
Exploring Chemokine Homodimer Stability: Structural Insights into CXC and CC Interfaces

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
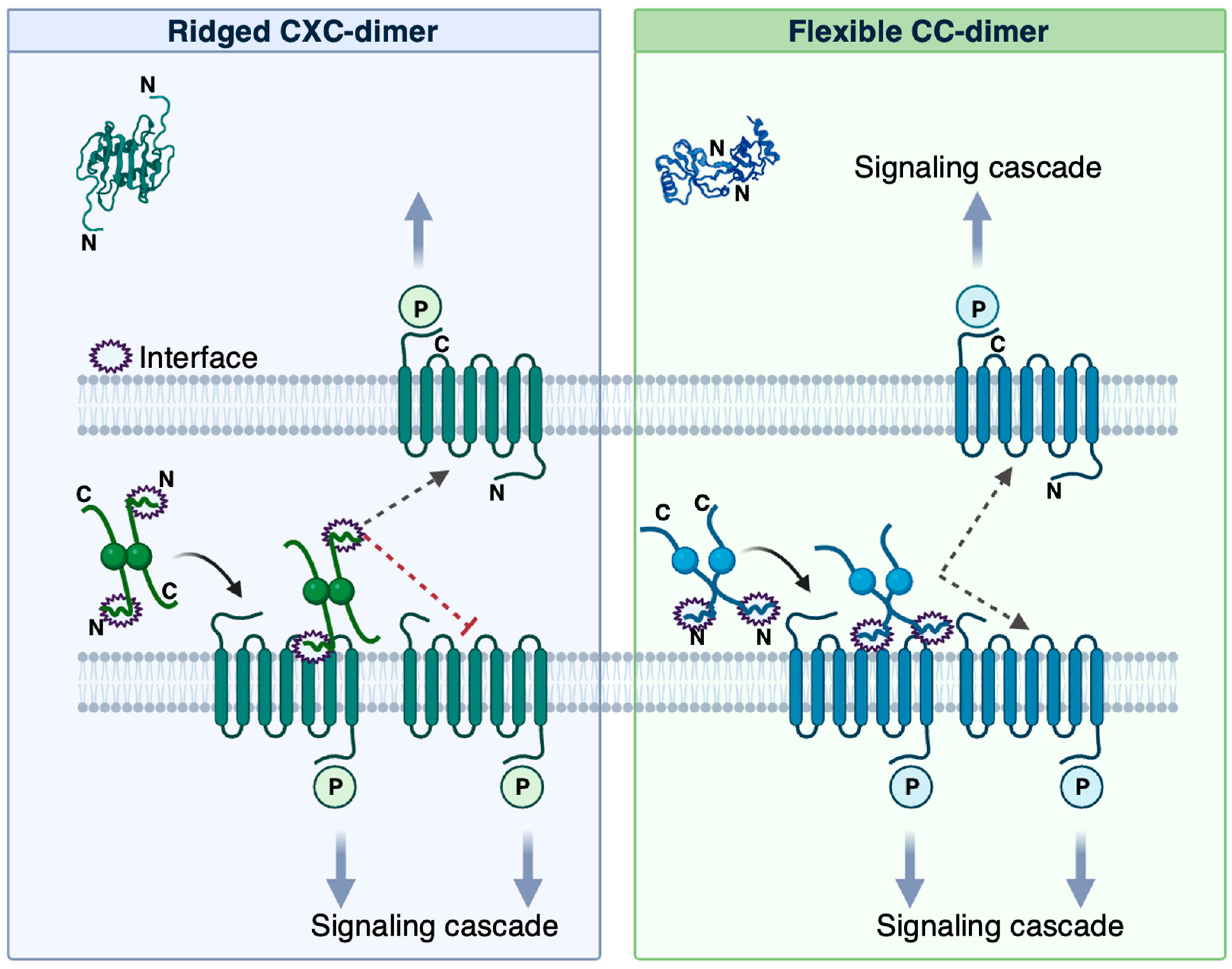
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sokol, C.L.; Luster, A.D. The chemokine system in innate immunity. Cold Spring Harb. Perspect. Biol. 2015, 7, a016303. [Google Scholar] [CrossRef] [PubMed]
- Laub, M.T.; Goulian, M. Specificity in two-component signal transduction pathways. Annu. Rev. Genet. 2007, 41, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef] [PubMed]
- Mellado, M.; Rodriguez-Frade, J.M.; Vila-Coro, A.J.; Fernandez, S.; Martin de Ana, A.; Jones, D.R.; Toran, J.L.; Martinez, A.C. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 2001, 20, 2497–2507. [Google Scholar] [CrossRef]
- Miller, M.C.; Mayo, K.H. Chemokines from a Structural Perspective. Int. J. Mol. Sci. 2017, 18, 2088. [Google Scholar] [CrossRef]
- Paavola, C.D.; Hemmerich, S.; Grunberger, D.; Polsky, I.; Bloom, A.; Freedman, R.; Mulkins, M.; Bhakta, S.; McCarley, D.; Wiesent, L.; et al. Monomeric monocyte chemoattractant protein-1 (MCP-1) binds and activates the MCP-1 receptor CCR2B. J. Biol. Chem. 1998, 273, 33157–33165. [Google Scholar] [CrossRef]
- Wedemeyer, M.J.; Mahn, S.A.; Getschman, A.E.; Crawford, K.S.; Peterson, F.C.; Marchese, A.; McCorvy, J.D.; Volkman, B.F. The chemokine X-factor: Structure-function analysis of the CXC motif at CXCR4 and ACKR3. J. Biol. Chem. 2020, 295, 13927–13939. [Google Scholar] [CrossRef]
- Wang, X.; Sharp, J.S.; Handel, T.M.; Prestegard, J.H. Chemokine oligomerization in cell signaling and migration. Prog. Mol. Biol. Transl. Sci. 2013, 117, 531–578. [Google Scholar] [CrossRef]
- Monneau, Y.; Arenzana-Seisdedos, F.; Lortat-Jacob, H. The sweet spot: How GAGs help chemokines guide migrating cells. J. Leukoc. Biol. 2016, 99, 935–953. [Google Scholar] [CrossRef]
- Gunasekaran, K.; Nussinov, R. How different are structurally flexible and rigid binding sites? Sequence and structural features discriminating proteins that do and do not undergo conformational change upon ligand binding. J. Mol. Biol. 2007, 365, 257–273. [Google Scholar] [CrossRef]
- Gunasekaran, K.; Ma, B.; Nussinov, R. Is allostery an intrinsic property of all dynamic proteins? Proteins 2004, 57, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Arimont, M.; Sun, S.L.; Leurs, R.; Smit, M.; de Esch, I.J.P.; de Graaf, C. Structural Analysis of Chemokine Receptor-Ligand Interactions. J. Med. Chem. 2017, 60, 4735–4779. [Google Scholar] [CrossRef] [PubMed]
- Penfield, J.; Zhang, L. Interaction and dynamics of chemokine receptor CXCR4 binding with CXCL12 and hBD-3. Commun. Chem. 2024, 7, 205. [Google Scholar] [CrossRef] [PubMed]
- Garton, M.; MacKinnon, S.S.; Malevanets, A.; Wodak, S.J. Interplay of self-association and conformational flexibility in regulating protein function. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170190. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Kurth, E.A.; Budean, D.; Momplaisir, N.; Qu, E.; Simien, J.M.; Orellana, G.E.; Brautigam, C.A.; Smrcka, A.V.; Haglund, E. Biophysical characterization of the CXC chemokine receptor 2 ligands. PLoS ONE 2024, 19, e0298418. [Google Scholar] [CrossRef]
- Bittrich, S.; Segura, J.; Duarte, J.M.; Burley, S.K.; Rose, Y. RCSB protein Data Bank: Exploring protein 3D similarities via comprehensive structural alignments. Bioinformatics 2024, 40, btae370. [Google Scholar] [CrossRef]
- Harder, M.E.; Deinzer, M.L.; Leid, M.E.; Schimerlik, M.I. Global analysis of three-state protein unfolding data. Protein Sci. 2004, 13, 2207–2222. [Google Scholar] [CrossRef]
- Ge, B.; Jiang, X.; Chen, Y.; Sun, T.; Yang, Q.; Huang, F. Kinetic and thermodynamic studies reveal chemokine homologues CC11 and CC24 with an almost identical tertiary structure have different folding pathways. BMC Biophys. 2017, 10, 7. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. BioRxiv 2022. [Google Scholar] [CrossRef]
- Lubkowski, J.; Bujacz, G.; Boque, L.; Domaille, P.J.; Handel, T.M.; Wlodawer, A. The structure of MCP-1 in two crystal forms provides a rare example of variable quaternary interactions. Nat. Struct. Biol. 1997, 4, 64–69. [Google Scholar] [CrossRef]
- Bhusal, R.P.; Aryal, P.; Devkota, S.R.; Pokhrel, R.; Gunzburg, M.J.; Foster, S.R.; Lim, H.D.; Payne, R.J.; Wilce, M.C.J.; Stone, M.J. Structure-guided engineering of tick evasins for targeting chemokines in inflammatory diseases. Proc. Natl. Acad. Sci. USA 2022, 119, e2122105119. [Google Scholar] [CrossRef] [PubMed]
- Fairbrother, W.J.; Reilly, D.; Colby, T.J.; Hesselgesser, J.; Horuk, R. The solution structure of melanoma growth stimulating activity. J. Mol. Biol. 1994, 242, 252–270. [Google Scholar] [CrossRef] [PubMed]
- Sepuru, K.M.; Poluri, K.M.; Rajarathnam, K. Solution structure of CXCL5--a novel chemokine and adipokine implicated in inflammation and obesity. PLoS ONE 2014, 9, e93228. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M.; Appella, E.; Yamada, M.; Matsushima, K.; Gronenborn, A.M. Three-dimensional structure of interleukin 8 in solution. Biochemistry 1990, 29, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Romero-Molina, S.; Ruiz-Blanco, Y.B.; Mieres-Perez, J.; Harms, M.; Münch, J.; Ehrmann, M.; Sanchez-Garcia, E. PPI-Affinity: A Web Tool for the Prediction and Optimization of Protein–Peptide and Protein–Protein Binding Affinity. J. Proteome Res. 2022, 21, 1829–1841. [Google Scholar] [CrossRef]
- Gangavarapu, P.; Rajagopalan, L.; Kolli, D.; Guerrero-Plata, A.; Garofalo, R.P.; Rajarathnam, K. The monomer-dimer equilibrium and glycosaminoglycan interactions of chemokine CXCL8 regulate tissue-specific neutrophil recruitment. J. Leukoc. Biol. 2012, 91, 259–265. [Google Scholar] [CrossRef]
- Herring, C.A.; Singer, C.M.; Ermakova, E.A.; Khairutdinov, B.I.; Zuev, Y.F.; Jacobs, D.J.; Nesmelova, I.V. Dynamics and thermodynamic properties of CXCL7 chemokine. Proteins 2015, 83, 1987–2007. [Google Scholar] [CrossRef]
- Rumfeldt, J.A.; Galvagnion, C.; Vassall, K.A.; Meiering, E.M. Conformational stability and folding mechanisms of dimeric proteins. Prog. Biophys. Mol. Biol. 2008, 98, 61–84. [Google Scholar] [CrossRef]
- Crump, M.P.; Rajarathnam, K.; Kim, K.S.; Clark-Lewis, I.; Sykes, B.D. Solution structure of eotaxin, a chemokine that selectively recruits eosinophils in allergic inflammation. J. Biol. Chem. 1998, 273, 22471–22479. [Google Scholar] [CrossRef]
- Kleist, A.B.; Getschman, A.E.; Ziarek, J.J.; Nevins, A.M.; Gauthier, P.A.; Chevigne, A.; Szpakowska, M.; Volkman, B.F. New paradigms in chemokine receptor signal transduction: Moving beyond the two-site model. Biochem. Pharmacol. 2016, 114, 53–68. [Google Scholar] [CrossRef]
- Allen, S.J.; Crown, S.E.; Handel, T.M. Chemokine: Receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 2007, 25, 787–820. [Google Scholar] [CrossRef] [PubMed]
- Mellado, M.; Vila-Coro, A.J.; Martinez, C.; Rodriguez-Frade, J.M. Receptor dimerization: A key step in chemokine signaling. Cell. Mol. Biol. (Noisy-le-grand) 2001, 47, 575–582. [Google Scholar] [PubMed]
- Liu, K.; Wu, L.; Yuan, S.; Wu, M.; Xu, Y.; Sun, Q.; Li, S.; Zhao, S.; Hue, T.; Liu, Z. Structural basis of CXC chemokine receptor 2 activation and signaling. Nature 2020, 585, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Dyer, D.P. Understanding the mechanisms that facilitate specificity, not redundancy, of chemokine-mediated leukocyte recruitment. Immunology 2020, 160, 336–344. [Google Scholar] [CrossRef]
- Stephens, B.; Handel, T.M. Chemokine receptor oligomerization and allostery. Prog. Mol. Biol. Transl. Sci. 2013, 115, 375–420. [Google Scholar] [CrossRef]
- Handl, H.L.; Vagner, J.; Han, H.; Mash, E.; Hruby, V.J.; Gillies, R.J. Hitting multiple targets with multimeric ligands. Expert. Opin. Ther. Targets 2004, 8, 565–586. [Google Scholar] [CrossRef]
- Fernando, H.; Chin, C.; Rosgen, J.; Rajarathnam, K. Dimer dissociation is essential for interleukin-8 (IL-8) binding to CXCR1 receptor. J. Biol. Chem. 2004, 279, 36175–36178. [Google Scholar] [CrossRef]
- Romantini, N.; Alam, S.; Dobitz, S.; Spillmann, M.; De Foresta, M.; Schibli, R.; Schertler, G.F.X.; Wennemers, H.; Deupi, X.; Behe, M.; et al. Exploring the signaling space of a GPCR using bivalent ligands with a rigid oligoproline backbone. Proc. Natl. Acad. Sci. USA 2021, 118, e2108776118. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tm1 °C | Tm2 °C | |
|---|---|---|
| CXCL1 a | 53.9 ± 0.6 | 76.8 ± 0.1 |
| CXCL5 a | 56.6 ± 0.3 | 77.6 ± 0.1 |
| CXCL8 a | 41.9 ± 0.3 | 80.0 ± 0.3 |
| CCL2 b | 64.8 ± 0.4 | 75.9 ± 0.1 |
| CCL8 c | n.d. | 56.9 ± 0.2 |
| CCL11 b | 41.0 ± 0.6 | 40.6 ± 2.3 |
| MP M | mD-N M−1 | ΔG kcal/mol | |
|---|---|---|---|
| CXCL1 a | 3.31 | 0.82 | −3.68 ± 0.03 |
| CXCL5 b | 3.32 | 1.23 | −5.57 ± 0.04 |
| CXCL8 b | 5.00 | 1.04 | −7.07 ± 0.10 |
| CCL2 a | 5.29 | 1.10 | −7.91 ± 0.10 |
| CCL8 a | 4.17 | 0.93 | −5.27 ± 0.06 |
| CCL11 b | 2.33 | 0.79 | −2.50 ± 0.08 |
| Score kcal/mol | |
|---|---|
| CXCL1(GROα) | −9.7 |
| CXCL5 (ENA78) | −10.4 |
| CXCL8 (IL-8) | −14.3 |
| CCL2 (MCP-1) | −12.6 |
| CCL8 (MCP-2) | −11.6 |
| CCL11 a (Eotaxin) | −10.9 |
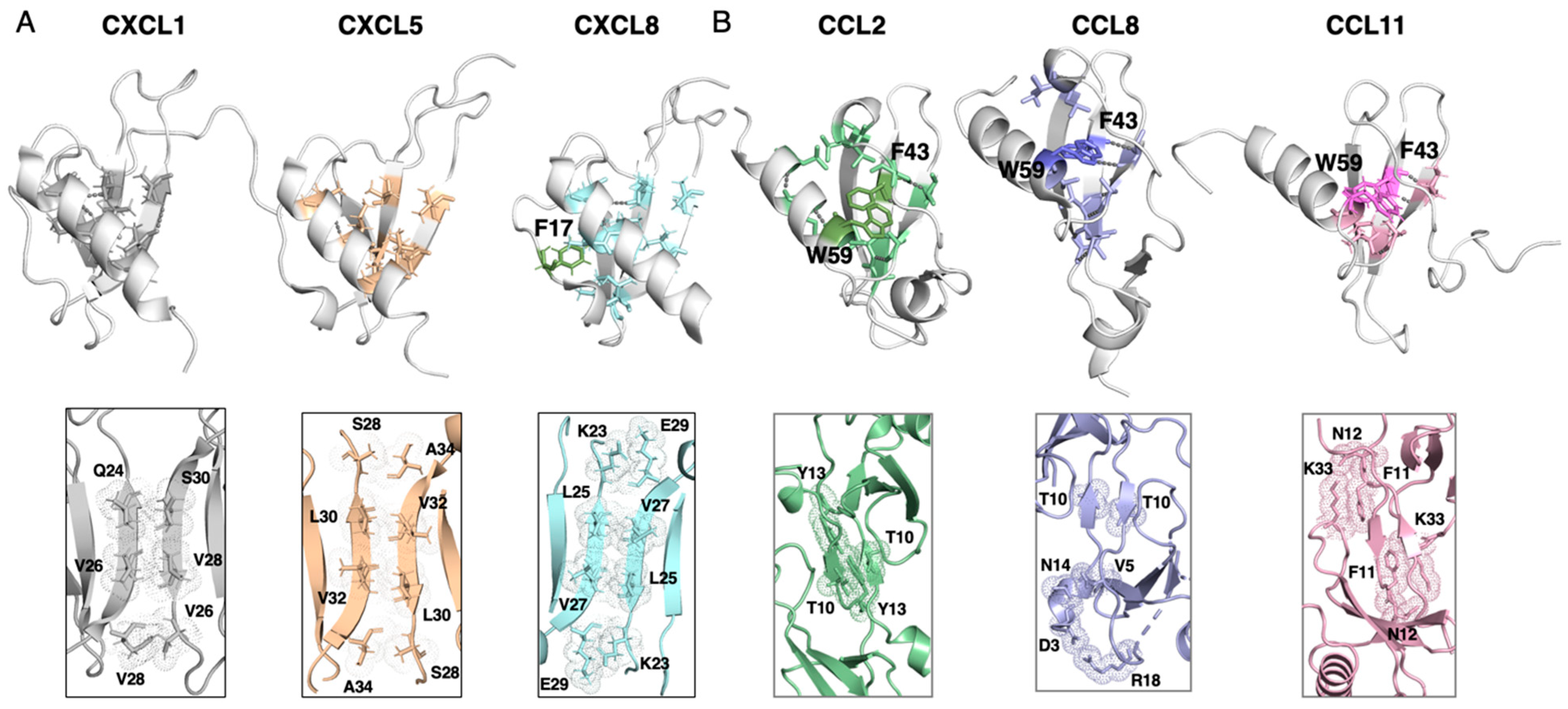
| Hydrophobic Core | Hydrogen Bonding | Aromatic Core | Dimer Interface | |
|---|---|---|---|---|
| CXCL1 | 9 | V40-L5 | - | Q24-S30, V26-V28, V28-V26 |
| CXCL5 | 8 | - | - | S28-A34, L30-V32, V32-L30, A34-S28 |
| CXCL8 | 11 | I34-L51 | F17 | K23-E29, L25-V27, V27-L25, E29-K23 |
| CCL2 | 12 | V41-A53, F43-I51 | F43, W59 | T10-T10, Y13-Y13 |
| CCL8 | 10 | I31-A40, V41-A53, F43-V51 | F43, W5 | T10-T10, N14-V5, D3-R18 |
| CCL11 | 6 | V39-A51, F41-I49 | F41, W57 | N12-K33-F11, K33-F11-N12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budean, D.; Almeida-Hernández, Y.; Pandey, J.; Pérez, J.M.; Sánchez García, E.; Haglund, E. Exploring Chemokine Homodimer Stability: Structural Insights into CXC and CC Interfaces. Biophysica 2024, 4, 561-572. https://doi.org/10.3390/biophysica4040037
Budean D, Almeida-Hernández Y, Pandey J, Pérez JM, Sánchez García E, Haglund E. Exploring Chemokine Homodimer Stability: Structural Insights into CXC and CC Interfaces. Biophysica. 2024; 4(4):561-572. https://doi.org/10.3390/biophysica4040037
Chicago/Turabian StyleBudean, David, Yasser Almeida-Hernández, Jitendra Pandey, Joel Mieres Pérez, Elsa Sánchez García, and Ellinor Haglund. 2024. "Exploring Chemokine Homodimer Stability: Structural Insights into CXC and CC Interfaces" Biophysica 4, no. 4: 561-572. https://doi.org/10.3390/biophysica4040037
APA StyleBudean, D., Almeida-Hernández, Y., Pandey, J., Pérez, J. M., Sánchez García, E., & Haglund, E. (2024). Exploring Chemokine Homodimer Stability: Structural Insights into CXC and CC Interfaces. Biophysica, 4(4), 561-572. https://doi.org/10.3390/biophysica4040037