

Gibbs Free Energy and Enthalpy–Entropy Compensation in Protein Folding


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
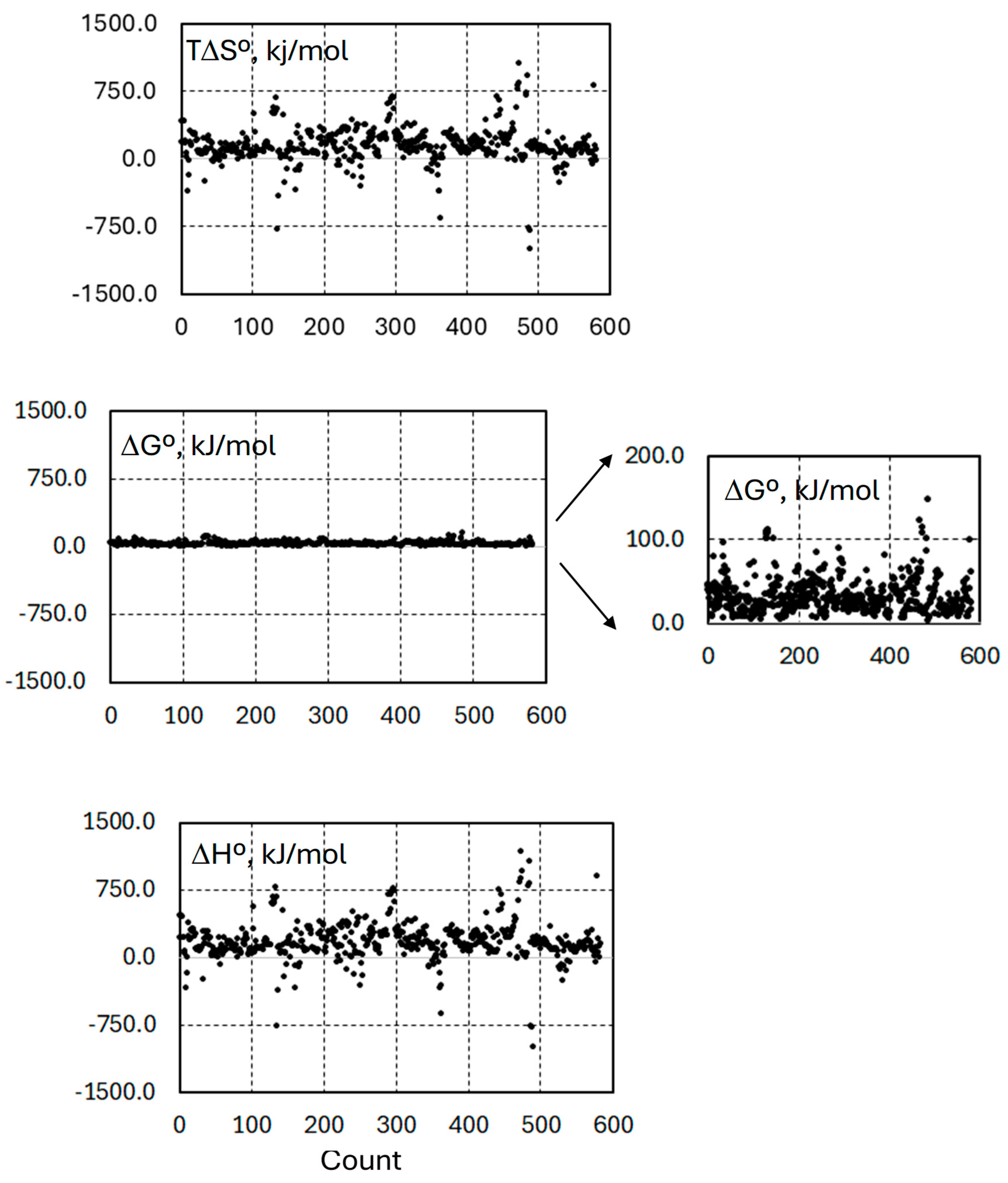
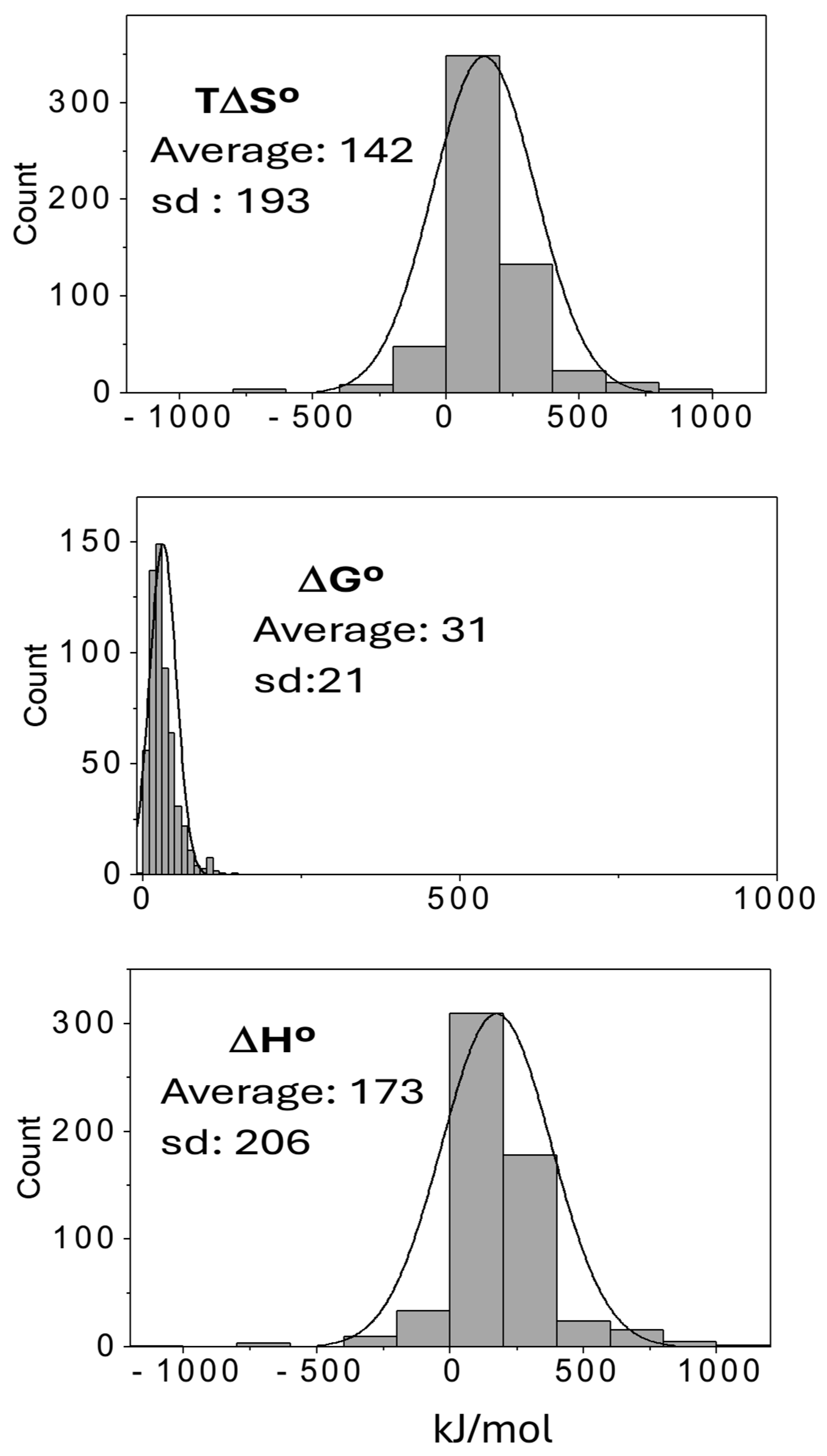
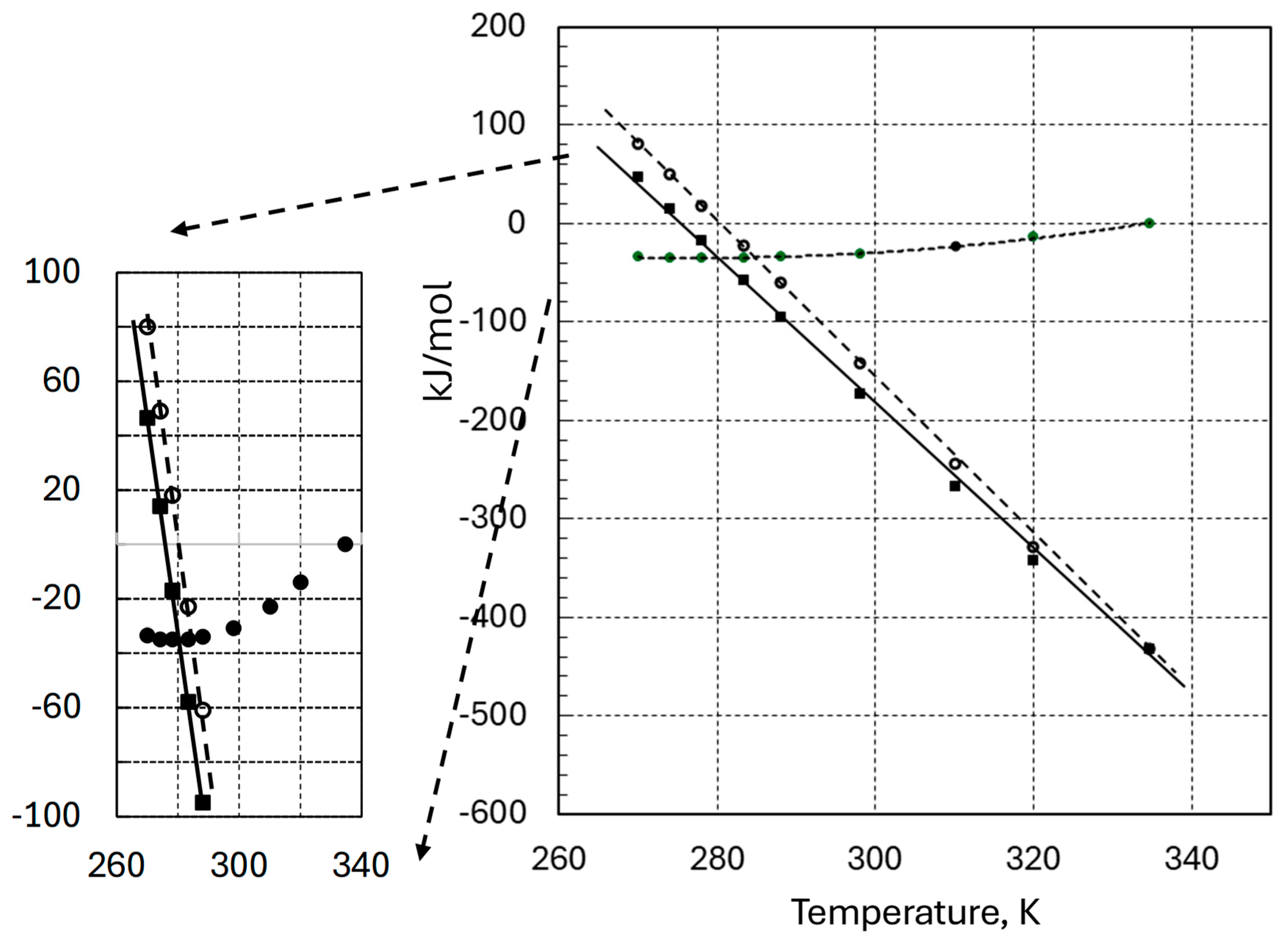
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lumry, R. Uses of enthalpy-entropy compensation in protein research. Biophys. Chem. 2003, 105, 545–557. [Google Scholar] [CrossRef]
- Cooper, A. Thermodynamic analysis of biomolecular interactions. Curr. Opin. Chem. Biol. 1999, 3, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Sharp, K. Entropy-Enthalpy compensation: Fact or artifact? Protein Sci. 2001, 10, 661–667. [Google Scholar] [CrossRef]
- Martin, S.F.; Clements, J.H. Correlating Structure and Energetics in Protein-Ligand Interactions: Paradigms and Paradoxes. Annu. Rev. Biochem. 2013, 82, 267–293. [Google Scholar] [CrossRef]
- Pan, A.; Kar, T.; Rakshit, A.K.; Moulik, S.P. Enthalpy–Entropy Compensation (EEC) Effect: Decisive Role of Free Energy. J. Phys. Chem. B 2016, 120, 10531–10539. [Google Scholar] [CrossRef]
- Fox, J.M.; Zhao, M.; Fink, M.J.; Kang, K.; Whitesides, G.M. The Molecular Origin of Enthalpy/Entropy Compensation in Biomolecular Recognition. Annu. Rev. Biophys. 2018, 47, 223–250. [Google Scholar] [CrossRef] [PubMed]
- Peccati, F.; Jiménez-Osés, G. Enthalpy–Entropy Compensation in Biomolecular Recognition: A Computational Perspective. ACS Omega 2021, 6, 11122–11130. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Z.-G. Using Implicit-Solvent Potentials to Extract Water Contributions to Enthalpy-Entropy Compensation in Biomolecular Associations. J. Phys. Chem. B 2023, 127, 6825–6832. [Google Scholar] [CrossRef]
- Jiménez, J.S.; Benítez, M.J. Gibbs Free Energy and Enthalpy–Entropy Compensation in Protein–Ligand Interactions. Biophysica 2024, 4, 298–309. [Google Scholar] [CrossRef]
- Kragelj, J.; Orand, T.; Delaforge, E.; Tengo, L.; Blackledge, M.; Palencia, A.; Jensen, M.R. Enthalpy-Entropy Compensation in the Promiscuous Interaction of an Intrinsically Disordered Protein with Homologous Protein Partners. Biomolecules 2021, 11, 1204. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, I.D.L.; Junior, F.H.X.; Magalhães, N.S.S.; Nogueira, M.C.d.B.L. Isothermal titration calorimetry (ITC) as a promising tool in pharmaceutical nanotechnology. Int. J. Pharm. 2023, 641, 123063. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.S.G.; Ladbury, J.E.; Pitt, W.R.; Williams, M.A. Extent of enthalpy–entropy compensation in protein–ligand interactions. Protein Sci. 2011, 20, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Nikam, R.; Kulandaisamy, A.; Harini, K.; Sharma, D.; Gromiha, M.M. ProThermDB: Thermodynamic database for proteins and mutants revisited after 15 years. Nucleic Acids Res. 2021, 49, D420–D424. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, C.; Guo, Q.-X. A study on the enthalpy-entropy compensation in protein unfolding. Biophys. Chem. 2000, 84, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Harano, Y.; Kinoshita, M. Translational-Entropy Gain of Solvent upon Protein Folding. Biophys. J. 2005, 89, 2701–2710. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M. Importance of Translational Entropy of Water in Biological Self-Assembly Processes like Protein Folding. Int. J. Mol.Sci. 2009, 10, 1064–1080. [Google Scholar] [CrossRef] [PubMed]
- Levine, N. Physical Chemistry, 5th ed.; McGraw Hill: New York, NY, USA, 2002; p. 866. [Google Scholar]
- Anderson, D.E.; Becktel, W.; Dahlquist, F.W. pH-induced denaturation of proteins: A single salt bridge contributes 3-5 kcal/mol to the free energy of folding of T4 lysozyme. Biochemistry 1990, 29, 2403–2408. [Google Scholar] [CrossRef]
- Gallivan, J.P.; Dougherty, D.A. Cation-pi interactions in structural biology. Proc. Natl. Acad. Sci. USA 1999, 96, 9459–9464. [Google Scholar] [CrossRef]
- Griko, Y.V.; Privalov, P.L.; Sturtevant, J.M.; Venyaminov, S.Y. Cold denaturation of staphylococcal nuclease. Proc. Natl. Acad. Sci. USA 1988, 85, 3343–3347. [Google Scholar] [CrossRef]
- Pyrpassopoulos, S.; Vlassi, M.; Tsortos, A.; Papanikolau, Y.; Petratos, K.; Vorgias, C.E.; Nounesis, G. Equilibrium heat-induced denaturation of chitinase 40 from Streptomyces thermoviolaceus. Proteins 2006, 64, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.Q.; Sturtevant, J.M. Thermodynamic study of yeast phosphoglycerate kinase. Biocemistry 1987, 26, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.; Kundu, A.; Kishore, N. 4-Chlorobutanol induces unusual reversible and irreversible thermal unfolding of ribonuclease A: Thermodynamic, kinetic, and conformational characterization. Int. J. Biol. Macrimol. 2004, 34, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Ikeuchi, H.; Tada, T.; Oyama, H.; Oda, K.; Kunugi, S. Synergetic effects of pressure and chemical denaturant on protein unfolding: Stability of a serine-type carboxyl protease, kumamolisin. Biochim. Biophys. Acta 2006, 1764, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zhang, B.; Zheng, H.; Zhuang, C.; Li, X.; Lu, X.; Quan, C.; Dong, Y.; Zheng, Z.; Xiu, Z. Trivaric acid, a new inhibitor of PTP1b with potent beneficial effect on diabetes. Life Sci. 2017, 169, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Ylilauri, M.; Mattila, E.; Nurminen, E.M.; Käpylä, J.; Niinivehmas, S.P.; Määttä, J.A.; Pentikäinen, U.; Ivaska, J.; Pentikäinen, O.T. Molecular mechanism of T-cell protein tyrosine phosphatase (TCPTP) activation by mitoxantrone. Biochim. Biophys. Acta 2013, 1834, 1988–1997. [Google Scholar] [CrossRef]
- Aggarwal, S.; Tanwar, N.; Singh, A.; Munde, M. Formation of Protamine and Zn–Insulin Assembly: Exploring Biophysical Consequences. ACS Omega 2022, 7, 41044–41057. [Google Scholar] [CrossRef] [PubMed]
- Crisalli, A.M.; Cai, A.; Cho, B.P. Probing the Interactions of Perfluorocarboxylic Acids of Various Chain Lengths with Human Serum Albumin: Calorimetric and Spectroscopic Investigations. Chem. Res. Toxicol. 2023, 36, 703–713. [Google Scholar] [CrossRef]
- Ueda, I.; Yamanaka, M. Titration calorimetry of anesthetic-protein interaction: Negative enthalpy of binding and anesthetic potency. Biophys. J. 1997, 72, 1812–1817. [Google Scholar] [CrossRef] [PubMed]
- Hinz, H.J.; Steininger, G.; Schmid, F.; Jaenide, R. Studies on an energy structure-function relationship of dehydrogenases. II. Calorimetric investigations on the interaction of coenzyme fragments with pig skeletal muscle lactate dehydrogenase. FEBS Lett. 1978, 87, 83–86. [Google Scholar] [CrossRef]
- Mateo, P.L.; Baron, C.; Lopez-Mayorga, O.; Jimenez, J.S.; Cortijo, M. AMP and IMP binding to glycogen phosphorylase b. A calorimetric and equilibrium dialysis study. J. Biol. Chem. 1984, 259, 9384–9389. [Google Scholar] [CrossRef]
- Camero, S.; Benítez, M.J.; Cuadros, R.; Hernández, F.; Ávila, J.; Jiménez, J.S. Thermodynamics of the Interaction between Alzheimer’s Disease Related Tau Protein and DNA. PLoS ONE 2014, 9, e104690. [Google Scholar] [CrossRef] [PubMed]
- Fukada, H.; Sturtevant, J.M.; Quiocho, F.A. Thermodynamics of the binding of L-arabinose and of D-galactose to the L-arabinose-binding protein of Escherichia coli. J. Biol. Chem. 1983, 258, 13193–13198. [Google Scholar] [CrossRef] [PubMed]
- Paketurytė, V.; Linkuvienė, V.; Krainer, G.; Chen, W.-Y.; Matulis, D. Repeatability, precision, and accuracy of the enthalpies and Gibbs energies of a protein-ligand binding reaction measured by isothermal titration calorimetry. Eur. Biophys. J. 2019, 48, 139–152. [Google Scholar] [CrossRef]
- Shi, J.-H.; Lou, Y.-Y.; Zhou, K.-L.; Pan, D.-Q. Elucidation of intermolecular interaction of bovine serum albumin with Fenhexamid: A biophysical prospect. J. Photochem. Photobiol. B Biol. 2018, 180, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, Y.; Panahi-Azar, V.; Barzegar, A.; Dolatabadi, J.E.N.; Dehghan, P. Spectroscopic, thermodynamic and molecular docking studies of bovine serum albumin interaction with ascorbyl palmitate food additive. BioImpacts 2017, 7, 241–246. [Google Scholar] [CrossRef]
- Sobhany, M.; Negishi, M. Characterization of specific donor binding to α1,4 Nacteylhexosaminyltransferase EXTL2 using Iso-thermal Titration Calorimetry. Methods Enzymol. 2006, 416, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.; Woods, R.J.; Gluska, J.; Cooper, A.; Nutley, M.A.; Boons, G.-J. Involvement of Water in Carbohydrate-Protein Binding. J. Am. Chem. Soc. 2001, 123, 12238–12247. [Google Scholar] [CrossRef]
- Hamilton, P.D.; Andley, U.P. In vitro interactions of histones and α-crystallin. Biochem. Biophys. Rep. 2018, 15, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Timmer, C.M.; Michmerhuizen, N.L.; Witte, A.B.; Van Winkle, M.; Zhou, D.; Sinniah, K. An Isothermal Titration and Differ-ential Scanning Calorimetry Study of the G-Quadruplex DNA–Insulin Interaction. J.Phys. Chem. B 2014, 118, 1784–1790. [Google Scholar] [CrossRef] [PubMed]
- Honnappa, S.; Cutting, B.; Jahnke, W.; Seelig, J.; Steinmetz, M.O. Thermodynamics of the Op18/Stathmin-Tubulin Interaction. J. Biol. Chem. 2003, 278, 38926–38934. [Google Scholar] [CrossRef]
- Danesh, N.; Sedighi, Z.N.; Beigoli, S.; Sharifi-Rad, A.; Saberi, M.R.; Chamani, J. Determining the binding site and binding affinity of estradiol to human serum albumin and holo-transferrin: Fluorescence spectroscopic, isothermal titration calorimetry and molecular modeling approaches. J. Biomol. Struct. Dyn. 2018, 36, 1747–1763. [Google Scholar] [CrossRef] [PubMed]
- Sturtevant, J.M. Heat capacity and entropy changes in processes involving proteins. Proc. Natl. Acad. Sci. USA 1977, 74, 2236–2240. [Google Scholar] [CrossRef]
- Wilfong, E.M.; Kogiso, Y.; Muthukrishnan, S.; Kowatz, T.; Du, Y.; Bowie, A.; Naismith, J.H.; Hadad, C.M.; Toone, E.J.; Gustafson, T.L. A Multidisciplinary Approach to Probing Enthalpy-Entropy Compensation and the Interfacial Mobility Model. J. Am. Chem. Soc. 2011, 133, 11515–11523. [Google Scholar] [CrossRef]
- Baldwin, R.L. Temperature dependence of the hydrophobic interaction in protein folding. Proc. Natl. Acad. Sci. USA. 1986, 83, 8069–8072. [Google Scholar] [CrossRef] [PubMed]
- Doig, A.J.; Williams, D.H. Why water-soluble, compact, globular proteins have similar specific enthalpies of unfolding at 110.degree.C. Biochemistry 1992, 31, 9371–9375. [Google Scholar] [CrossRef]
- Privalov, P.L.; Makhatadze, G.I. Contribution of hydration to protein folding thermodynamics. II. The entropy and Gibbs energy of hydration. J. Mol. Biol. 1993, 232, 660–676. [Google Scholar] [CrossRef]
- Dragan, A.I.; Read, C.M.; Crane-Robinson, C. Enthalpy-entropy compensation: The role of solvation. Eur. Biophys. J. 2017, 46, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, J.D. Win some, lose some: Enthalpy-entropy compensation in weak intermolecular interactions. Chem. Biol. 1995, 2, 709–712. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benítez, M.J.; Jiménez, J.S. Gibbs Free Energy and Enthalpy–Entropy Compensation in Protein Folding. Biophysica 2025, 5, 2. https://doi.org/10.3390/biophysica5010002
Benítez MJ, Jiménez JS. Gibbs Free Energy and Enthalpy–Entropy Compensation in Protein Folding. Biophysica. 2025; 5(1):2. https://doi.org/10.3390/biophysica5010002
Chicago/Turabian StyleBenítez, María J., and Juan S. Jiménez. 2025. "Gibbs Free Energy and Enthalpy–Entropy Compensation in Protein Folding" Biophysica 5, no. 1: 2. https://doi.org/10.3390/biophysica5010002
APA StyleBenítez, M. J., & Jiménez, J. S. (2025). Gibbs Free Energy and Enthalpy–Entropy Compensation in Protein Folding. Biophysica, 5(1), 2. https://doi.org/10.3390/biophysica5010002