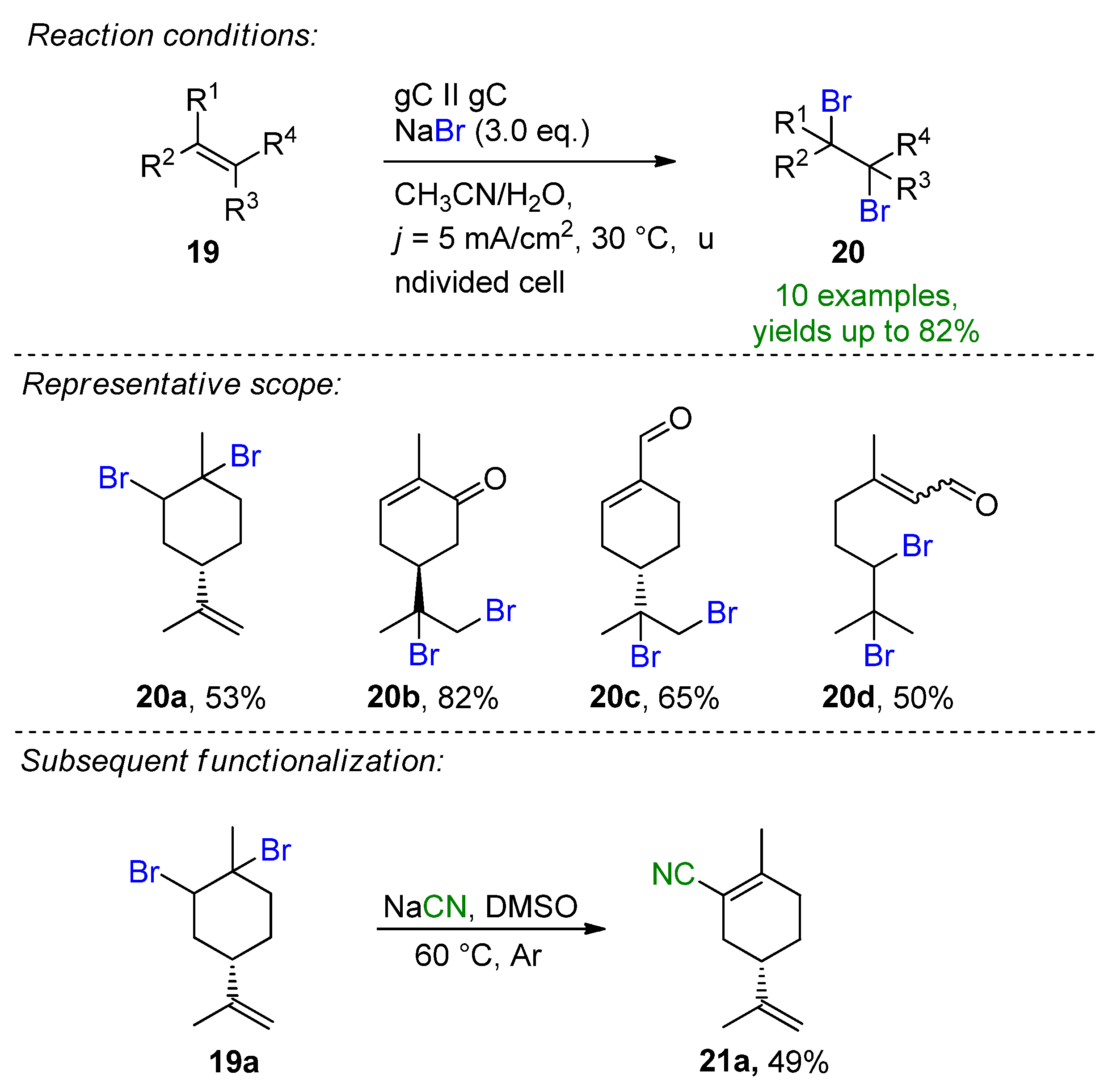
- Gombos, L.G.; Werner, L.; Schollmeyer, D.; Martínez-Huitle, C.A.; Waldvogel, S.R. Selective Electrochemical Dibromination of Terpenes and Naturally Derived Olefins. Eur. J. Org. Chem. 2022. accepted. [Google Scholar] [CrossRef]
- Yuan, Y.; Yao, A.; Zheng, Y.; Gao, M.; Zhou, Z.; Qiao, J.; Hu, J.; Ye, B.; Zhao, J.; Wen, H.; et al. Electrochemical Oxidative Clean Halogenation Using HX/NaX with Hydrogen Evolution. iScience 2019, 12, 293–303. [Google Scholar] [CrossRef]
- Seitz, J.; Wirth, T. Electrochemical bromofunctionalization of alkenes in a flow reactor. Org. Biomol. Chem. 2021, 19, 6892–6896. [Google Scholar] [CrossRef]
- Wan, C.; Song, R.; Li, J.H. Electrooxidative 1,2-Bromoesterification of Alkenes with Acids and N-Bromosuccinimide. J. Org. Lett. 2019, 21, 2800–2803. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Ouyang, X.; Hu, M.; Xie, Y.; Li, J. Copper-Catalyzed Intermolecular Aminoalkylation of Alkenes with α-Bromoalkyl Esters and Amines toward Pyrrolidin-2-ones. Adv. Synth. Catal. 2017, 359, 2564–2570. [Google Scholar] [CrossRef]
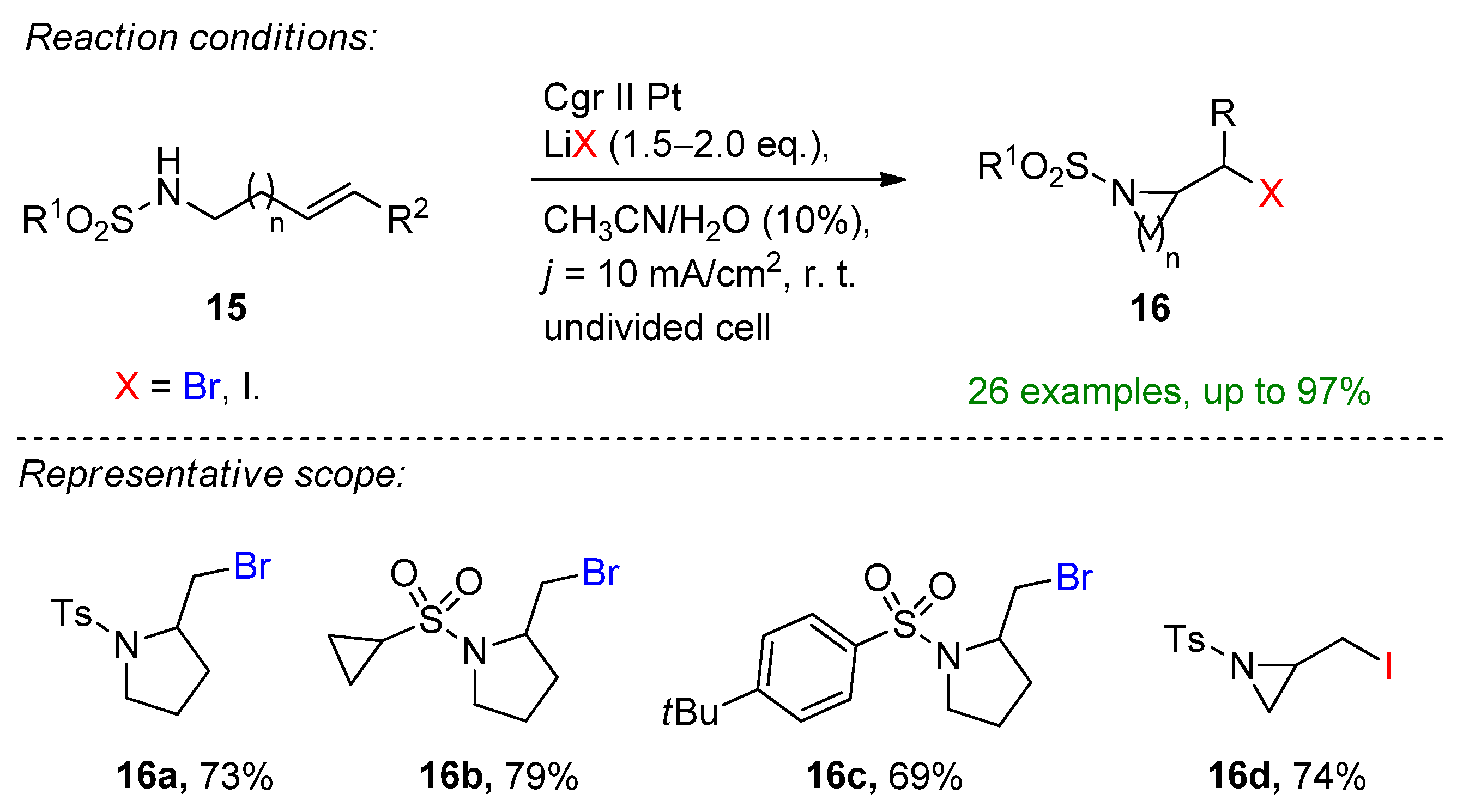
- Zhang, T.; Luo, M.; Li, Y.; Song, R.; Li, J. Electrochemical Alkoxyhalogenation of Alkenes with Organohalides as the Halide Sources via Dehalogenation. Org. Lett. 2020, 22, 7250–7254. [Google Scholar] [CrossRef] [PubMed]
- Bortolami, M.; Petrucci, R.; Rocco, D.; Scarano, V.; Chiarotto, I. Alkynes as Building Blocks, Intermediates and Products in the Electrochemical Procedures Since 2000. ChemElectroChem 2021, 8, 3604–3613. [Google Scholar] [CrossRef]
- Chung, W.; Vanderwal, C.D. Stereoselective Halogenation in Natural Product Synthesis. Angew. Chem. Int. Ed. 2016, 55, 4365–4434. [Google Scholar] [CrossRef]
- Sadhukhan, S.; Santhi, J.; Baire, B. The α,α-Dihalocarbonyl Building Blocks: An Avenue for New Reaction Development in Organic Synthesis. Chem. Eur. J. 2020, 26, 7145–7175. [Google Scholar] [CrossRef]
- Li, Z.; Sun, Q.; Qian, P.; Hu, K.; Zha, Z.; Wang, Z. Electrochemical synthesis of α,α-dihaloacetophenones from terminal alkyne derivatives. Chin. Chem. Lett. 2020, 31, 1855–1858. [Google Scholar] [CrossRef]
- Wang, D.; Wan, Z.; Zhang, H.; Lei, A. Electrochemical Oxidative Functionalization of Arylalkynes: Access to α,α-Dibromo Aryl Ketones. Adv. Synth. Catal. 2021, 363, 1022–1027. [Google Scholar] [CrossRef]
- Zhang, T.; Hao, W.; Wang, R.; Wang, S.; Tu, S.; Jiang, B. Electrocatalytic three-component annulation-halosulfonylation of 1,6-enynes toward 1-indanones using sodium halides as both halogen sources and electrolytes. Green Chem. 2020, 22, 4259–4269. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, S.; Hao, W.; Dong, G.; Tu, S.; Jiang, B. Tunable Electrocatalytic Annulations of o-Arylalkynylanilines: Green and Switchable Syntheses of Skeletally Diverse Indoles. J. Org. Chem. 2021, 86, 15886–15896. [Google Scholar] [CrossRef]
- Reddy, M.B.; Peri, R.; Bhagavathiachari, M.; Anandhan, R. Electrochemical synthesis of isobenzofuran-1-imines using oxidative halocyclization of o-alkynylbenzamides. Org. Biomol. Chem. 2021, 19, 6792–6796. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Kong, X.; Yang, J.; Li, G.; Xu, B.; Chen, Q. Electrochemical Oxidative Halogenation of N-Aryl Alkynamides for the Synthesis of Spiro[4.5]trienones. J. Org. Chem. 2021, 86, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhang, Y.; Luo, J.; Wang, F.; Cao, X.; Huang, S. Electrochemical Oxidative Oxydihalogenation of Alkynes for the Synthesis of α,α-Dihaloketones. Org. Lett. 2020, 22, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Gütz, C.; Selt, M.; Bäzinger, M.; Bucher, C.; Römelt, C.; Hecken, N.; Gallou, F.; Galvão, T.R.; Waldvogel, S.R. Novel cathode material for cathodic dehalogenation of 1,1,-dibromo cyclopropane derivatives. Chem. Eur. J. 2015, 21, 13878–13882. [Google Scholar] [CrossRef] [PubMed]
- Gütz, C.; Bäzinger, M.; Bucher, C.; Galvão, T.R.; Waldvogel, S.R. Development and scale-up of electrochemical dehalogenation for the synthesis of a key intermediate for NS5A inhibitors. Org. Process Res. Dev. 2015, 19, 1428–1433. [Google Scholar] [CrossRef]
- Gütz, C.; Grimaudo, V.; Holtkamp, M.; Hartmer, M.; Werra, J.; Frensemeier, L.; Kehl, A.; Karst, U.; Broekmann, P.; Waldvogel, S.R. Leaded Bronze—An Innovative Lead Substitute for Cathodic Electrosynthesis. ChemElectroChem 2018, 5, 247–252. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagents 1 | Bulk Price 2 | Molar Price 3 |
|---|---|---|
| KBr | €95/kg | €11.31/mol |
| NaBr | €135/kg | €13.93/mol |
| LiBr | €294/kg | €25.53/mol |
| NH4Br | €220/kg | €21.55/mol |
| NEt4Br | €121/kg | €25.43/mol |
| NBu4Br | €831/kg | €267.89/mol |
| N-Bromosuccinimide | €140/kg | €24.92/mol |
| Pyridinium tribromide | €362/kg | €115.77/mol |
| HBr (48%) | €151/L | €8.20/mol |
| Bromine | €264/L | €13.57/mol * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gombos, L.G.; Waldvogel, S.R. Electrochemical Bromofunctionalization of Alkenes and Alkynes—To Sustainability and Beyond. Sustain. Chem. 2022, 3, 430-454. https://doi.org/10.3390/suschem3040027
Gombos LG, Waldvogel SR. Electrochemical Bromofunctionalization of Alkenes and Alkynes—To Sustainability and Beyond. Sustainable Chemistry. 2022; 3(4):430-454. https://doi.org/10.3390/suschem3040027
Chicago/Turabian StyleGombos, Lilla G., and Siegfried R. Waldvogel. 2022. "Electrochemical Bromofunctionalization of Alkenes and Alkynes—To Sustainability and Beyond" Sustainable Chemistry 3, no. 4: 430-454. https://doi.org/10.3390/suschem3040027
APA StyleGombos, L. G., & Waldvogel, S. R. (2022). Electrochemical Bromofunctionalization of Alkenes and Alkynes—To Sustainability and Beyond. Sustainable Chemistry, 3(4), 430-454. https://doi.org/10.3390/suschem3040027