
Investigation of the Impact of Lipid Acyl Chain Saturation on Fusion Peptide Interactions with Lipid Bilayers



Abstract
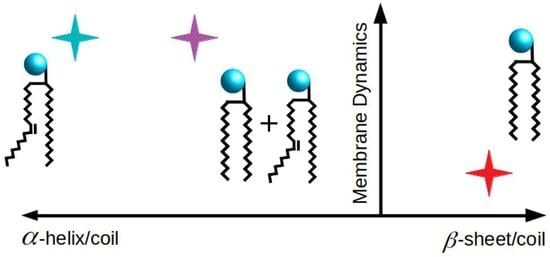
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Vesicle Preparation
2.3. CD Spectroscopy
2.4. Small-Angle Neutron Scattering Experiments
2.5. Small-Angle Neutron Scattering Data Analysis
2.6. Neutron Spin Echo Spectroscopy
2.7. Neutron Spin Echo Spectroscopy Data Analysis
2.8. Molecular Dynamics Simulations
3. Results
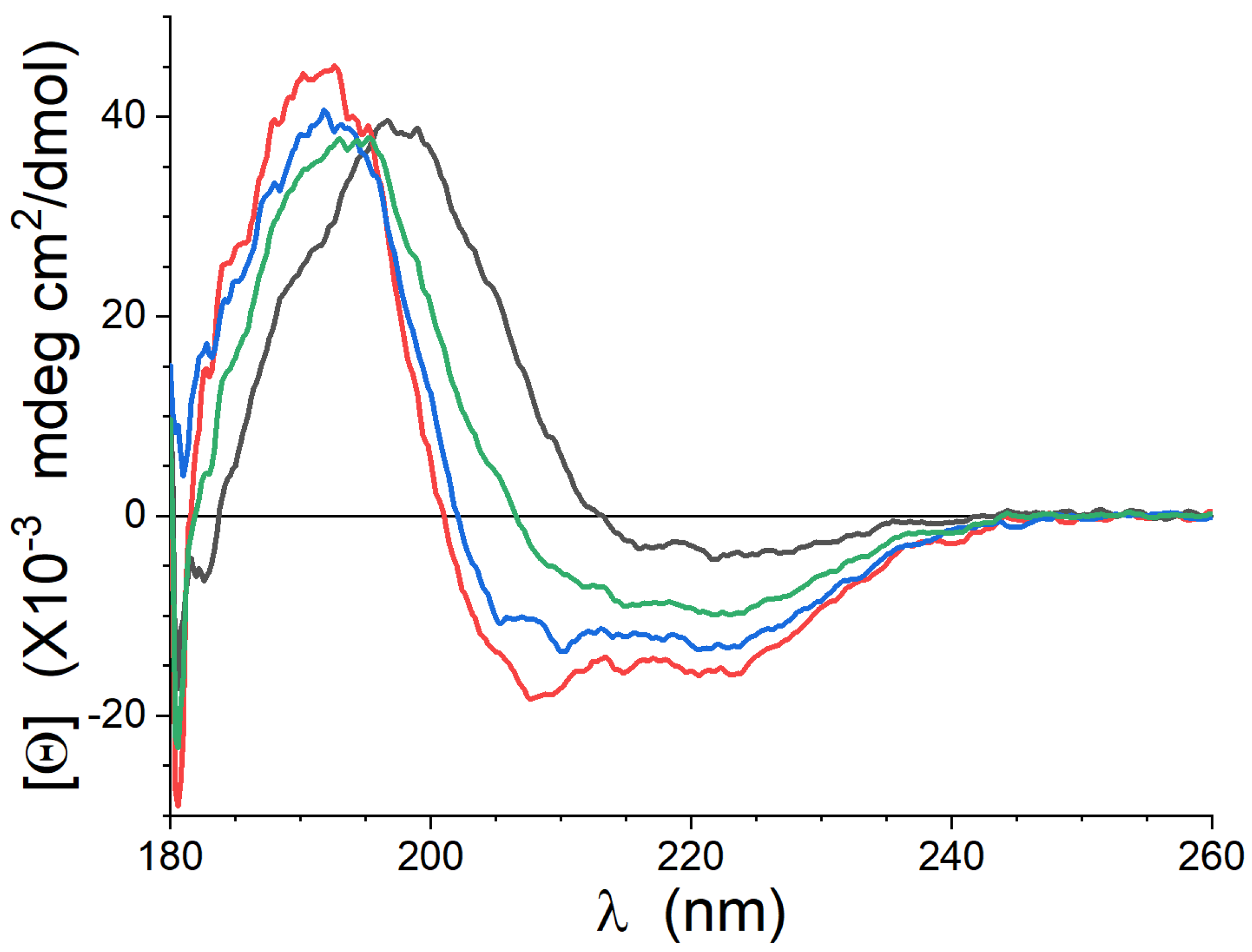
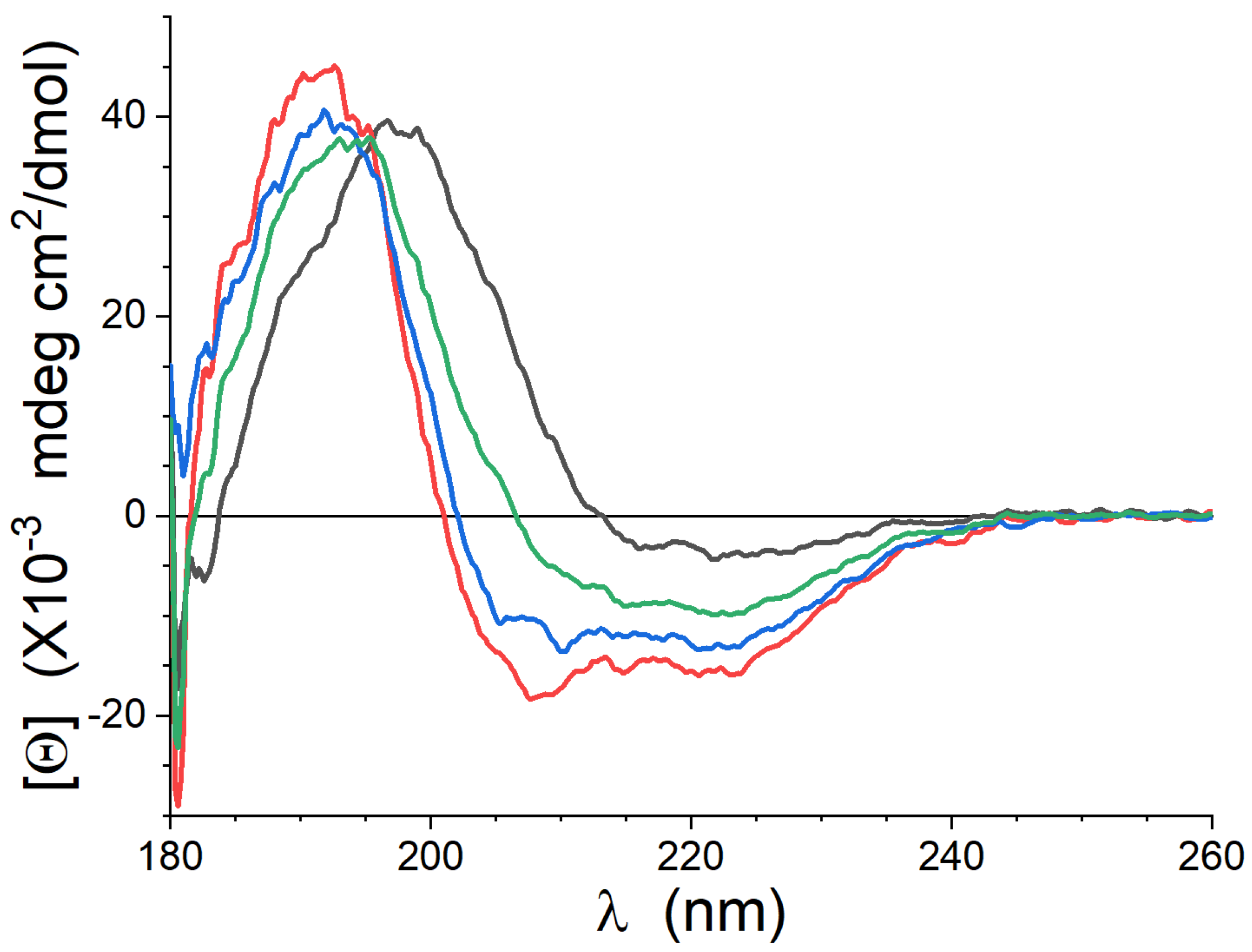
3.1. CD
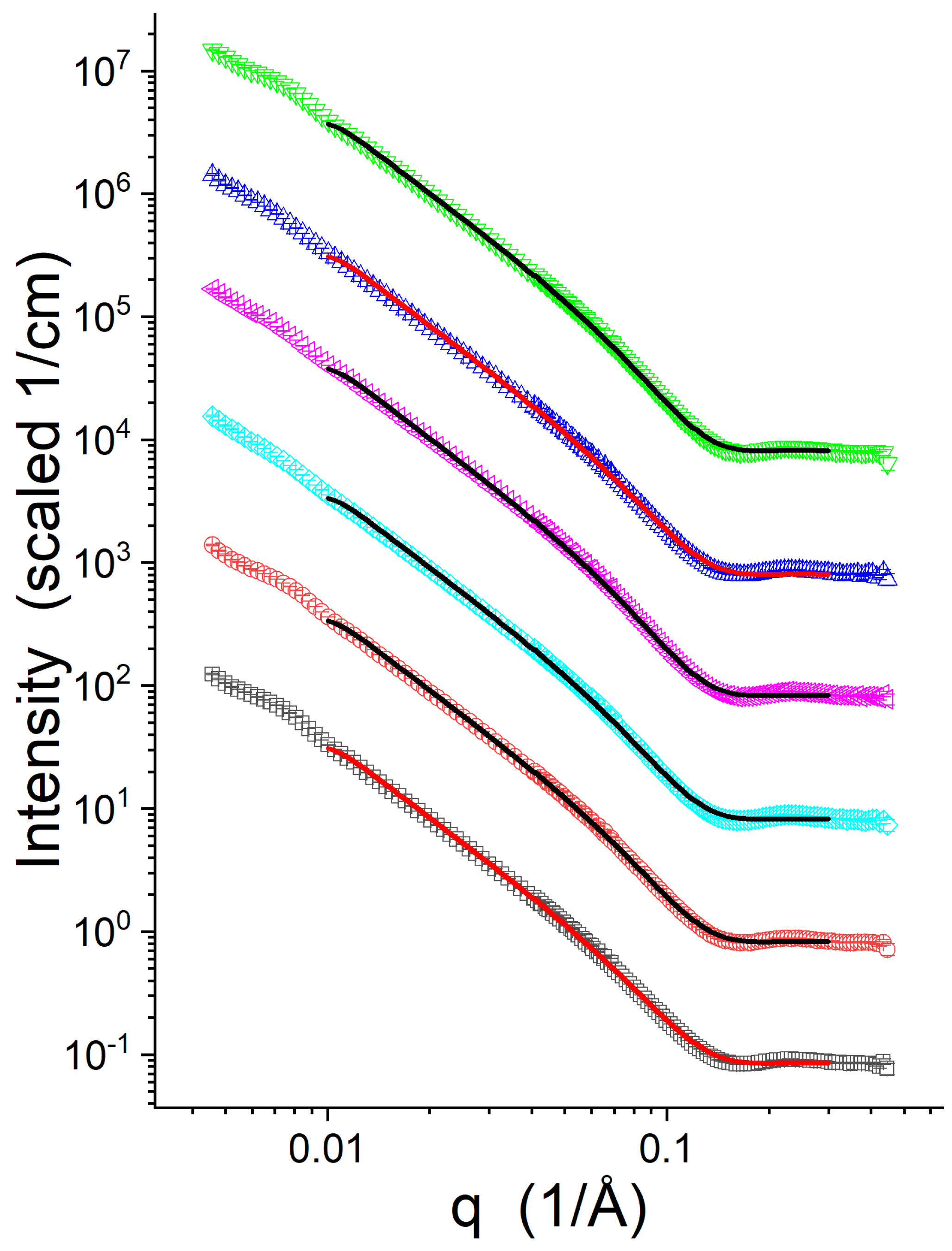
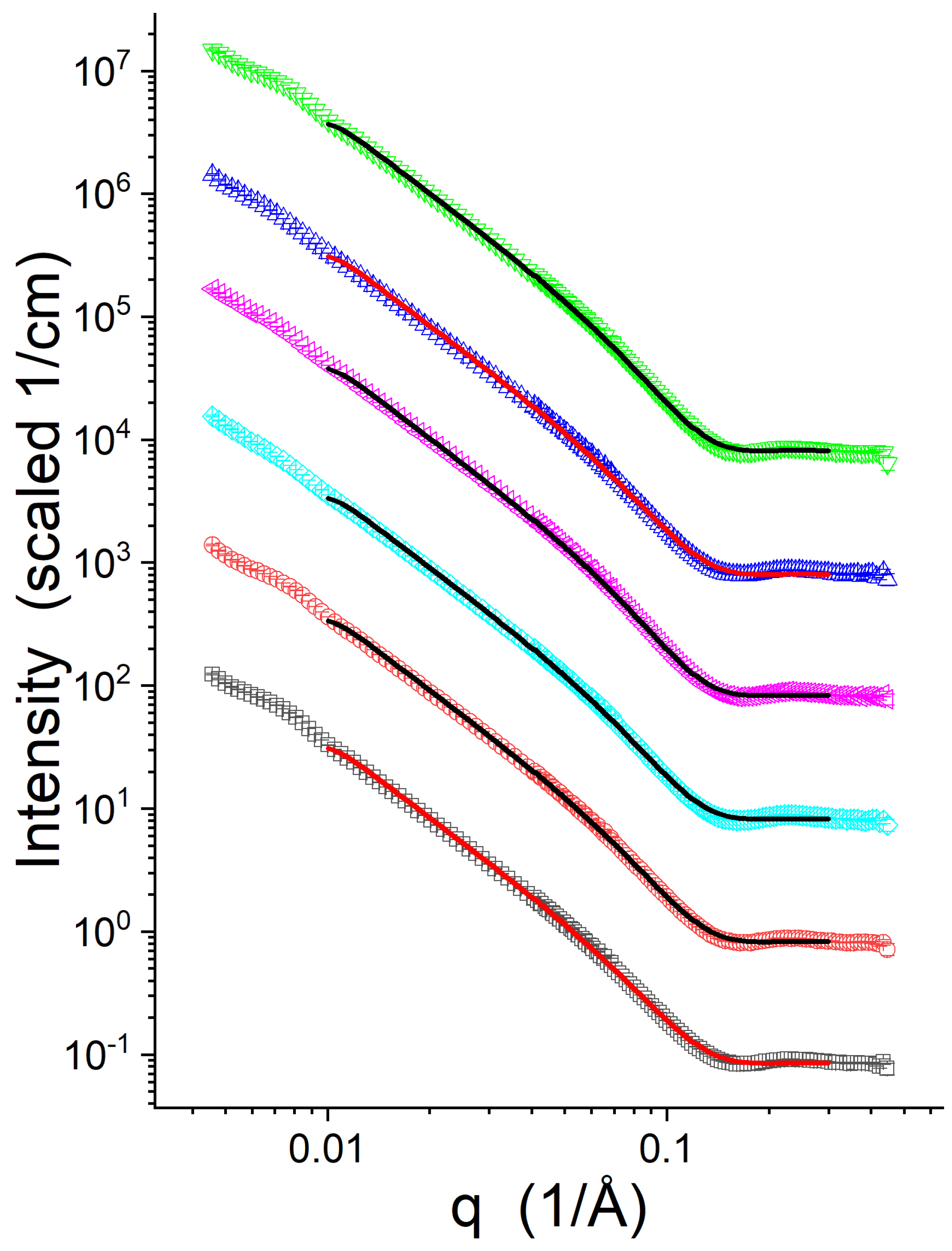
3.2. SANS
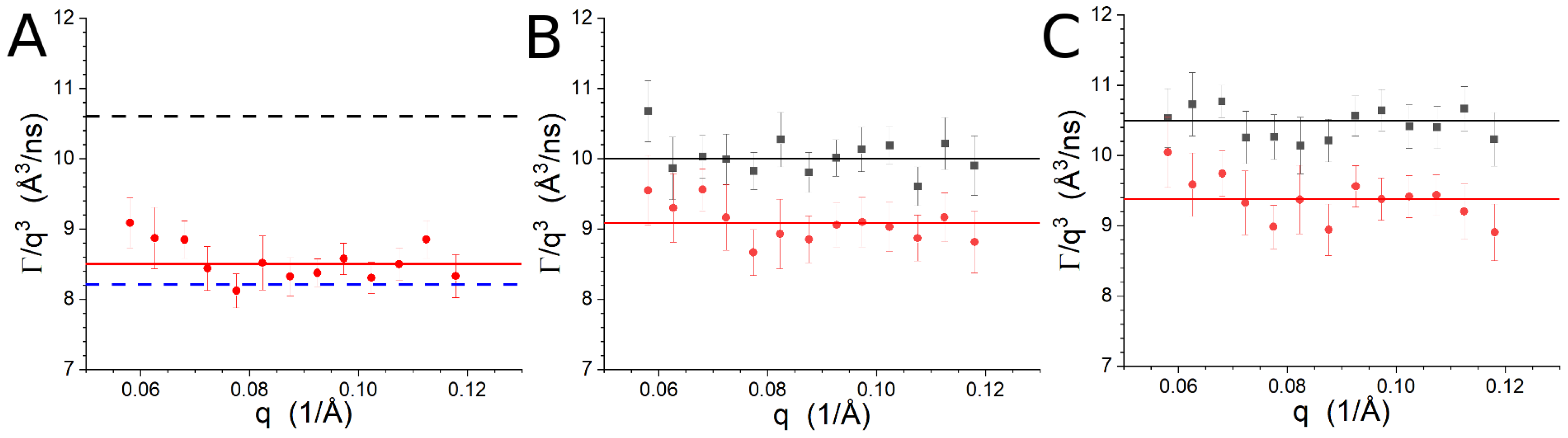
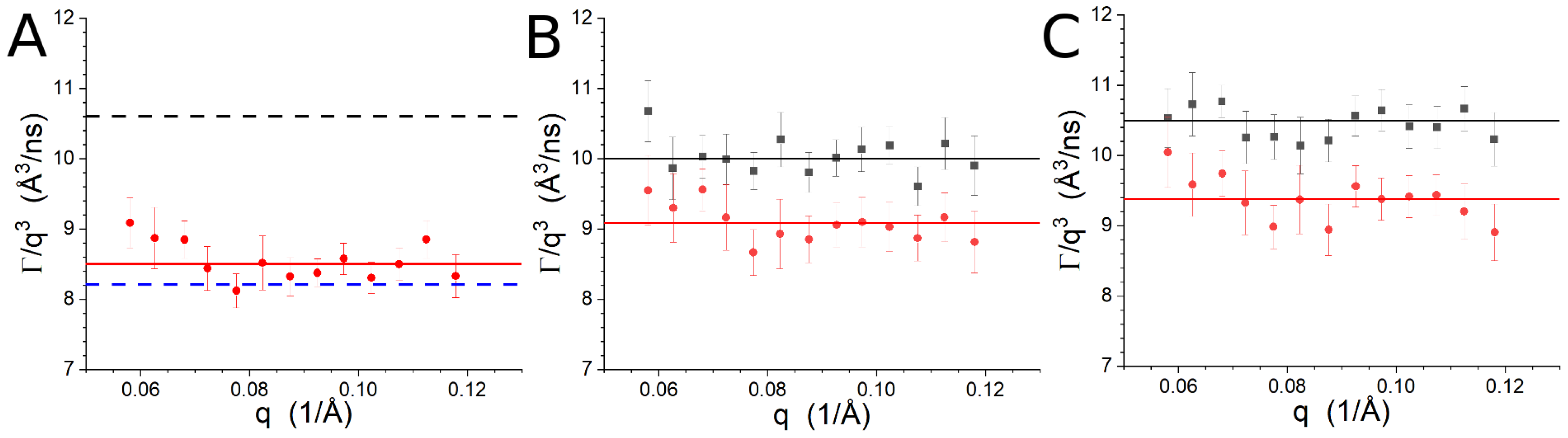
3.3. NSE
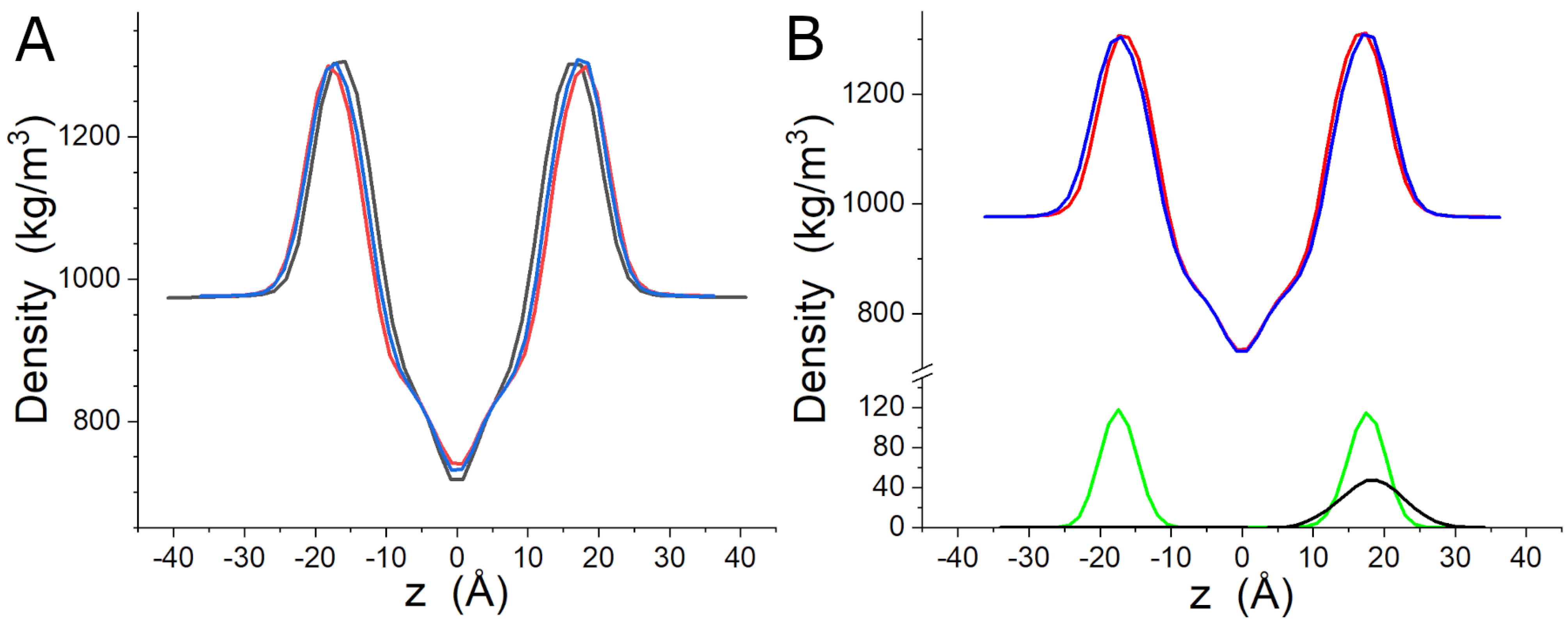
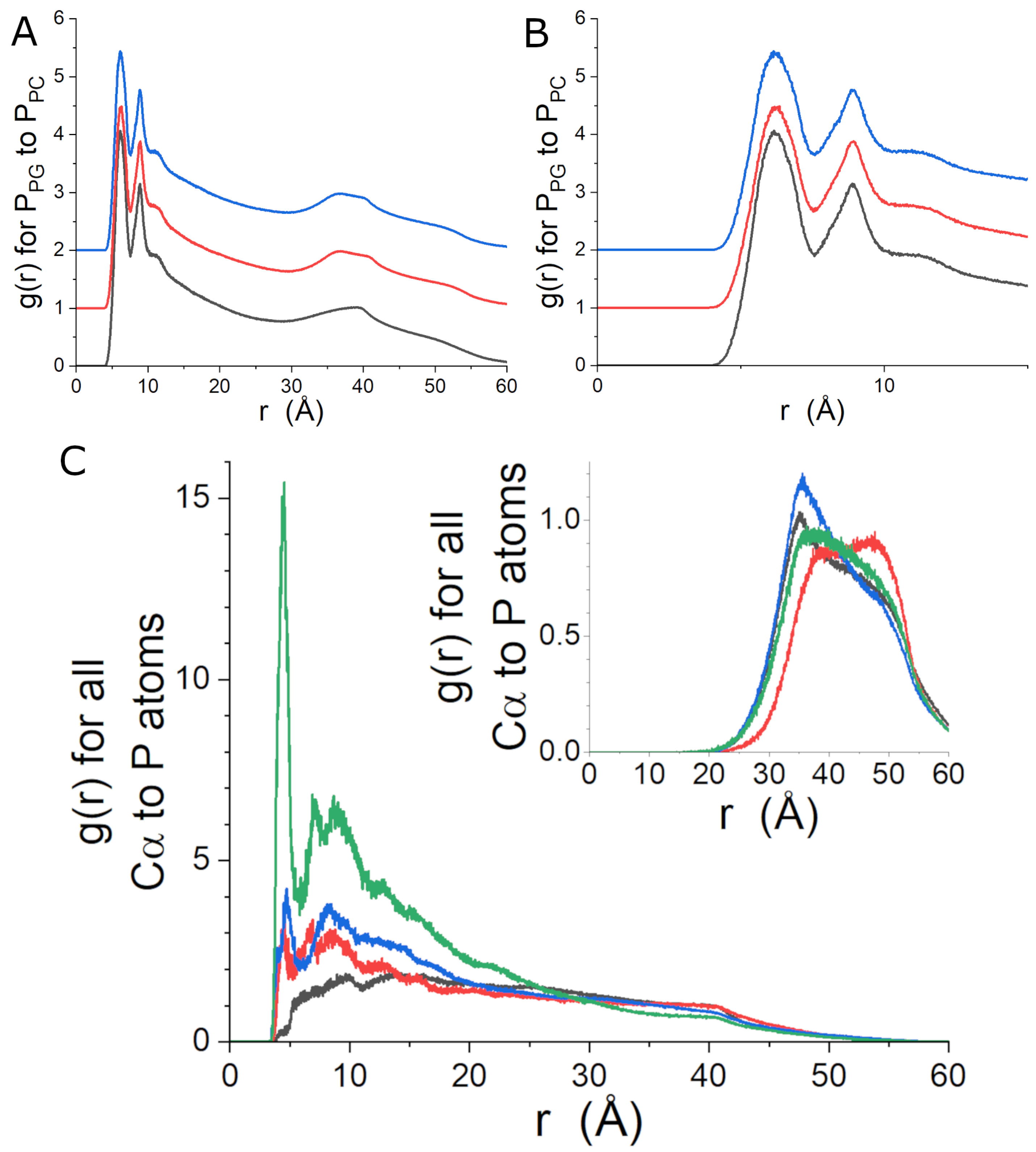
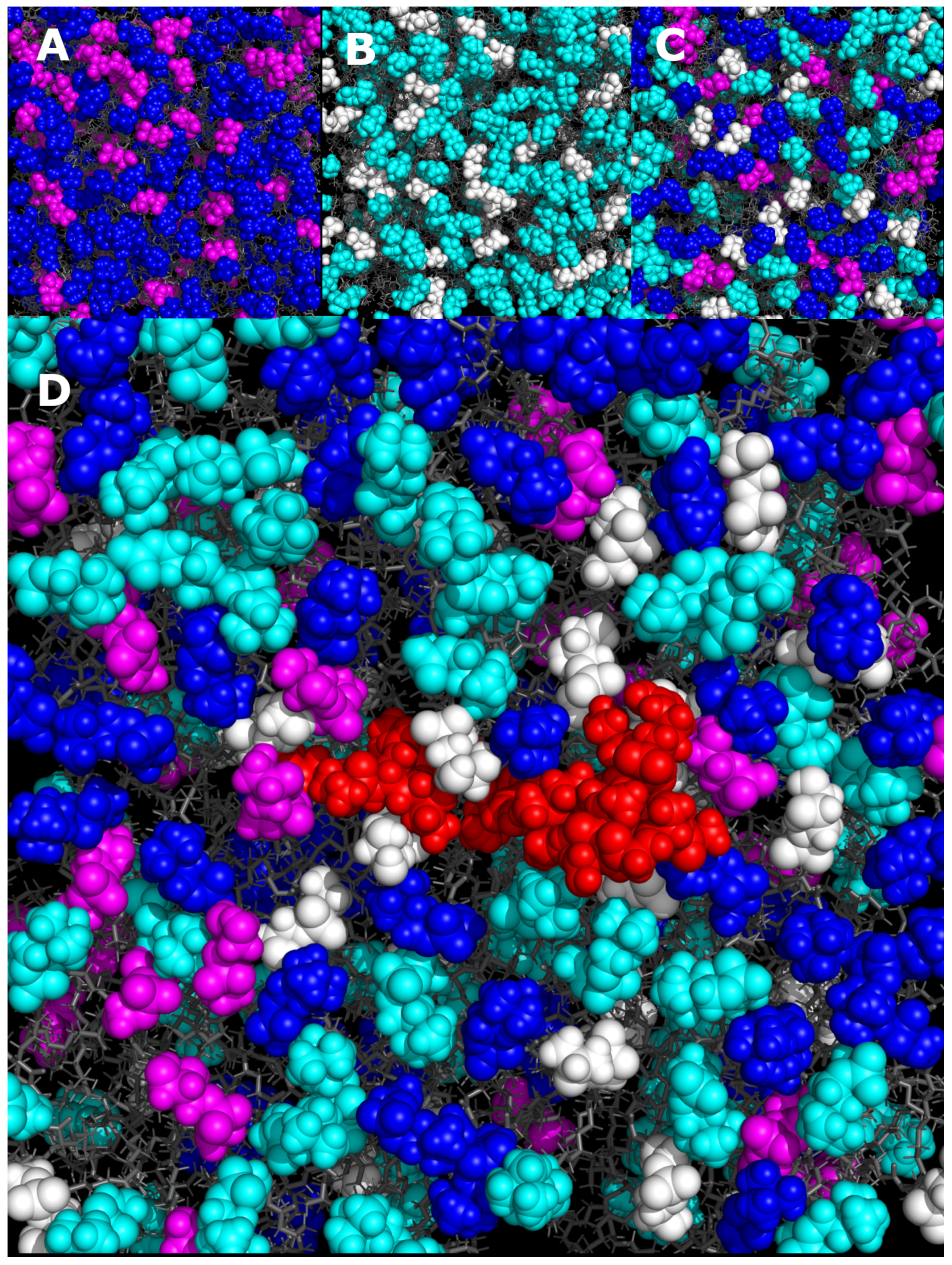
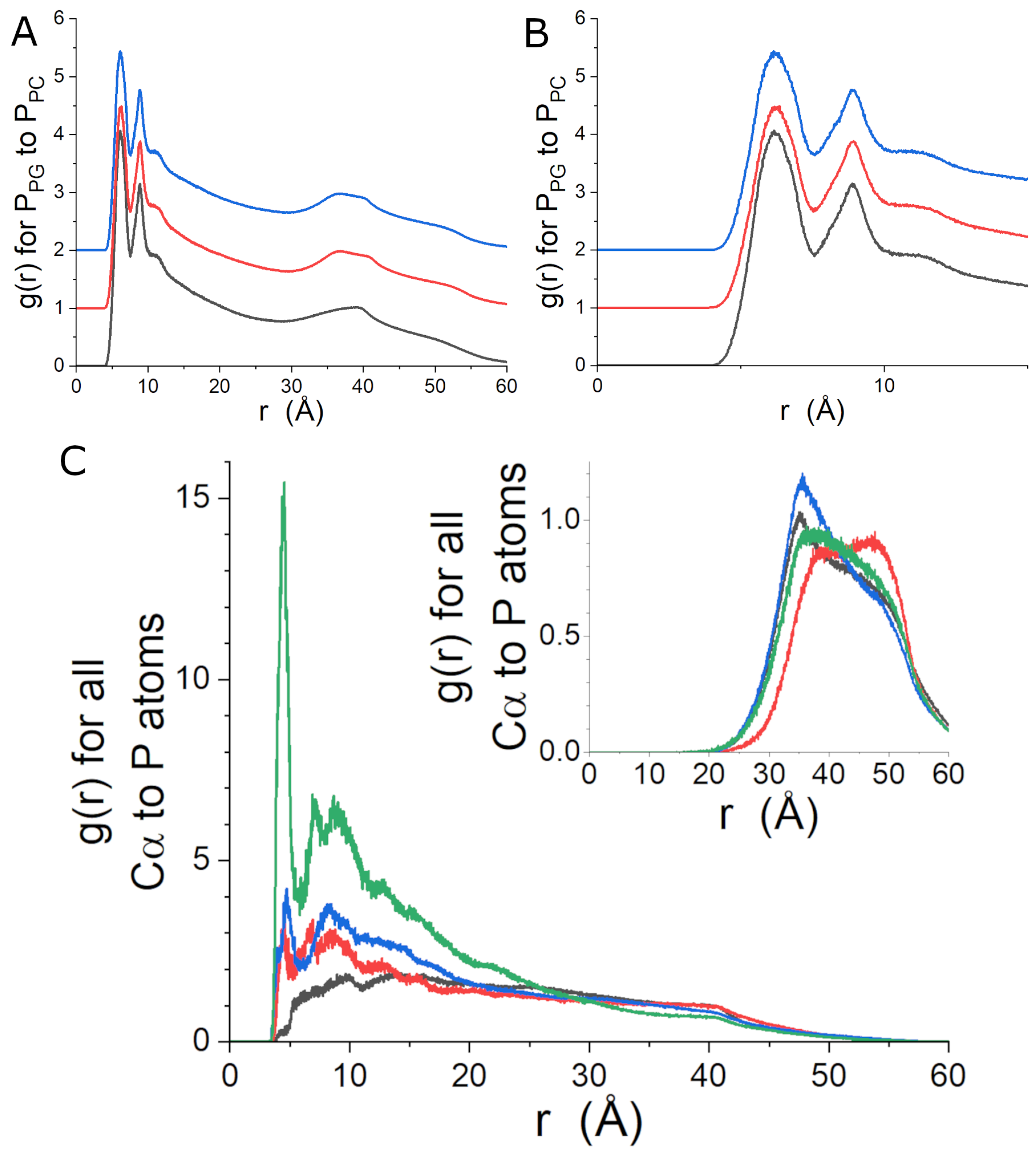
3.4. MD Simulations
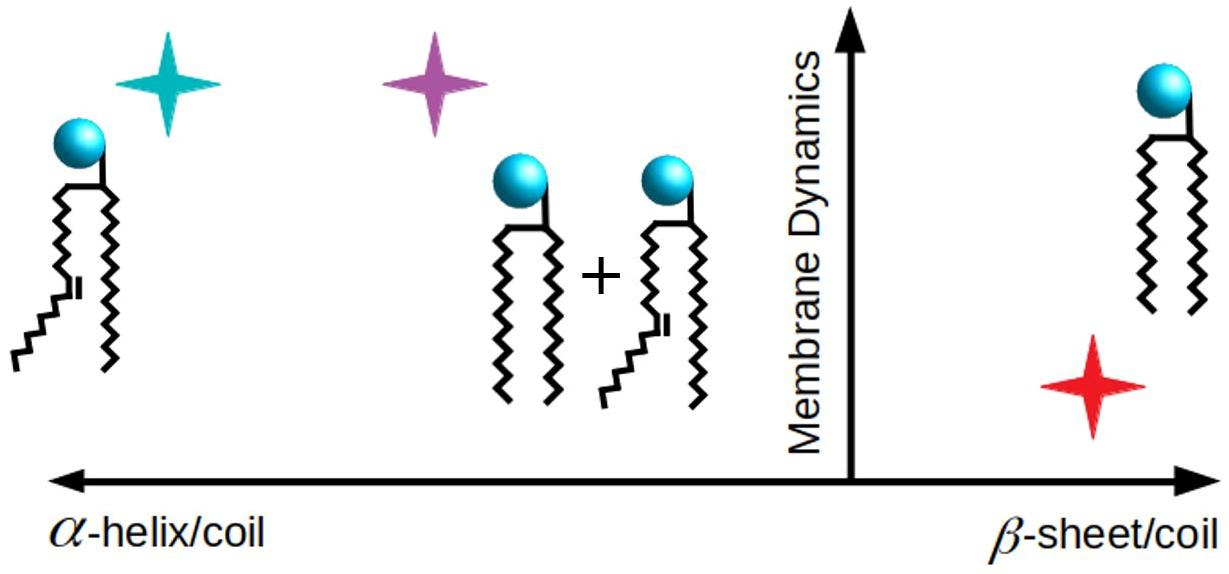
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FP | fusion peptide |
| SANS | small-angle neutron scattering |
| NSE | neutron spin echo spectroscopy |
| CD | circular dichroism |
| MD | molecular dynamics |
| PME | particle mesh Ewald |
| RDF | radial density function |
| RMSF | root mean square fluctuation |
| MSD | mean square displacement |
| DMPC | 1,2-dimyristoyl-sn-glycero-3-phosphocholine |
| DMPG | 1,2-dimyristoyl-sn-glycero-3-phospho-(1’-rac-glycerol) sodium salt |
| POPC | 1-palmityol-2-oleoyl-sn-glycero-3-phosphocholine |
| POPG | 1-palmityol-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) sodium salt |
| POPS | 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine sodium salt |
| DOPC | 1,2-dioleoyl-sn-glycero-3-phosphocholine |
| diCPC | 1,2-dierucoyl-sn-glycero-3-phosphocholine |
| TFE | 2,2,2-trifluoroethanol |
References
- Freed, E.O.; Martin, M.A. The Role of Human-Immunodeficiency-Virus Type-1 Envelope Glycoproteins in Virus Infection. J. Biol. Chem. 1995, 270, 23883–23886. [Google Scholar] [CrossRef] [Green Version]
- Rafalski, M.; Lear, J.D.; Degrado, W.F. Phospholipid Interactions of Synthetic Peptides Representing the N-Terminus of Hiv Gp41. Biochemistry 1990, 29, 7917–7922. [Google Scholar] [CrossRef]
- Nieva, J.L.; Nir, S.; Muga, A.; Goni, F.M.; Wilschut, J. Interaction of the HIV-1 Fusion Peptide with Phospholipid Vesicles: Different Structural Requirements for Fusion and Leakage. Biochemistry 1994, 33, 3201–3209. [Google Scholar] [CrossRef]
- Martin, I.; Schaal, H.; Scheid, A.; Ruysschaert, J.M. Lipid Membrane Fusion Induced by the Human Immunodeficiency Virus Type 1 GP41 N-Terminal Extremity is Determined by its Orientation in the Lipid Bilayer. J. Virol. 1996, 70, 298–304. [Google Scholar] [CrossRef] [Green Version]
- Pereira, F.B.; Goni, F.M.; Muga, A.; Nieva, J.L. Permeabilization and Fusion of Uncharged Lipid Vesicles Induced by the HIV-1 Fusion Peptide Adopting an Extended Conformation: Dose and Sequence Effects. Biophys. J. 1997, 73, 1977–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, W.T.; Rai, D.K. Changes in Lipid Bilayer Structure Caused by the Helix-to-Sheet Transition of an HIV-1 GP41 Fusion Peptide Derivative. Chem. Phys. Lipids 2017, 203, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, W.T.; Zolnierczuk, P.A. The Helix-to-Sheet Transition of an HIV-1 Fusion Peptide Derivative Changes the Mechanical Properties of Lipid Bilayer Membranes. Biochim. Biophys. Acta-Biomembr. 2019, 1861, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Mobley, P.W.; Waring, A.J.; Sherman, M.A.; Gordon, L.M. Membrane Interactions of the Synthetic N-Terminal Peptide of HIV-1 GP41 and its Structural Analogs. Biochim. Biophys. Acta-Biomembr. 1999, 1418, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Parkanzky, P.D.; Khunte, B.A.; Canlas, C.G.; Yang, R.; Gabrys, C.M.; Weliky, D.P. Solid State NMR Measurements of Conformation and Conformational Distributions in the Membrane-Bound HIV-1 Fusion Peptide. J. Mol. Graph. Model. 2001, 19, 129–135. [Google Scholar] [CrossRef]
- Qiang, W.; Yang, J.; Weliky, D.P. Solid-State Nuclear Magnetic Resonance Measurements of HIV Fusion Peptide to Lipid Distances Reveal the Intimate Contact of Beta Strand Peptide with Membranes and the Proximity of the Ala-14-Gly-16 Region with Lipid Headgroups. Biochemistry 2007, 46, 4997–5008. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.H.; Liang, S.; Sackett, K.; Xie, L.; Ghosh, U.; Weliky, D.P. REDOR Solid-State NMR as a Probe of the Membrane Locations of Membrane-Associated Peptides and Proteins. J. Magn. Reson. 2015, 253, 154–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiang, W.; Weliky, D.P. HIV Fusion Peptide and Its Cross-Linked Oligomers: Efficient Syntheses, Significance of the Trimer in Fusion Activity, Correlation of beta Strand Conformation with Membrane Cholesterol, and Proximity to Lipid Headgroups. Biochemistry 2009, 48, 289–301. [Google Scholar] [CrossRef] [Green Version]
- Heller, W.T. A Small-Angle Neutron Scattering Study of the Physical Mechanism that Drives the Action of a Viral Fusion Peptide. Chem. Phys. Lipids 2021, 234, 105022. [Google Scholar] [CrossRef]
- Lai, A.L.; Moorthy, A.E.; Li, Y.L.; Tamm, L.K. Fusion Activity of HIV GP41 Fusion Domain Is Related to Its Secondary Structure and Depth of Membrane Insertion in a Cholesterol-Dependent Fashion. J. Mol. Biol. 2012, 418, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Lai, A.L.; Freed, J.H. HIV GP41 Fusion Peptide Increases Membrane Ordering in a Cholesterol-Dependent Fashion. Biophys. J. 2014, 106, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Gabrys, C.M.; Weliky, D.P. Solid-State Nuclear Magnetic Resonance Evidence for an Extended β Strand Conformation of the Membrane-Bound HIV-1 Fusion Peptide. Biochemistry 2001, 40, 8126–8137. [Google Scholar] [CrossRef]
- Yang, J.; Weliky, D.P. Solid-State Nuclear Magnetic Resonance Evidence for Parallel and Antiparallel Strand Arrangements in the Membrane-Associated HIV-1 Fusion Peptide. Biochemistry 2003, 42, 11879–11890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasniewski, C.M.; Parkanzky, P.D.; Bodner, M.L.; Weliky, D.P. Solid-State Nuclear Magnetic Resonance Studies of HIV and Influenza Fusion Peptide Orientations in Membrane Bilayers using Stacked Glass Plate Samples. Chem. Phys. Lipids 2004, 132, 89–100. [Google Scholar] [CrossRef]
- Sackett, K.; Shai, Y. The HIV Fusion Peptide Adopts Intermolecular Parallel Beta-Sheet Structure in Membranes when Stabilized by the Adjacent N-Terminal Heptad Repeat: A C-13 FTIR Study. J. Mol. Biol. 2005, 350, 790–805. [Google Scholar] [CrossRef] [PubMed]
- Buzón, V.; Cladera, J. Effect of Cholesterol on the Interaction of the HIV GP41 Fusion Peptide with Model Membranes. Importance of the Membrane Dipole Potential. Biochemistry 2006, 45, 15768–15775. [Google Scholar] [CrossRef]
- Bodner, M.L.; Gabrys, C.M.; Struppe, J.O.; Weliky, D.P. C-13-C-13 and N-15-C-13 Correlation Spectroscopy of Membrane-Associated and Uniformly Labeled Human Immunodeficiency Virus and Influenza Fusion Peptides: Amino Acid-Type Assignments and Evidence for Multiple Conformations. J. Chem. Phys. 2008, 128, 052319. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Bodner, M.L.; Weliky, D.P. Solid-State NMR Spectroscopy of Human Immunodeficiency Virus Fusion Peptides Associated with Host-Cell-like Membranes: 2D Correlation Spectra and Distance Measurements Support a Fully Extended Conformation and Models for Specific Antiparallel Strand Registries. J. Am. Chem. Soc. 2008, 130, 5459–5471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sackett, K.; Nethercott, M.J.; Epand, R.F.; Epand, R.M.; Kindra, D.R.; Shai, Y.; Weliky, D.P. Comparative Analysis of Membrane-Associated Fusion Peptide Secondary Structure and Lipid Mixing Function of HIV GP41 Constructs that Model the Early Pre-Hairpin Intermediate and Final Hairpin Conformations. J. Mol. Biol. 2010, 397, 301–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmick, S.D.; Weliky, D.P. Major Antiparallel and Minor Parallel beta Sheet Populations Detected in the Membrane-Associated Human Immunodeficiency Virus Fusion Peptide. Biochemistry 2010, 49, 10623–10635. [Google Scholar] [CrossRef] [Green Version]
- Sackett, K.; Nethercott, M.J.; Zheng, Z.X.; Weliky, D.P. Solid-State NMR Spectroscopy of the HIV gp41 Membrane Fusion Protein Supports Intermolecular Antiparallel 13 Sheet Fusion Peptide Structure in the Final Six-Helix Bundle State. J. Mol. Biol. 2014, 426, 1077–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meher, G.; Sinha, S.; Pattnaik, G.P.; Dastidar, S.G.; Chakraborty, H. Cholesterol Modulates Membrane Properties and the Interaction of gp41 Fusion Peptide To Promote Membrane Fusion. J. Phys. Chem. B 2019, 123, 7113–7122. [Google Scholar] [CrossRef]
- Seelig, J.; Waespe-Sarcevic, N. Molecular Order in Cis and Trans Unsaturated Phospholipid Bilayers. Biochemistry 1978, 17, 3310–3315. [Google Scholar] [CrossRef]
- Holte, L.; Peter, S.; Sinnwell, T.; Gawrisch, K. 2H Nuclear Magnetic Resonance Order Parameter Profiles suggest a Change of Molecular Shape for Phosphatidylcholines Containing a Polyunsaturated Acyl Chain. Biophys. J. 1995, 68, 2396–2403. [Google Scholar] [CrossRef] [Green Version]
- Binder, H.; Gawrisch, K. Effect of Unsaturated Lipid Chains on Dimensions, Molecular Order and Hydration of Membranes. J. Phys. Chem. B 2001, 105, 12378–12390. [Google Scholar] [CrossRef]
- Vermeer, L.S.; de Groot, B.L.; Reat, V.; Milon, A.; Czaplicki, J. Acyl chain order parameter profiles in phospholipid bilayers: Computation from molecular dynamics simulations and comparison with H-2 NMR experiments. Eur. Biophys. J. Biophys. Lett. 2007, 36, 919–931. [Google Scholar] [CrossRef] [Green Version]
- Subczynski, W.K.; Wisniewska, A.; Yin, J.J.; Hyde, J.S.; Kusumi, A. Hydrophobic Barriers of Lipid Bilayer Membranes Formed by Reduction of Water Penetration by Alkyl Chain Unsaturation and Cholesterol. Biochemistry 1994, 33, 7670–7681. [Google Scholar] [CrossRef]
- Marsh, D. Polarity and permeation profiles in lipid membranes. Proc. Natl. Acad. Sci. USA 2001, 98, 7777–7782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, W.C.; Lee, M.T.; Chen, F.Y.; Huang, H.W. The Condensing Effect of Cholesterol in Lipid Bilayers. Biophys. J. 2007, 92, 3960–3967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Mills, T.T.; Tristram-Nagle, S.; Nagle, J.F. Cholesterol Perturbs Lipid Bilayers Nonuniversally. Phys. Rev. Lett. 2008, 100, 198103. [Google Scholar] [CrossRef] [PubMed]
- Rawicz, W.; Smith, B.; McIntosh, T.; Simon, S.; Evans, E. Elasticity, Strength, and Water Permeability of Bilayers that Contain Raft Microdomain-Forming Lipids. Biophys. J. 2008, 94, 4725–4736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Tristram-Nagle, S.; Nagle, J.F. Effect of cholesterol on structural and mechanical properties of membranes depends on lipid chain saturation. Phys. Rev. E 2009, 80, 021931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, A.; Capraro, B.R.; Esposito, C.; Baumgart, T. Bending stiffness depends on curvature of ternary lipid mixture tubular membranes. Biophys. J. 2009, 97, 1636–1646. [Google Scholar] [CrossRef] [Green Version]
- Gracià, R.S.; Bezlyepkina, N.; Knorr, R.L.; Lipowsky, R.; Dimova, R. Effect of cholesterol on the rigidity of saturated and unsaturated membranes: Fluctuation and electrodeformation analysis of giant vesicles. Soft Matter 2010, 6, 1472–1482. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.H.; Goto, Y.; Refregiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [Green Version]
- Micsonai, A.; Wien, F.; Bulyaki, E.; Kun, J.; Moussong, E.; Lee, Y.H.; Goto, Y.; Refregiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef]
- Heller, W.T.; Cuneo, M.; Debeer-Schmitt, L.; Do, C.; He, L.; Heroux, L.; Littrell, K.; Pingali, S.V.; Qian, S.; Stanley, C.; et al. The Suite of Small-Angle Neutron Scattering Instruments at Oak Ridge National Laboratory. J. Appl. Crystallogr. 2018, 51, 242–248. [Google Scholar] [CrossRef]
- Heller, W.T.; Hetrick, J.; Bilheux, J.; Borreguero Calvo, J.M.; Chen, W.R.; DeBeer-Schmitt, L.; Do, C.; Doucet, M.; Fitzsimmons, M.R.; Godoy, W.F.; et al. drtsans: The data reduction toolkit for small-angle neutron scattering at Oak Ridge National Laboratory. SoftwareX 2022, 19, 101101. [Google Scholar] [CrossRef]
- Arnold, O.; Bilheux, J.C.; Borreguero, J.M.; Buts, A.; Campbell, S.I.; Chapon, L.; Doucet, M.; Draper, N.; Leal, R.F.; Gigg, M.A.; et al. Mantid-Data Analysis and Visualization Package for Neutron Scattering and μ SR Experiments. Nucl. Instrum. Methods Phys. Res. Sect. A-Accel. Spectrometers Detect. Assoc. Equip. 2014, 764, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Wignall, G.D.; Bates, F.S. Absolute Calibration of Small-Angle Neutron Scattering Data. J. Appl. Crystallogr. 1987, 20, 28–40. [Google Scholar] [CrossRef]
- Heller, W.T.; Doucet, M.; Archibald, R.K. Sas-temper: Software for fitting small-angle scattering data that provides automated reproducibility characterization. SoftwareX 2021, 16, 100849. [Google Scholar] [CrossRef]
- Jacrot, B. The study of biological structures by neutron scattering from solution. Rep. Prog. Phys. 1976, 39, 911–953. [Google Scholar] [CrossRef]
- Marra, J. Direct measurement of the interaction between phosphatidylglycerol bilayers in aqueous electrolyte solutions. Biophys. J. 1986, 50, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Armen, R.S.; Uitto, O.D.; Feller, S.E. Phospholipid Component Volumes: Determination and Application to Bilayer Structure Calculations. Biophys. J. 1998, 75, 734–744. [Google Scholar] [CrossRef] [Green Version]
- Nagle, J.F.; Tristram-Nagle, S. Structure of Lipid Bilayers. Biochim. Biophys. Acta (BBA)-Rev. Biomembr. 2000, 1469, 159–195. [Google Scholar] [CrossRef] [Green Version]
- Kučerka, N.; Liu, Y.; Chu, N.; Petrache, H.I.; Tristram-Nagle, S.; Nagle, J.F. Structure of fully hydrated fluid phase DMPC and DLPC lipid bilayers using X-ray scattering from oriented multilamellar arrays and from unilamellar vesicles. Biophys. J. 2005, 88, 2626–2637. [Google Scholar] [CrossRef] [Green Version]
- Kučerka, N.; Holland, B.W.; Gray, C.G.; Tomberli, B.; Katsaras, J. Scattering Density Profile Model of POPG Bilayers As Determined by Molecular Dynamics Simulations and Small-Angle Neutron and X-ray Scattering Experiments. J. Phys. Chem. B 2012, 116, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Heberle, F.A.; Tristram-Nagle, S.; Szymanski, M.; Koepfinger, M.; Katsaras, J.; Kučerka, N. Molecular Structures of Fluid Phase Phosphatidylglycerol Bilayers as Determined by Small Angle Neutron and X-ray Scattering. Biochim. Biophys. Acta (BBA) Biomembr. 2012, 1818, 2135–2148. [Google Scholar] [CrossRef] [PubMed]
- Do, C.; Heller, W.T.; Stanley, C.; Gallmeier, F.X.; Doucet, M.; Smith, G.S. Understanding Inelastically Scattered Neutrons from Water on a Time-of-Flight Small-Angle Neutron Scattering (SANS) Instrument. Nucl. Instrum. Methods Phys. Res. Sect. A-Accel. Spectrometers Detect. Assoc. Equip. 2014, 737, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Ohl, M.; Monkenbusch, M.; Arend, N.; Kozielewski, T.; Vehres, G.; Tiemann, C.; Butzek, M.; Soltner, H.; Giesen, U.; Achten, R.; et al. The Spin-Echo Spectrometer at the Spallation Neutron Source (SNS). Nucl. Instrum. Methods Phys. Res. Sect. A-Accel. Spectrometers Detect. Assoc. Equip. 2012, 696, 85–99. [Google Scholar] [CrossRef]
- Zolnierczuk, P.A.; Holderer, O.; Pasini, S.; Kozielewski, T.; Stingaciu, L.R.; Monkenbusch, M. Efficient Data Extraction from Neutron Time-of-Flight Spin-Echo Raw Data. J. Appl. Crystallogr. 2019, 52, 1022–1034. [Google Scholar] [CrossRef] [PubMed]
- Zilman, A.G.; Granek, R. Undulations and Dynamic Structure Factor of Membranes. Phys. Rev. Lett. 1996, 77, 4788–4791. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.C.; Brown, F.L.H. Interpreting Membrane Scattering Experiments at the Mesoscale: The Contribution of Dissipation within the Bilayer. Biophys. J. 2010, 98, L9–L11. [Google Scholar] [CrossRef] [Green Version]
- Hardy, R.C.; Cottington, R.L. Viscosity OF Deuterium Oxide and Water in the Range 5-Degrees-C TO 125-Degrees-C. J. Res. Natl. Bur. Stand. 1949, 42, 573–578. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, S.M.; Doe, C.; Faraone, A.; Pincus, P.A.; Kline, S.R. Thermal Fluctuation and Elasticity of Lipid Vesicles Interacting with Pore-Forming Peptides. Phys. Rev. Lett. 2010, 105, 038101. [Google Scholar] [CrossRef]
- Woodka, A.C.; Butler, P.D.; Porcar, L.; Farago, B.; Nagao, M. Lipid Bilayers and Membrane Dynamics: Insight into Thickness Fluctuations. Phys. Rev. Lett. 2012, 109, 058102. [Google Scholar] [CrossRef] [Green Version]
- Nagao, M.; Kelley, E.G.; Ashkar, R.; Bradbury, R.; Butler, P.D. Probing Elastic and Viscous Properties of Phospholipid Bilayers Using Neutron Spin Echo Spectroscopy. J. Phys. Chem. Lett. 2017, 8, 4679–4684. [Google Scholar] [CrossRef] [PubMed]
- Bloom, M.; Evans, E.; Mouritsen, O.G. Physical-Properties of the Fluid Lipid-Bilayer Component of Cell-Membranes—A Perspective. Q. Rev. Biophys. 1991, 24, 293–397. [Google Scholar] [CrossRef] [PubMed]
- Kelley, E.G.; Butler, P.D.; Nagao, M. Collective dynamics in lipid membranes containing transmembrane peptides. Soft Matter 2021, 17, 5671–5681. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1-2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, E.; Abraham, M.J.; Hess, B.; van der Spoel, D. GROMACS 2021.4 Manual. Available online: https://manual.gromacs.org/2021.4/reference-manual/index.html (accessed on 3 November 2022). [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jämbeck, J.P.M.; Lyubartsev, A.P. Derivation and Systematic Validation of a Refined All-Atom Force Field for Phosphatidylcholine Lipids. J. Phys. Chem. B 2012, 116, 3164–3179. [Google Scholar] [CrossRef]
- Jämbeck, J.P.M.; Lyubartsev, A.P. An Extension and Further Validation of an All-Atomistic Force Field for Biological Membranes. J. Chem. Theory Comput. 2012, 8, 2938–2948. [Google Scholar] [CrossRef]
- Jämbeck, J.P.M.; Lyubartsev, A.P. Another Piece of the Membrane Puzzle: Extending Slipids Further. J. Chem. Theory Comput. 2013, 9, 774–784. [Google Scholar] [CrossRef]
- Grote, F.; Lyubartsev, A.P. Optimization of Slipids Force Field Parameters Describing Headgroups of Phospholipids. J. Phys. Chem. B 2020, 124, 8784–8793. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins-Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Knight, C.J.; Hub, J.S. MemGen: A general web server for the setup of lipid membrane simulation systems. Bioinformatics 2015, 31, 2897–2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.H.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef]
- Frielinghaus, H. Small-angle scattering model for multilamellar vesicles. Phys. Rev. E 2007, 76, 051603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takechi-Haraya, Y.; Goda, Y.; Sakai-Kato, K. Atomic Force Microscopy Study on the Stiffness of Nanosized Liposomes Containing Charged Lipids. Langmuir 2018, 34, 7805–7812. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Zolnierczuk, P.A. Interaction of a short antimicrobial peptide on charged lipid bilayer: A case study on aurein 1.2 peptide. BBA Adv. 2022, 2, 100045. [Google Scholar] [CrossRef]
- Schrödinger, LLC. The PyMOL Molecular Graphics System; Version 2.5.2; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Koller, D.; Lohner, K. The role of spontaneous lipid curvature in the interaction of interfacially active peptides with membranes. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1838, 2250–2259. [Google Scholar] [CrossRef] [Green Version]
- Tristram-Nagle, S.; Nagle, J.F. HIV-1 Fusion Peptide Decreases Bending Energy and Promotes Curved Fusion Intermediates. Biophys. J. 2007, 93, 2048–2055. [Google Scholar] [CrossRef] [Green Version]
- Tristram-Nagle, S.; Chan, R.; Kooijman, E.; Uppamoochikkal, P.; Qiang, W.; Weliky, D.P.; Nagle, J.F. HIV Fusion Peptide Penetrates, Disorders, and Softens T-Cell Membrane Mimics. J. Mol. Biol. 2010, 402, 139–153. [Google Scholar] [CrossRef] [Green Version]
- Shchelokovskyy, P.; Tristram-Nagle, S.; Dimova, R. Effect of the HIV-1 Fusion Peptide on the Mechanical Properties and Leaflet Coupling of Lipid Bilayers. New J. Phys. 2011, 13, 025004. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
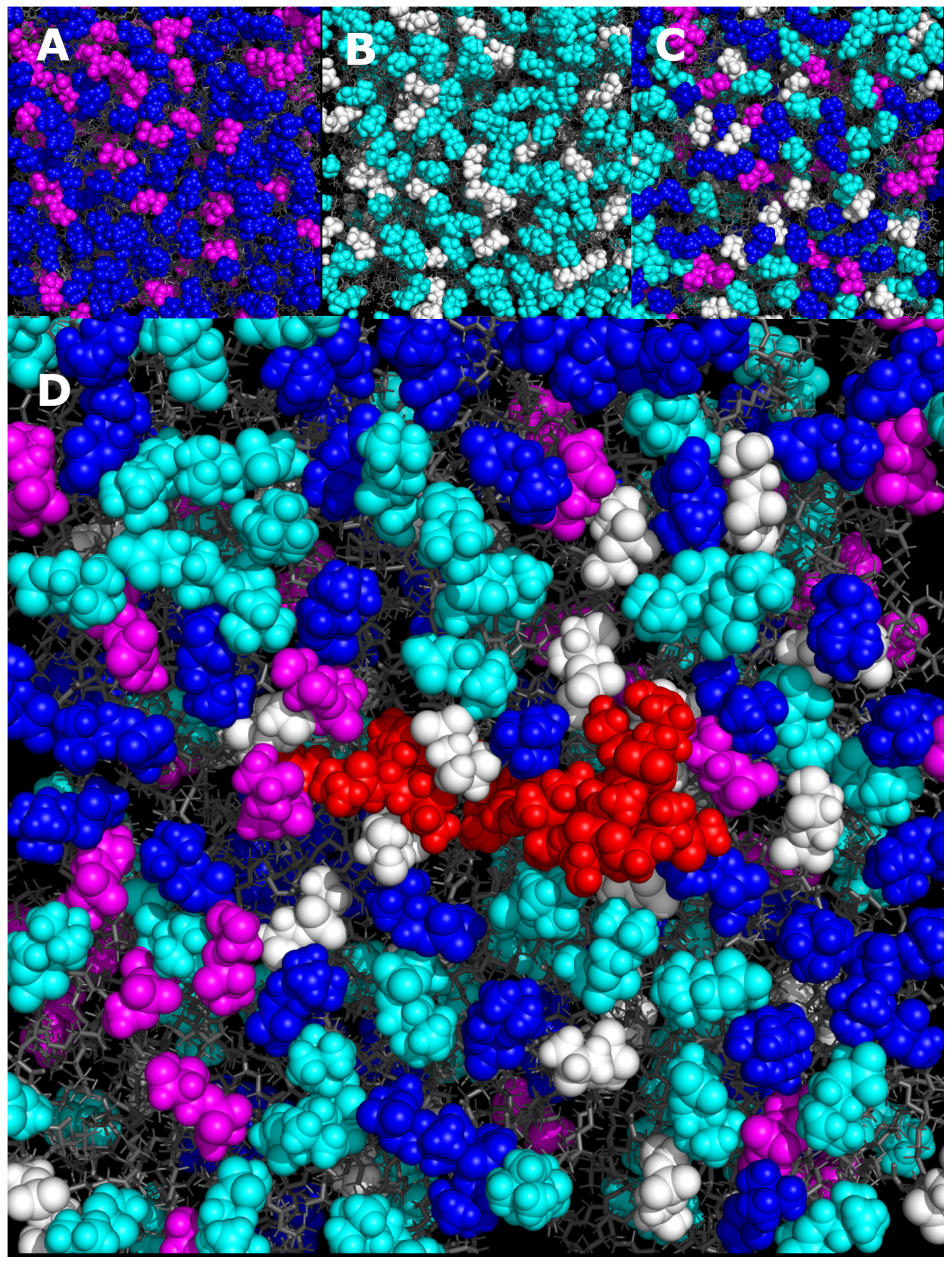{kind=link}
| Lipid | -Helix | Anti-Parallel -Sheet | Turns | “Other” |
|---|---|---|---|---|
| Lipid-1 | 5% | 61% | 14% | 20% |
| Lipid-2 | 40% | 5% | 5% | 50% |
| Lipid-3 | 42% | 11% | 13% | 34% |
| Sample | (Å) | (Å) | (Å) | ||
|---|---|---|---|---|---|
| Lipid-1, P/L = 0 | 59.2 ± 0.1 | 0 | 13.2 ± 0.1 | 44.4 ± 0.2 | 7.3 ± 0.1 |
| Lipid-1, P/L = 1/50 | 59.7 ± 0.1 | 0.48 ± 0.24 | 13.1 ± 0.1 | 44.1 ± 0.2 | 6.6 ± 0.4 |
| Lipid-2, P/L = 0 | 65.1 ± 0.1 | 0 | 14.4 ± 0.1 | 46.8 ± 0.2 | 9.1 ± 0.1 |
| Lipid-2, P/L = 1/50 | 66.8 ± 0.1 | 0.35 ± 0.19 | 14.0 ± 0.1 | 46.0 ± 0.2 | 8.9 ± 0.4 |
| Lipid-3, P/L = 0 | 62.9 ± 0.1 | 0 | 13.6 ± 0.1 | 45.3 ± 0.2 | 8.4 ± 0.1 |
| Lipid-3, P/L = 1/50 | 63.7 ± 0.1 | 0.45 ± 0.27 | 13.5 ± 0.1 | 44.9 ± 0.2 | 7.8 ± 0.5 |
| Sample | /ns) | |
|---|---|---|
| Lipid-1, P/L = 0 from [7] | 10.6 ± 0.4 | 150 ± 10 |
| Lipid-1, P/L = 1/50 from [7] | 8.2 ± 0.3 | 240 ± 30 |
| Lipid-1, P/L = 1/50 | 8.5 ± 0.4 | 230 ± 10 |
| Lipid-2, P/L = 0 | 10.0 ± 0.2 | 160 ± 10 |
| Lipid-2, P/L = 1/50 | 9.1 ± 0.4 | 200 ± 10 |
| Lipid-3, P/L = 0 | 10.5 ± 0.2 | 150 ± 10 |
| Lipid-3, P/L = 1/50 | 9.4 ± 0.3 | 190 ± 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heller, W.T.; Zolnierczuk, P.A. Investigation of the Impact of Lipid Acyl Chain Saturation on Fusion Peptide Interactions with Lipid Bilayers. Biophysica 2023, 3, 121-138. https://doi.org/10.3390/biophysica3010009
Heller WT, Zolnierczuk PA. Investigation of the Impact of Lipid Acyl Chain Saturation on Fusion Peptide Interactions with Lipid Bilayers. Biophysica. 2023; 3(1):121-138. https://doi.org/10.3390/biophysica3010009
Chicago/Turabian StyleHeller, William T., and Piotr A. Zolnierczuk. 2023. "Investigation of the Impact of Lipid Acyl Chain Saturation on Fusion Peptide Interactions with Lipid Bilayers" Biophysica 3, no. 1: 121-138. https://doi.org/10.3390/biophysica3010009
APA StyleHeller, W. T., & Zolnierczuk, P. A. (2023). Investigation of the Impact of Lipid Acyl Chain Saturation on Fusion Peptide Interactions with Lipid Bilayers. Biophysica, 3(1), 121-138. https://doi.org/10.3390/biophysica3010009