

Gated Ethidium- and Bleomycin-Loading in Phage T4 That Is Subsequently Purified Leak-Free



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phage T4 Propagation and Initial Purification for Preparative Fractionation
2.2. Phage T4 Preparation for Analytical Fractionation
2.3. Fractionation by Rate Zonal Centrifugation in a Sucrose Gradient
2.4. Native Agarose Gel Electrophoresis
2.5. Electron Microscopy
2.6. Loading of Ethidium and Bleomycin
3. Results
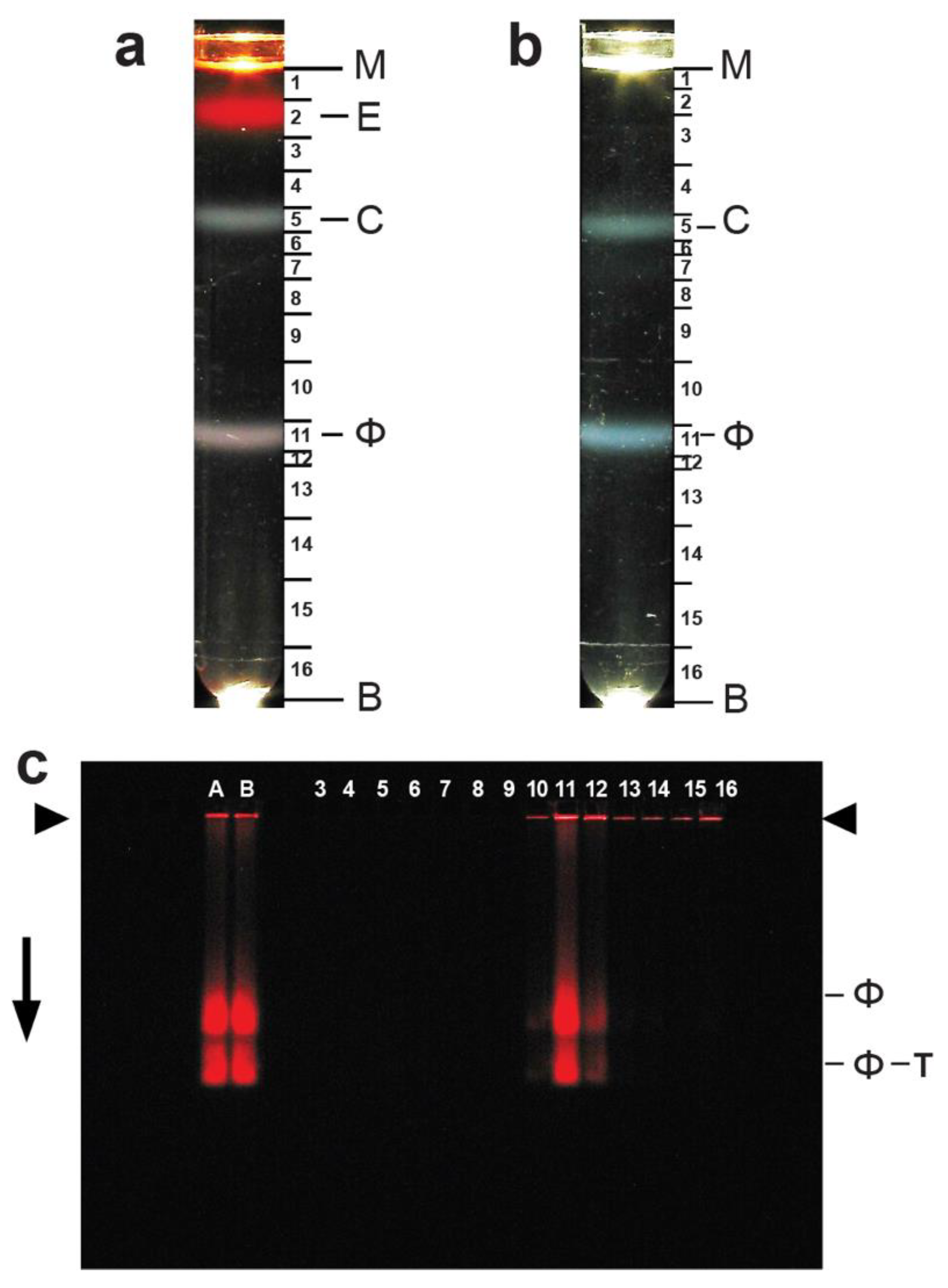
3.1. Production of Phage T4 and Initial T4 Purification
3.2. Ethidium Loading and Purification of Loaded Phage T4
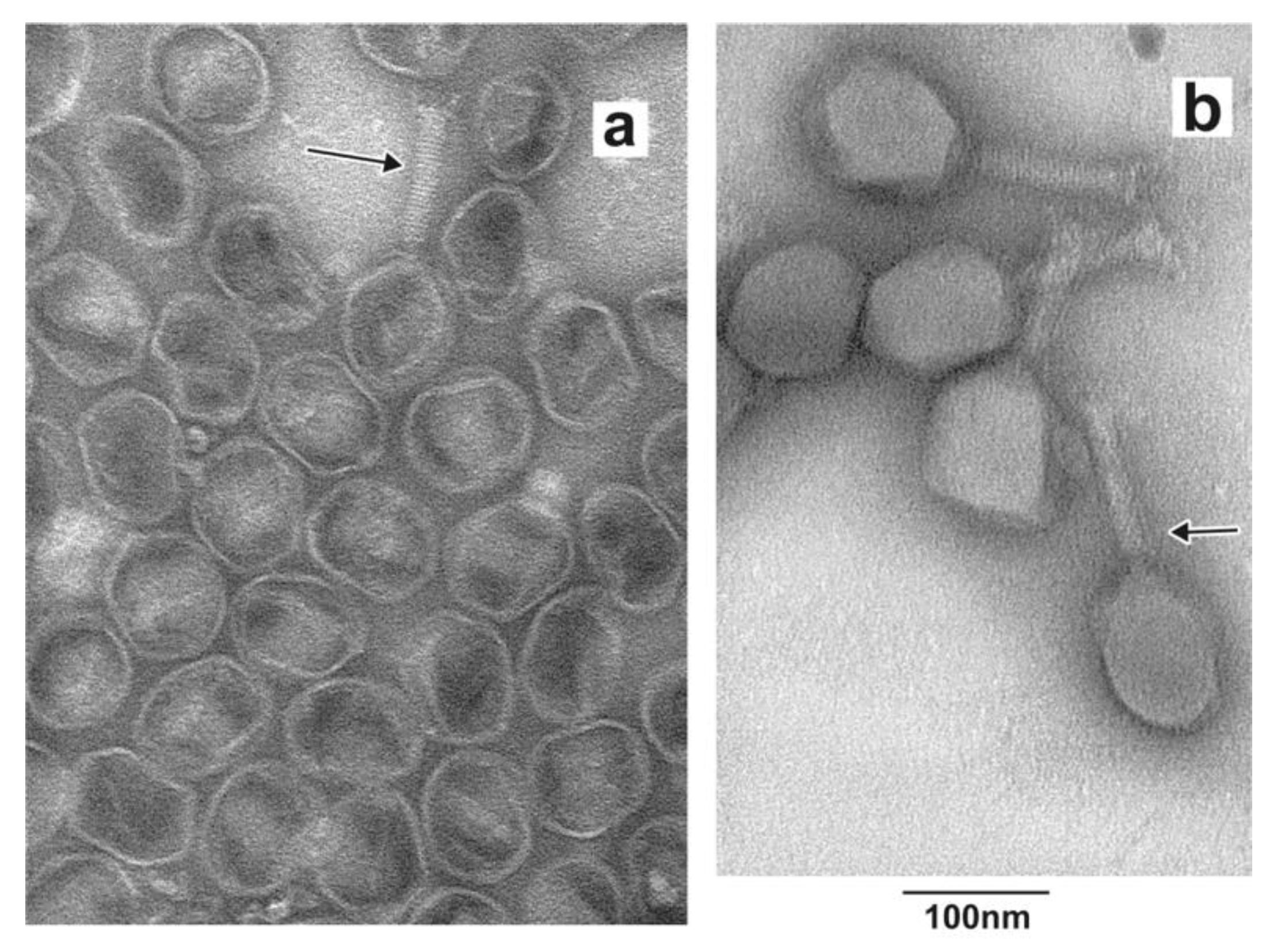
3.3. Identity of Band-Forming Particles: Electron Microscopy
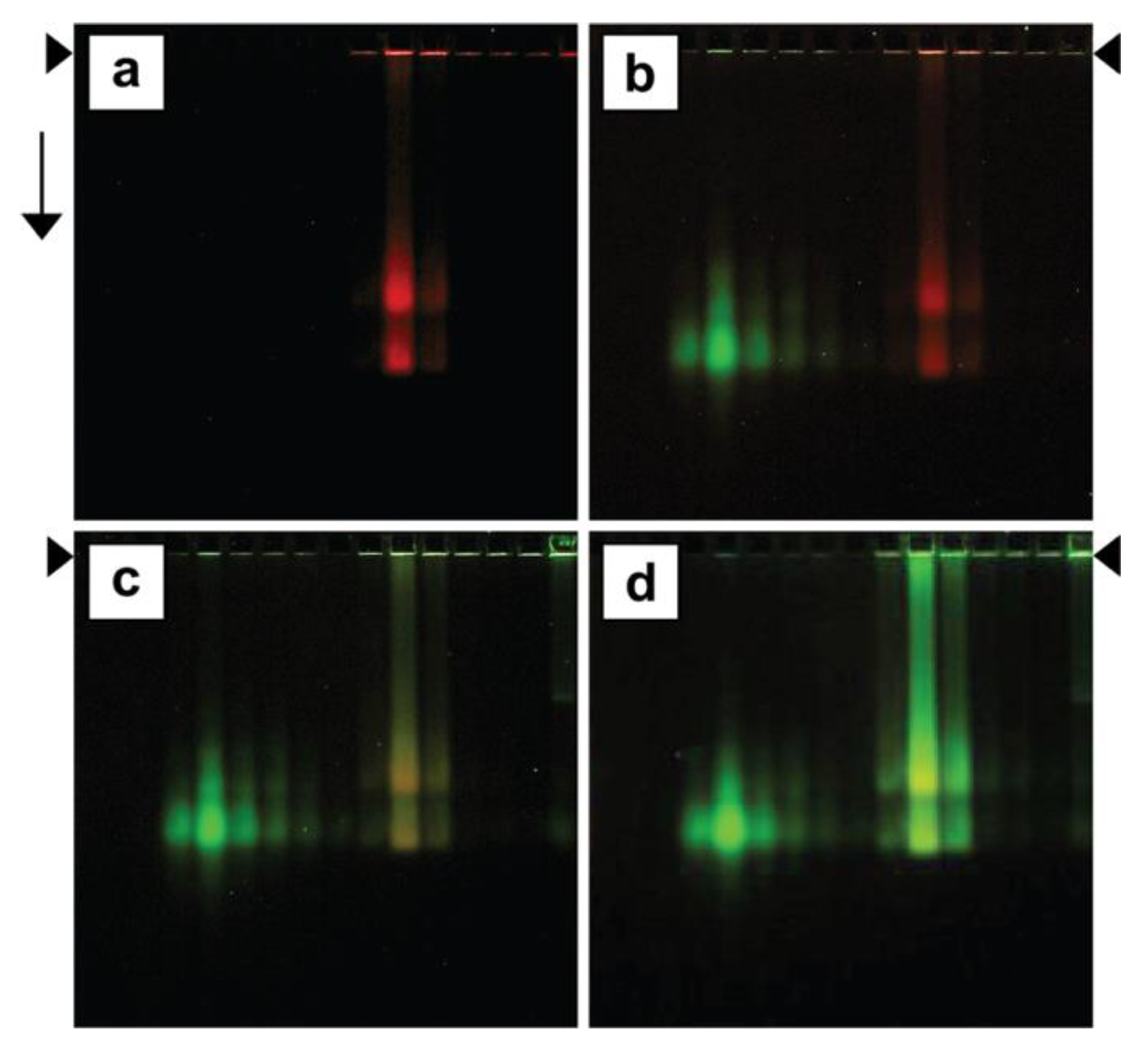
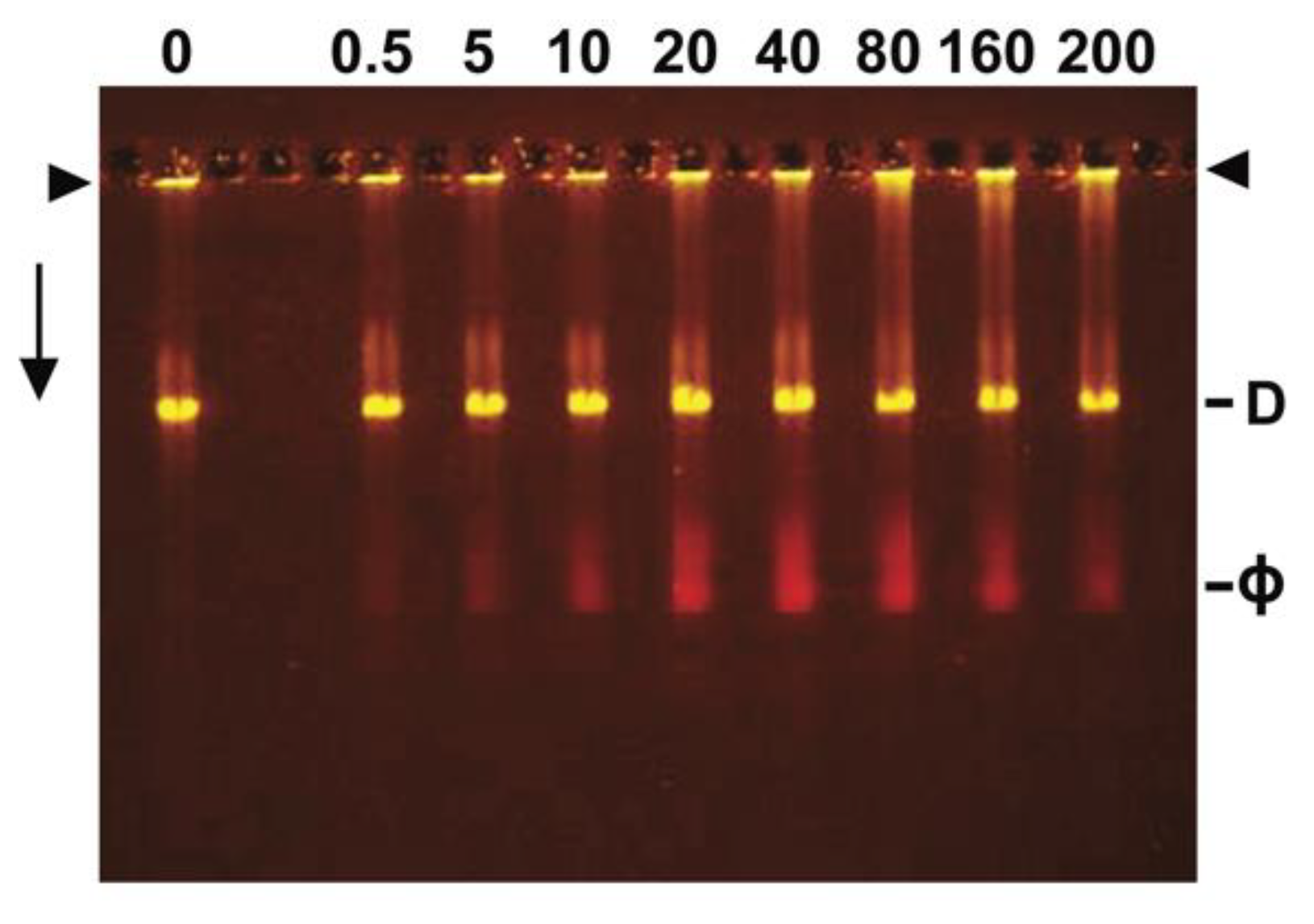
3.4. Ethidium Loading Assayed by Native Agarose Gel Electrophoresis (AGE): Stability of Loading
3.5. Stability of Loading
3.6. Bleomycin Loading
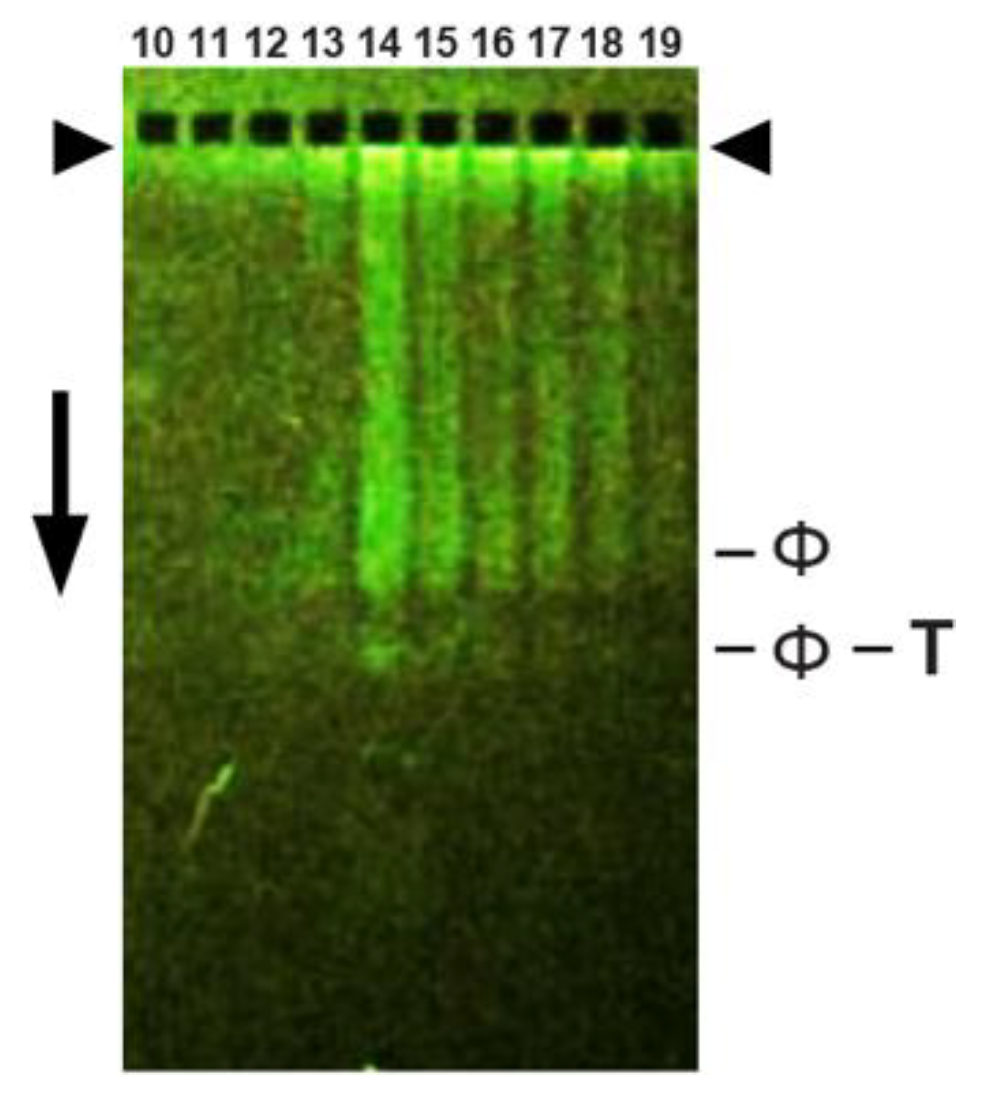
3.7. Loading Details; Analytical AGE without the Sucrose Gradient
4. Discussion
4.1. Advantages of the Preparative and Analytical Procedures Used Here
4.2. Gating of Phage T4: DDV Potential
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, G.T.; Choi, K.C. Advanced new strategies for metastatic cancer treatment by therapeutic stem cells and oncolytic virotherapy. Oncotarget 2016, 7, 58684–58695. [Google Scholar] [CrossRef] [Green Version]
- Leaf, C. The Truth in Small Doses; Simon and Schuster: New York, NY, USA, 2014. [Google Scholar]
- Bergamo, A.; Sava, G. Chemical and molecular approach to tumor metastases. Int. J. Mol. Sci. 2018, 19, 843. [Google Scholar] [CrossRef] [Green Version]
- Mamdouhi, T.; Twomey, J.D.; McSweeney, K.M.; Zhang, B. Fugitives on the run: Circulating tumor cells (CTCs) in metastatic disease. Cancer Metastasis Rev. 2019, 38, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 4 July 2022).
- National Cancer Institute, US. Understanding Cancer. Available online: https://www.cancer.gov/about-cancer/understanding/statistics (accessed on 4 July 2022).
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The different mechanisms of cancer drug resistance: A brief review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The challenge of drug resistance in cancer treat-ment: A current overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Beyaz, H.; Uludag, H.; Kavaz, D.; Rizaner, N. Mechanisms of drug resistance and use of nanoparticle delivery to overcome resistance in breast cancers. Adv. Exp. Med. Biol. 2021, 1347, 163–181. [Google Scholar] [CrossRef]
- Serwer, P.; Wright, E.T.; Liu, Z.; Jiang, W. Length quantization of DNA partially expelled from heads of a bacterio-phage T3 mutant. Virology 2014, 456–457, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Unsoy, G.; Gunduz, U. Smart drug-delivery systems in cancer therapy. Curr. Drug Targets 2018, 19, 202–212. [Google Scholar] [CrossRef]
- Shen, H.; Aggarwal, N.; Wun, K.S.; Lee, Y.S.; Hwang, I.Y.; Chang, M.W. Engineered microbial systems for advanced drug delivery. Adv. Drug Deliv. Rev. 2022, 187, 114364. [Google Scholar] [CrossRef]
- Liu, P.; Chen, G.; Zhang, J. A review of liposomes as a drug delivery system: Current status of approved products, regulatory environments, and future perspectives. Molecules 2022, 27, 1372. [Google Scholar] [CrossRef]
- Anchordoquoy, T.J.; Barenholz, Y.; Boraschi, D.; Chorny, M.; Decuzzi, P.; Dobrovolskaia, M.A.; Farhangrazi, Z.S.; Farrell, D.; Gabizon, A.; Ghandehari, H.; et al. Mechanisms and barriers in cancer nanomedicine: Ad-dressing challenges, looking for solutions. ACS Nano 2017, 11, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Silverman, L.; Barenholz, B. In vitro experiments showing enhanced release of doxorubicin from Doxil® in the presence of ammonia may explain drug release at tumor site. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1841–1850. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Dedrick, R.M.; Schooley, R.T. Phage therapy for antibiotic-resistant bacterial infections. Annu. Rev. Med. 2022, 73, 197–211. [Google Scholar] [CrossRef]
- São-José, C.; Costa, A.R.; Melo, L.D.R. Editorial: Bacteriophages and their lytic enzymes as alternative antibacterial therapies in the age of antibiotic resistance. Front. Microbiol. 2022, 13, 884176. [Google Scholar] [CrossRef]
- Górski, A.; Borysowski, J.; Miȩdzybrodzki, R. Bacteriophage interactions with epithelial cells: Therapeutic implica-tions. Front. Microbiol. 2021, 11, 631161. [Google Scholar] [CrossRef]
- Zaczek, M.; Górski, A.; Szkaradzinska, A.; Lusiak-Szelachowska, M.; Weber-Dabrowska, B. Phage penetration of eukaryotic cells: Practical implications. Future Virol. 2020, 11, 745–760. [Google Scholar] [CrossRef]
- Serwer, P.; Wright, E.T.; Gonzales, C.B. Phage capsids as gated, long-persistence, uniform drug delivery vehicles. In Current and Future Aspects of Nanomedicine [Internet]; Khalil, I.A.H., Ed.; IntechOpen: London, UK, 2020; Available online: https://www.intechopen.com/chapters/70945 (accessed on 4 July 2022). [CrossRef]
- Serwer, P.; Wright, E.T.; De La Chapa, J.; Gonzales, C.B. Basics for improved use of phages for therapy. Antibiotics 2021, 10, 723. [Google Scholar] [CrossRef]
- Wood, W.B.; Revel, H.R. The genome of bacteriophage T4. Bacteriol. Rev. 1976, 40, 847–868. [Google Scholar] [CrossRef]
- Griess, G.A.; Khan, S.A.; Serwer, P. Variation of the permeability of bacteriophage T4: Analysis by use of a protein-specific probe for the T4 interior. Biopolymers 1991, 31, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Bull, H.B. Introduction to Physical Biochemistry, 2nd ed.; F.A. Davis Company: Philadelphia, PA, USA, 1971; Chapter 10. [Google Scholar]
- LePecq, J.B.; Paoletti, C. A fluorescent complex between ethidium bromide and nucleic acids. Physical-chemical characterization. J. Mol. Biol. 1967, 27, 87–106. [Google Scholar] [CrossRef]
- Motlagh, N.S.; Parvin, P.; Ghasemi, F.; Atyabi, F. Fluorescence properties of several chemotherapy drugs: Doxorubi-cin, paclitaxel and bleomycin. Biomed. Opt. Express 2016, 7, 2400–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkinson, J.S.; Huskey, R.J. Deletion mutants of bacteriophage lambda. I. Isolation and initial characterization. J. Mol. Biol. 1971, 56, 369–384. [Google Scholar] [CrossRef]
- Griess, G.A.; Serwer, P.; Horowitz, P.M. Binding of ethidium to bacteriophage T7 and T7 deletion mutants. Biopolymers 1985, 24, 1635–1646. [Google Scholar] [CrossRef]
- Serwer, P.; Wright, E.T. ATP-driven contraction of phage T3 capsids with DNA incompletely packaged in vivo. Viruses 2017, 9, 119. [Google Scholar] [CrossRef] [Green Version]
- Wiehe, A.; O’Brien, J.M.; Senge, M.O. Trends and targets in antiviral phototherapy. Photochem. Photobiol. Sci. 2019, 18, 2565–2612. [Google Scholar] [CrossRef]
- Bartolomeu, M.; Oliveira, C.; Pereira, C.; Neves, M.G.P.M.S.; Faustino, M.A.F.; Almeida, A. Antimicrobial photody-namic approach in the inactivation of viruses in wastewater: Influence of alternative adjuvants. Antibiotics 2021, 10, 767. [Google Scholar] [CrossRef]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Rüge, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef]
- Kuhn, A.; Thomas, J.A. The beauty of bacteriophage T4 research: Lindsay W. Black and the T4 head assembly. Viruses 2022, 14, 700. [Google Scholar] [CrossRef]
- Wood, W.B.; Conley, M.P. Attachment of tail fibers in bacteriophage T4 assembly: Role of the phage whiskers. J. Mol. Biol. 1979, 127, 15–29. [Google Scholar] [CrossRef]
- Hyman, P.; van Raaij, M. Bacteriophage T4 long tail fiber domains. Biophys. Rev. 2018, 10, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, H.A.; Gorin, M.H.; Ponder, E. Electrophoresis and the chemistry of cell surfaces. Cold Spring Harb. Symp. Quant. Biol. 1940, 8, 72–79. [Google Scholar] [CrossRef]
- Bachrach, U.; Friedmann, A. Practical procedures for the purification of bacterial viruses. Appl. Microbiol. 1971, 22, 706–715. [Google Scholar] [CrossRef]
- Medscape. Bleomycin (Rx). Available online: https://reference.medscape.com/drug/bleomycin-342113 (accessed on 8 September 2022).
- Drugs.com. Bleomycin Dosage. Available online: https://www.drugs.com/dosage/bleomycin.html (accessed on 8 September 2022).
- Dawson, N.J. The surface-area/body-weight relationship in mice. Aust. J. Biol. Sci. 1967, 20, 687–690. [Google Scholar] [CrossRef]
- Heller, D.P.; Greenstock, C.L. Fluorescence lifetime analysis of DNA intercalated ethidium bromide and quenching by free dye. Biophys. Chem. 1994, 50, 305–312. [Google Scholar] [CrossRef]
- Nasukawa, T.; Uchiyama, J.; Taharaguchi, S.; Ota, S.; Ujihara, T.; Matsuzaki, S.; Murakami, H.; Mizukami, K.; SSakaguchi, M. Virus purification by CsCl density gradient using general centrifugation. Arch. Virol. 2017, 162, 3523–3528. [Google Scholar] [CrossRef]
- Liu, D.; Van Belleghem, J.D.; de Vries, C.R.; Burgener, E.; Chen, Q.; Manasherob, R.; Aronson, J.R.; Amanatullah, D.F.; Tamma, P.D.; Suh, G.A. The safety and toxicity of phage therapy: A review of animal and clinical studies. Viruses 2021, 13, 1268. [Google Scholar] [CrossRef]
- Serwer, P.; Masker, W.E.; Allen, J.L. Stability and in vitro DNA packaging of bacteriophages: Effects of dextrans, sugars, and polyols. J. Virol. 1983, 45, 665–671. [Google Scholar] [CrossRef]
- Leibo, S.P.; Kellenberger, E.; Kellenberger-van der Kamp, C.; Frey, T.G.; Steinberg, C.M. Gene 24-controlled osmotic shock resistance in bacteriophage T4: Probable multiple gene functions. J. Virol. 1979, 30, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Leibo, S.P.; Mazur, P. Effect of osmotic shock and low salt concentration on survival and density of bacteriophages T4B and T4Bo1. Biophys. J. 1966, 6, 747–772. [Google Scholar] [CrossRef] [Green Version]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine 2008, 3, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Huang, G.; Li, Y.; Yang, S.; Ramezani, S.; Lin, Z.; Wang, Y.; Ma, X.; Zeng, Z.; Luo, M.; et al. A transistor-like pH nanoprobe for tumour detec-tion and image-guided surgery. Nat. Biomed. Eng. 2016, 1, 0006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodyra-Stefaniak, K.; Miernikiewicz, P.; Drapała, J.; Drab, M.; Jończyk-Matysiak, E.; Lecion, D.; Kaźmierczak, Z.; Beta, W.; Majewska, J.; Harhala, M.; et al. Mammalian host-versus-phage immune response determines phage fate in vivo. Sci. Rep. 2015, 5, 14802. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Ananthaswamy, N.; Jain, S.; Batra, H.; Tang, W.C.; Lewry, D.A.; Richards, M.L.; David, S.A.; Kilgore, P.B.; Sha, J.; et al. A universal bacteriophage T4 nanoparticle platform to design multiplex SARS-CoV-2 vaccine candidates by CRISPR engineering. Sci. Adv. 2021, 7, eabh1547. [Google Scholar] [CrossRef]
- Zhu, J.; Tao, P.; Mahalingam, M.; Sha, J.; Kilgore, P.; Chopra, A.K.; Rao, V. A prokaryotic-eukaryotic hybrid viral vector for delivery of large cargos of genes and proteins into human cells. Sci. Adv. 2019, 5, eaax0064. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serwer, P.; Wright, E.T. Gated Ethidium- and Bleomycin-Loading in Phage T4 That Is Subsequently Purified Leak-Free. Biophysica 2022, 2, 366-380. https://doi.org/10.3390/biophysica2040033
Serwer P, Wright ET. Gated Ethidium- and Bleomycin-Loading in Phage T4 That Is Subsequently Purified Leak-Free. Biophysica. 2022; 2(4):366-380. https://doi.org/10.3390/biophysica2040033
Chicago/Turabian StyleSerwer, Philip, and Elena T. Wright. 2022. "Gated Ethidium- and Bleomycin-Loading in Phage T4 That Is Subsequently Purified Leak-Free" Biophysica 2, no. 4: 366-380. https://doi.org/10.3390/biophysica2040033
APA StyleSerwer, P., & Wright, E. T. (2022). Gated Ethidium- and Bleomycin-Loading in Phage T4 That Is Subsequently Purified Leak-Free. Biophysica, 2(4), 366-380. https://doi.org/10.3390/biophysica2040033