
Analysis of the Substituent Distribution in Carboxymethyl-1,4-glucans on Different Structural Levels—An Ongoing Challenge


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Substituent Distribution in the Glucosyl Unit
1.2. Analysis of the Intact Polymer
1.3. Substituent Distribution along and across the Polysaccharide Chains
2. Materials and Methods
2.1. Materials
2.2. Capillary Electrophoresis
2.3. Partial Depolymerization of CMG and Acylation for MS Analysis
2.3.1. Partial Methanolysis
2.3.2. Acylation of Methanolysates
2.3.3. Partial Hydrolysis
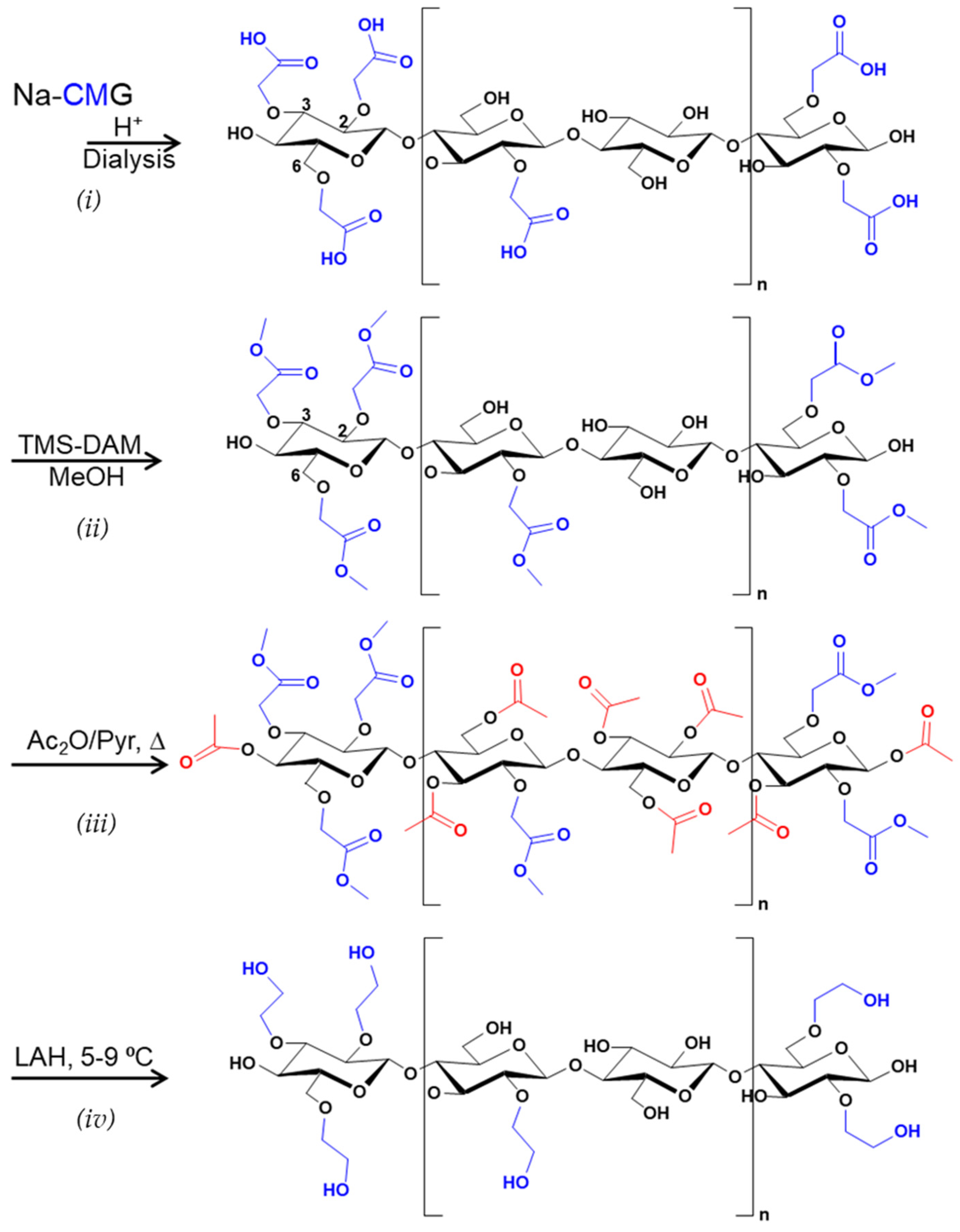
2.4. Reduction of CMG Zu HEG
2.4.1. Transformation of Na-CMG to H-CMG
2.4.2. Methyl Esterification of H-CMG
2.4.3. Acetylation of CMG Methyl Esters
2.4.4. Reduction of CMG-Methyl Ester/Acetate With LAH
2.5. ESI-MS Analysis
2.6. ATR-IR Spectroscopy (Attenuated Total Reflection-Infrared)
3. Results and Discussion
3.1. Distribution of CM in the Glucosyl Units
3.2. Distribution of CM over and across the Polysaccharide Chains
3.2.1. Methanolysis
3.2.2. Production of CM Oligosaccharides by Acid Hydrolysis of CMC and Labeling
3.3. Transformation of Carboxymethyl Glucans to Hydroxyethyl Glucans
3.3.1. Reduction with NaBH4 after Activation by Carbodiimide
3.3.2. Reduction with LiAlH4
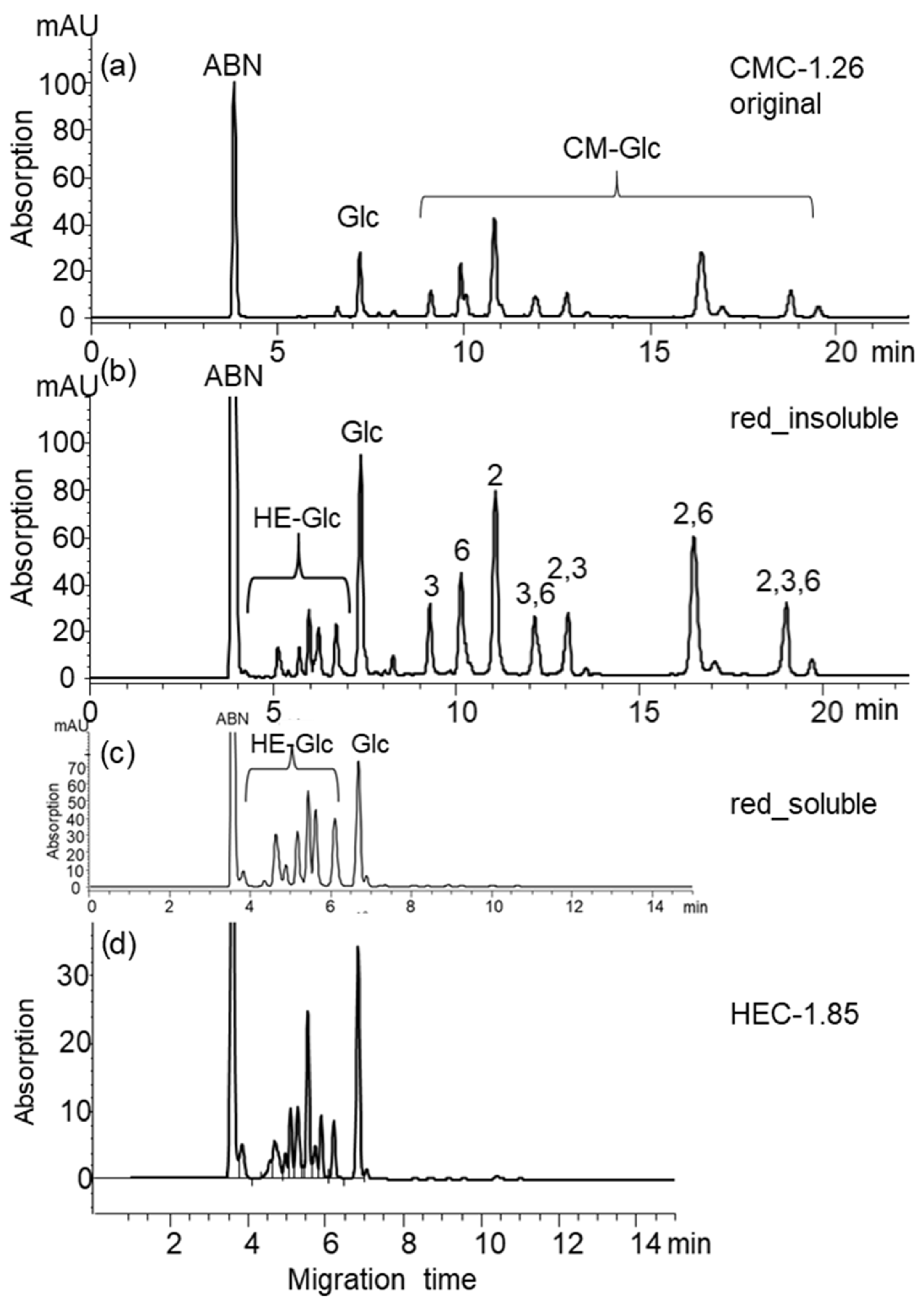
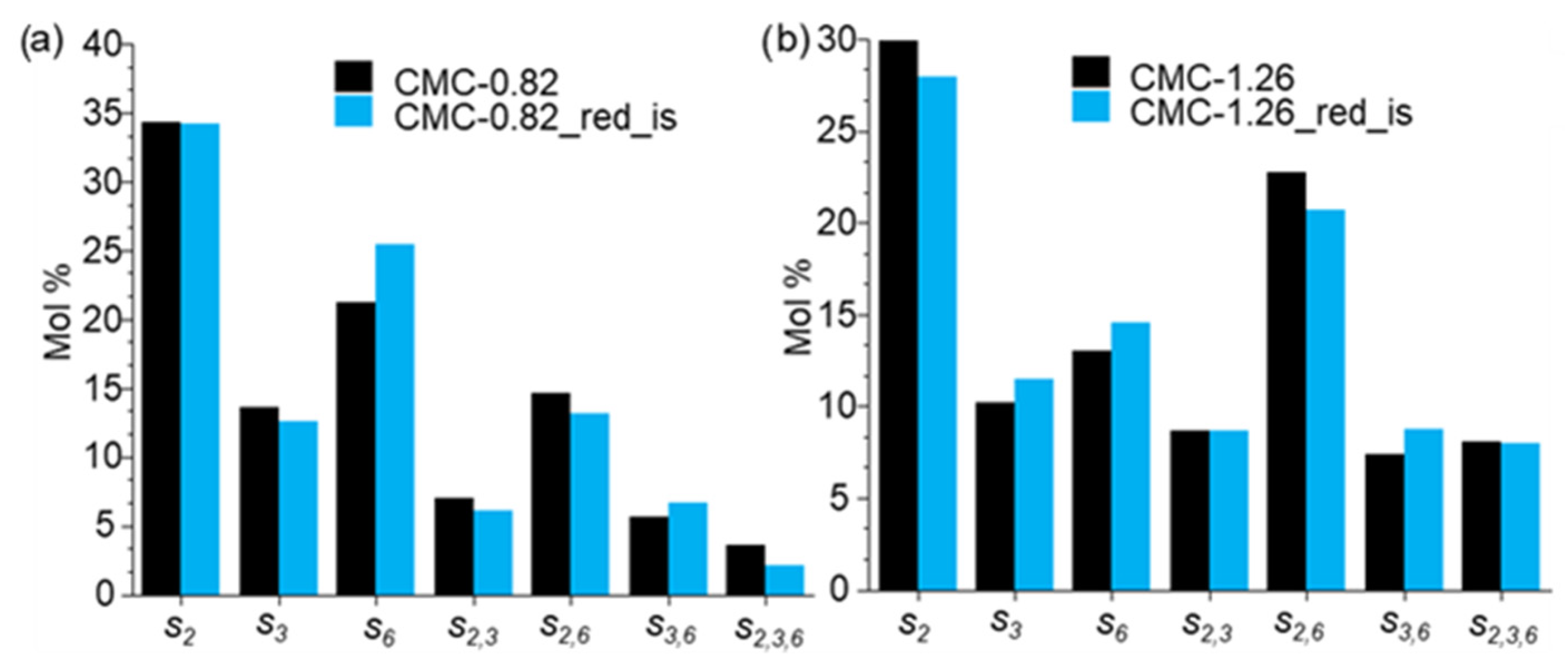
3.3.3. CE Analysis of Reduced CMG
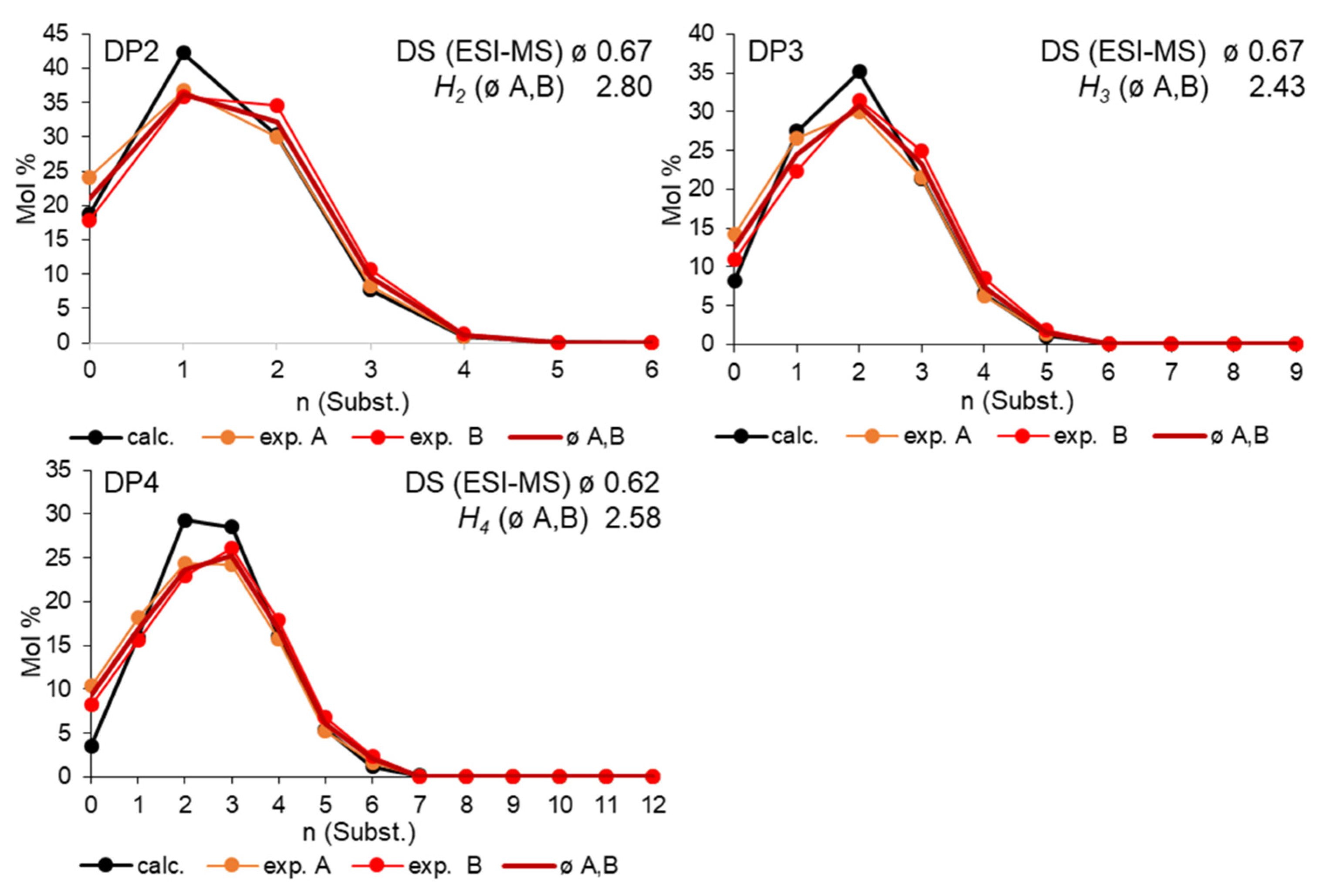
3.4. Quantitative MS Oligosaccharide Analysis of Hydroxyethyl Glucans Derived from Carboxymethyl Glucans
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chowdhury, J.K. Über Äther von Polysacchariden mit Oxysäuren. Biochem. Ztschr. 1924, 148, 76–97. [Google Scholar]
- Heinze, T.; Koschella, A. Carboxymethyl ethers of cellulose and starch—A review. Macromol. Symp. 2005, 223, 13–40. [Google Scholar] [CrossRef]
- Heinze, T.; El Seoud, O.A.; Koschella, A. Chapter 6.1.1. Anionic cellulose ethers. In Cellulose Derivatives—Synthesis, Structure and Properties, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 431–444. [Google Scholar] [CrossRef]
- Spurlin, H.M. Arrangement of substituents in cellulose derivatives. J. Am. Chem. Soc. 1939, 61, 2222–2227. [Google Scholar] [CrossRef]
- Mischnick, P.; Voiges, K.; Cuers-Dammann, J.; Unterieser, I.; Sudwischer, P.; Wubben, A.; Hashemi, P. Analysis of the heterogeneities of first and second order of cellulose derivatives: A complex challenge. Polysaccharides 2021, 2, 843–865. [Google Scholar] [CrossRef]
- Mischnick, P.; Momcilovic, D. Chemical Structure Analysis of Starch and Cellulose Derivatives. Adv. Carbohydr. Chem. Biochem. 2010, 64, 117–210. [Google Scholar] [CrossRef]
- Cuers, J.; Rinken, M.; Adden, R.; Mischnick, P. Critical investigation of the substituent distribution in the polymer chains of hydroxypropyl methylcelluloses by (LC-)ESI-MS. Anal. Bioanal. Chem. 2013, 405, 9021–9032. [Google Scholar] [CrossRef]
- Adden, R.; Müller, R.; Mischnick, P. Fractionation of methyl cellulose according to polarity—A tool to differentiate first and second order heterogeneity of the substituent distribution. Macromol. Chem. Phys. 2006, 207, 954–965. [Google Scholar] [CrossRef]
- Croon, I.; Purves, C.B. The distributioin of substituents in partially substituted carboxymethyl celluloses. Svensk Papperstidn. 1959, 62, 876–882. [Google Scholar]
- Heinze, T. Ionische Funktionspolymers aus Cellulose: Neue Synthesekonzepte, Strukturaufklärung und Eigenschaften; Habilitation, Universität Jena, Shaker: Aachen, Germany, 1998; pp. 62–66. [Google Scholar]
- Crössmann, F.; Kupfer, H.; Soucid, S.W.; Mergenthaler, E. Zur physikalischen und chemischen Charakterisierung der als Lebensmittelzusatzstoffe verwendeten Celluloseäther, IV. Mitteilung. Z. Lbm. Forsch. 1970, 142, 410–418. [Google Scholar] [CrossRef]
- Ho, F.F.L.; Klosiewicz, D.W. Proton nuclear magnetic resonance spectrometry for determination of substituents and their distribution in carboxymethylcellulose. Anal. Chem. 1980, 52, 913–916. [Google Scholar] [CrossRef]
- Reuben, J.; Conner, H.T. Analysis of the carbon-13 n.m.r. spectrum of hydrolyzed O-(carboxymethyl)cellulose. Monomer composition and substitution patterns. Carbohydr. Res. 1983, 115, 1–13. [Google Scholar] [CrossRef]
- Baar, A.; Kulicke, W.M.; Szablikowski, K.; Kiesewetter, R. Nuclear magnetic resonance spectroscopic characterization of carboxymethylcellulose. Macromol. Chem. Phys. 1994, 195, 1483–1492. [Google Scholar] [CrossRef]
- Käuper, P.; Kulicke, W.-M.; Horner, S.; Saake, B.; Puls, J.; Kunze, J.; Fink, H.-P.; Heinze, U.; Heinze, T.; Klohr, E.-A.; et al. Development and evaluation of methods for determining the pattern of funtionalization in sodium carboxymethylcelluloses. Angew. Makromol. Chem. 1998, 260, 53–62. [Google Scholar] [CrossRef]
- Saake, B.; Horner, S.; Puls, J.; Heinze, T.; Koch, W. A New Approach in the Analysis of the Substituent Distribution of Carboxymethyl Celluloses. Cellulose 2001, 8, 59–67. [Google Scholar] [CrossRef]
- Lazik, W.; Heinze, T.; Pfeiffer, K.; Albrecht, G.; Mischnick, P. Starch derivatives of high degree of functionalization—6. Multi step carboxymethylation. J. Appl. Polym. Sci. 2002, 86, 743–752. [Google Scholar] [CrossRef]
- Reuben, J. Analysis of the 13C-N.M.R. spectra of hydrolyzed and methanolyzed O-methylcelluloses: Monomer compositions and models for their description. Carbohydr. Res. 1986, 157, 201–213. [Google Scholar] [CrossRef]
- Kono, H.; Oshima, K.; Hashimoto, H.; Shimizu, Y.; Tajima, K. NMR characterization of sodium carboxymethyl cellulose. Substituent distribution and mole fraction of monomers in the polymer chains. Carbohydr. Polym. 2016, 146, 1–9. [Google Scholar] [CrossRef]
- Heinze, T.; Erler, U.; Nehls, I.; Klemm, D. Determination of the substituent pattern of heterogeneously and homogeneously synthesized carboxymethyl cellulose by using high-performance liquid chromatography. Angew. Makromol. Chem. 1994, 215, 93–106. [Google Scholar] [CrossRef]
- Niemelä, K.; Sjöström, E. Identification of the products of hydrolysis of carboxymethylcellulose. Carbohydr. Res. 1988, 180, 43–52. [Google Scholar] [CrossRef]
- Zeller, S.G.; Greisgraber, G.W.; Gray, G.R. Analysis of positions of substitution of O-carboxymethyl groups in partially O-carboxymethylated cellulose by the reductive-cleavage method. Carbohydr. Res. 1991, 211, 41–45. [Google Scholar] [CrossRef]
- Kragten, E.A.; Kamerling, J.P.; Vliegenthart, J.F.G. Composition analysis of carboxymethylcellulose by high-pH anion-exchange chromatography with pulsed amperometric detection. J. Chromatogr. A 1992, 623, 49–53. [Google Scholar] [CrossRef]
- Tüting, W.; Albrecht, G.; Volkert, B.; Mischnick, P. Structure Analysis of Carboxymethyl Starch by Enzymic Degradation and Capillary Electrophoresis. Starch/Stärke 2004, 56, 315–321. [Google Scholar] [CrossRef]
- Ramos, L.A.; Frollini, E.; Heinze, T. Carboxymethylations of cellulose in the new solvent dimethylsulfoxide/tetrabutyl ammonium fluoride. Carbohydr. Pol. 2005, 60, 259–267. [Google Scholar] [CrossRef]
- Volkert, B.; Loth, F.; Lazik, W.; Engelhardt, J. Highly carboxymethylated starch. Starch/Stärke 2004, 56, 307–314. [Google Scholar] [CrossRef]
- Jardeby, K.; Lennholm, H.; Germgård, U. Characterisation of the undissolved residuals in CMC-solutions. Cellulose 2004, 11, 195–202. [Google Scholar] [CrossRef]
- Jardeby, K.; Germgård, U.; Kreutz, B.; Heinze, T.; Heinze, U.; Lennholm, H. The influence of fibre wall thickness on the undissolved residuals in CMC solutions. Cellulose 2005, 12, 165–167. [Google Scholar] [CrossRef]
- Olaru, N.; Staimberg, S.; Bontea, D.; Asandei, N. Research on optimization of some reaction parameters in synthesis of sodium carboxymethylcellulose. Cell. Chem. Technol. 1980, 14, 713–717. [Google Scholar]
- Olaru, N.; Olaru, L.; Stoleriu, A.; Timpu, D. Carboxymethyl cellulose synthesis in organic media containing ethanol and/or acetone. J. Appl. Polym. Sci. 1998, 67, 481–486. [Google Scholar] [CrossRef]
- Olaru, N.; Olaru, L. Influence of organic diluents on cellulose carboxymethylation. Macromol. Chem. Phys. 2001, 202, 207–211. [Google Scholar] [CrossRef]
- Stigsson, V.; Kloow, G.; Germgård, U. The influence oft he solvent system used during manufacturing of CMC. Cellulose 2006, 13, 705–712. [Google Scholar] [CrossRef]
- Mann, G.; Kunze, J.; Loth, F.; Fink, H.-P. Cellulose ethers with a block-like distribution of the substituents by structure-selective derivatization of cellulose. Polymer 1998, 39, 3155–3165. [Google Scholar] [CrossRef]
- Steeneken, P.A.M.; Ebbe, S. Topochemical effects in the methylation of starch. Carbohydr. Res. 1991, 209, 239–249. [Google Scholar] [CrossRef]
- Fannon, J.E.; Gray, J.A.; Gunawan, N.; Huber, K.C.; BeMiller, J.N. Heterogeneity of starch granules and the effect of granule channelization on starch modification. Cellulose 2004, 11, 247–254. [Google Scholar] [CrossRef]
- Oudhoff, K.A.; Buijtenhuijs, F.A.; Wijnen, P.H.; Schoenmakers, P.J.; Kok, W.T. Determination of the degree of substitution and its distribution of carboxymethylcelluloses by capillary zone electrophoresis. Carbohydr. Res. 2004, 339, 1917–1924. [Google Scholar] [CrossRef]
- Shakun, M.; Heinze, T.; Radke, W. Determination of the DS distribution of non-degraded sodium carboxymethyl cellulose by gradient chromatography. Carbohydr. Polym. 2013, 98, 943–950. [Google Scholar] [CrossRef]
- Shakun, M.; Heinze, T.; Radke, W. Characterization of sodium carboxymethyl cellulose by comprehensive two-dimensional liquid chromatography. Carbohydr. Polym. 2015, 130, 77–86. [Google Scholar] [CrossRef]
- Wirick, M.G. A study of the enzymic degradation of CMC and other cellulose ethers. J. Polym. Sci. Polm. Chem. 1968, 6, 1965–1974. [Google Scholar] [CrossRef]
- Gelman, R.A. Characterization of carboxymethylcellulose: Distribution of substituent groups along the chain. J. Appl. Polym. Sci. 1982, 27, 2957–2964. [Google Scholar] [CrossRef]
- Saake, B.; Horner, S.; Kruse, T.; Puls, J.; Liebert, T.; Heinze, T. Detailed investigation on the molecular structure of carboxymethyl cellulose with unusual substitution pattern by means of an enzyme-supported analysis. Macromol. Chem. Phys. 2000, 201, 1996–2002. [Google Scholar] [CrossRef]
- Olaru, L.; Popa, V.I.; Olaru, N. Kinetic study of rapid step CMC enzymatic hydrolysis. Cell. Chem. Technol. 2004, 38, 165–173. [Google Scholar]
- Enebro, J.; Momcilovic, D.; Siika-Aho, M.; Karlsson, S. Investigation of endoglucanase selectivity on carboxymethyl cellulose by mass spectrometric techniques. Cellulose 2009, 16, 271–280. [Google Scholar] [CrossRef]
- Enebro, J.; Momcilovic, D.; Siika-Aho, M.; Karlsson, S. Liquid chromatography combined with mass spectrometry for the investigation of endoglucanase selectivity on carboxymethyl cellulose. Carbohydr. Res. 2009, 344, 2173–2181. [Google Scholar] [CrossRef]
- Enebro, J.; Momcilovic, D.; Siika-Aho, M.; Karlsson, S. A New Approach for Studying Correlations between the Chemical Structure and the Rheological Properties in Carboxymethyl Cellulose. Biomacromolecules 2007, 8, 3253–3257. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, D. Characteristics of the aqueous solution of carboxymethyl starch ether. J. Appl. Polym. Sci. 1992, 46, 369–374. [Google Scholar] [CrossRef]
- Arisz, P.W.; Kauw, H.J.J.; Boon, J.J. Substituent distribution along the cellulose backbone in O-methylcelluloses using GC and FAB MS for monomer and oligomer analysis. Carbohydr. Res. 1995, 271, 1–14. [Google Scholar] [CrossRef]
- Mischnick, P.; Kühn, G. Correlation between reaction conditions and primary structure: Model studies on methyl amylose. Carbohydr. Res. 1996, 290, 199–207. [Google Scholar] [CrossRef]
- Adden, R.; Niedner, W.; Müller, R.; Mischnick, P. Comprehensive analysis of the substituent distribution in the glucosyl units and along the polymer chain of hydroxyethylmethylcelluloses and statistical evaluation. Anal. Chem. 2006, 78, 1146–1157. [Google Scholar] [CrossRef]
- Mischnick, P.; Unterieser, I.; Voiges, K.; Cuers, J.; Rinken, M.; Adden, R. A new method for the analysis of the substitution pattern of hydroxyethyl(methyl)-celluloses along the polysaccharide chain. Macromol. Chem. Phys. 2013, 214, 1363–1374. [Google Scholar] [CrossRef]
- Gohdes, M.; Mischnick, P. Determination of the substitution pattern in the polymer chain of cellulose sulfates. Carbohydr. Res. 1998, 309, 109–115. [Google Scholar] [CrossRef]
- Voiges, K.; Lämmerhardt, N.; Distelrath, C.; Mischnick, P. Substituent effects on the kinetics of acid-catalyzed hydrolysis of methyl cellulose. Cellulose 2017, 24, 555–569. [Google Scholar] [CrossRef]
- Voiges, K.; Lämmerhardt, N.; Mischnick, P. Kinetic studies of acid-catalyzed hydrolysis of mixed cellulose ethers. Cellulose 2017, 24, 627–639. [Google Scholar] [CrossRef]
- Schleicher, S.; Lottje, I.-R.; Mischnick, P. Impact of instrumental settings in electrospray ionization ion trap mass spectrometry on the analysis of multi-CH3/CD3-isotopologs in cellulose ether analysis: A quantitative evaluation. Anal. Bioanal. Chem. 2022, 414, 1279–1296. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, S.; O’Connor, G.; Mischnick, P. Comparing 13C methyl and deuterated methyl isotopic labeling for the quantification of methyl cellulose patterns using mass spectrometry. Anal. Bioanal. Chem. 2023, 415, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, S.; Horoba, D.; Krafzig, P.; Mischnick, P. Impact of instrumental settings in electrospray ionization ion trap mass spectrometry on the analysis of O-methoxyethyl-O-methyl cellulose: A comprehensive quantitative evaluation. Anal. Bioanal. Chem. 2022, 414, 4727–4743. [Google Scholar] [CrossRef] [PubMed]
- Stojanović, Ž.; Jeremić, K.; Jovanović, S. Molecular structure of carboxymethyl starch in dilute aqueous sodium chloride solutions. Starch/Stärke 2013, 65, 902–911. [Google Scholar] [CrossRef]
- Tang, L.; Kebarle, P. Dependence of ion intensity in electrospray mass spectrometry on the concentration of the analytes in the electrosprayed solution. Anal Chem. 1993, 65, 3654–3668. [Google Scholar] [CrossRef]
- Cech, N.B.; Enke, C.G. Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom. Rev. 2002, 20, 362–387. [Google Scholar] [CrossRef] [PubMed]
- Bol, M.; Sakellaris, C.N.; Jacob, C.R.; Mischnick, P. Differences in the complexation of sodium with methyl esterified carboxymethyl/methoxyacetyl-O-glucans in electrospray ionization-mass spectrometry. Int. J. Mass Spectrom. 2017, 419, 20–28. [Google Scholar] [CrossRef]
- Bol, M.; Dobos, M.A.; Lebioda, S.; Saake, B.; Mischnick, P. Methanolysis of Carboxymethyl Cellulose—A Comprehensive Study. Cellulose 2019, 26, 383–397. [Google Scholar] [CrossRef]
- Schulz, L.; Seger, B.; Burchard, W. Structures of cellulose in solution. Macromol. Chem. Phys. 2000, 201, 2008–2022. [Google Scholar] [CrossRef]
- Enebro, J.; Karlsson, S. Improved matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry of carboxymethyl cellulose. Rapid Commun. Mass Spectrom. 2006, 20, 3693–3698. [Google Scholar] [CrossRef]
- Naven, T.J.P.; Harvey, D.J. Cationic derivatization of oligosaccharides with Girard’s T reagent for improved performance in Matrix-Assisted Laser Desorption Ionization and Electrospray Mass Spectrometry. Rapid Commun. Mass. Spectrom. 1996, 10, 829–834. [Google Scholar] [CrossRef]
- Unterieser, I.; Mischnick, P. Labeling of Oligosaccharides for Quantitative Mass Spectrometry. Carbohydr. Res. 2011, 346, 68–75. [Google Scholar] [CrossRef]
- Trivedi, H.C.; Patel, C.K.; Patel, R.D. Studies on carboxymethyl cellulose—Potentiometric titrations, 3. Macromol. Chem. 1981, 82, 243–245. [Google Scholar] [CrossRef]
- Dogsa, I.; Tomšič, M.; Orehek, J.; Benigar, E.; Jamnik, A.; Stopar, D. Amorphous supramolecular structure of carboxymethyl cellulose in aqueous solution at different pH values as determined by rheology, small angle X-ray and light scattering. Carbohydr. Polym. 2014, 111, 492–504. [Google Scholar] [CrossRef]
- Taylor, R.L.; Conrad, H.E. Stoichiometric depolymerization of polyuronides and glycosaminoglycans to monosaccharides following reduction of their carbodiimide-activated carboxyl groups. Biochemistry 1972, 11, 1383–1388. [Google Scholar] [CrossRef]
- Ukai, S.; Honda, A.; Nagai, K.; Kiho, T. Gas-liquid chromatographic analysis of carboxymethyl cellulose and carboxymethyl starch. J. Chromoatogr. 1990, 513, 338–342. [Google Scholar] [CrossRef]
- De Vries, J.A.; Den Uijl, C.H.; Voragen, A.G.J.; Rombouts, F.M.; Pilnik, W. Structural features of the neutral sugar side chains of apple pectic substances. Carbohydr. Polym. 1983, 3, 193–205. [Google Scholar] [CrossRef]
- Shyluk, W.P.; Timell, T.E. A new method for preparing hydroxyethyl ethers of glucose. Can. J. Chem. 1956, 34, 571–574. [Google Scholar] [CrossRef]
- Höök, J.E.; Lindberg, B.; Nielsen, P.H.; Schwieter, U.; Paasivirta, J. Acid Hydrolysis of Mono-hydroxyethyl Ethers of Methyl alpha- and beta-D-Glucopyranoside. Acta Chem. Scand. 1968, 22, 921–926. [Google Scholar] [CrossRef]
- Aspinall, G.O.; Craig, J.W.T.; Whyte, J.L. Lemon-peel pectin: Part I. Fractionation and partial hydrolysis of water-soluble pectin. Carbohydr. Res. 1968, 7, 442–452. [Google Scholar] [CrossRef]
- Kühnel, E.; Laffan, D.D.P.; Lloyd-Jones, G.C.; Del Martínez Campo, T.; Shepperson, I.R.; Slaughter, J.L. Mechanism of methyl esterification of carboxylic acids by trimethylsilyldiazomethane. Angew. Chem. Int. Ed. Engl. 2007, 46, 7075–7078. [Google Scholar] [CrossRef] [PubMed]
- Tot, I.; Müller, Y.; Werner, C.; Rosenau, T.; Potthast, A. A novel, mild and selective methylation of carboxyl groups in cellulosic pulps 10th EWLP, Stockholm, Sweden, August 25–28, 2008. Holzforschung 2009, 63, 657–663. [Google Scholar] [CrossRef]
- Garcia, J.G.; Voll, R.J.; Younathan, E.S. Reductions of Carbohydrate Vicinal Diacetates with Lithium Aluminum Hydride. J. Carbohydr. Chem. 1993, 119–124. [Google Scholar] [CrossRef]
- Karrasch, A.; Jäger, C.; Saake, B.; Potthast, A.; Rosenau, T. Solid-state NMR studies of methyl celluloses. Part 2: Determination of degree of substitution and O-6 vs. O-2/O-3 substituent distribution in commercial methyl cellulose samples. Cellulose 2009, 16, 1159–1166. [Google Scholar] [CrossRef]
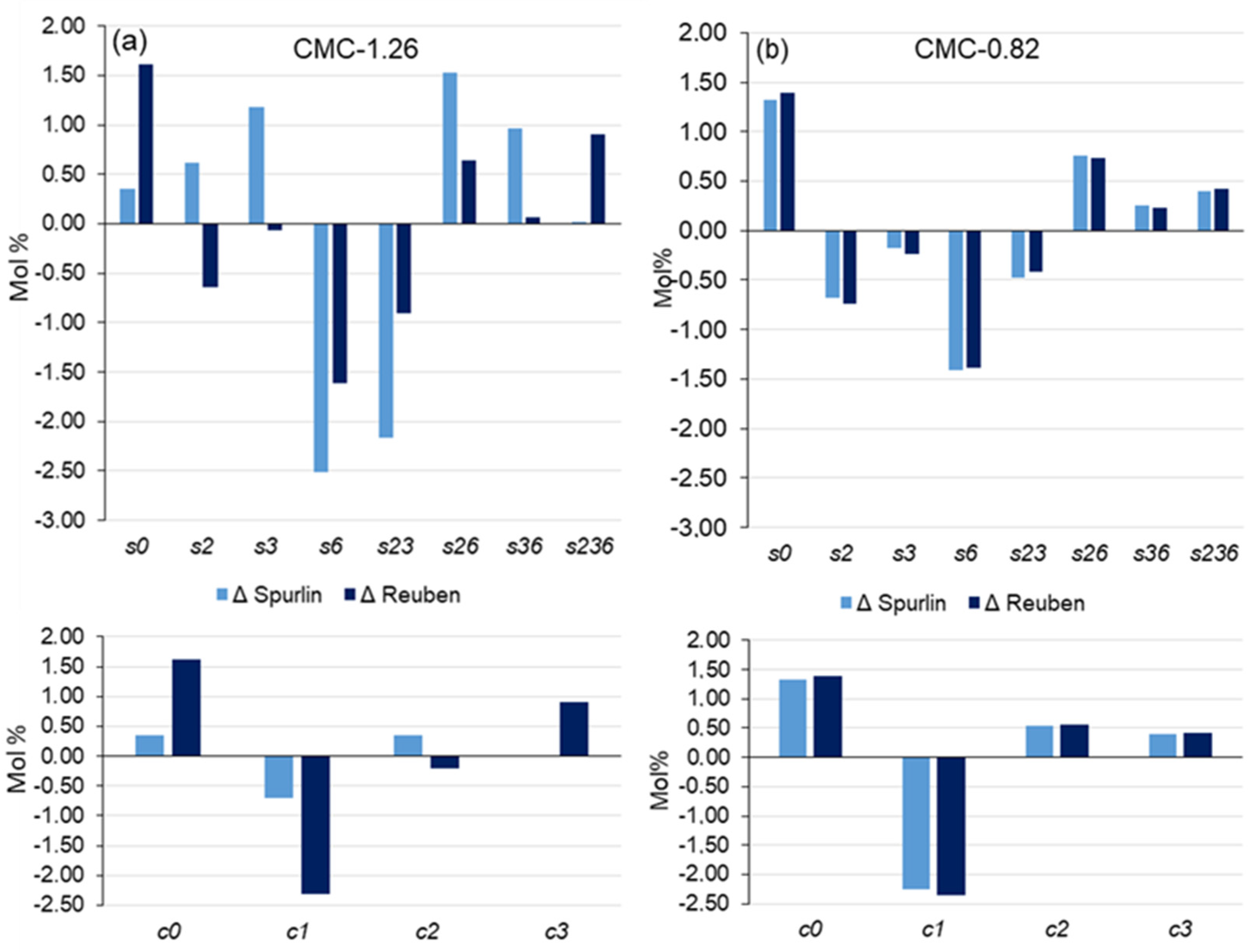
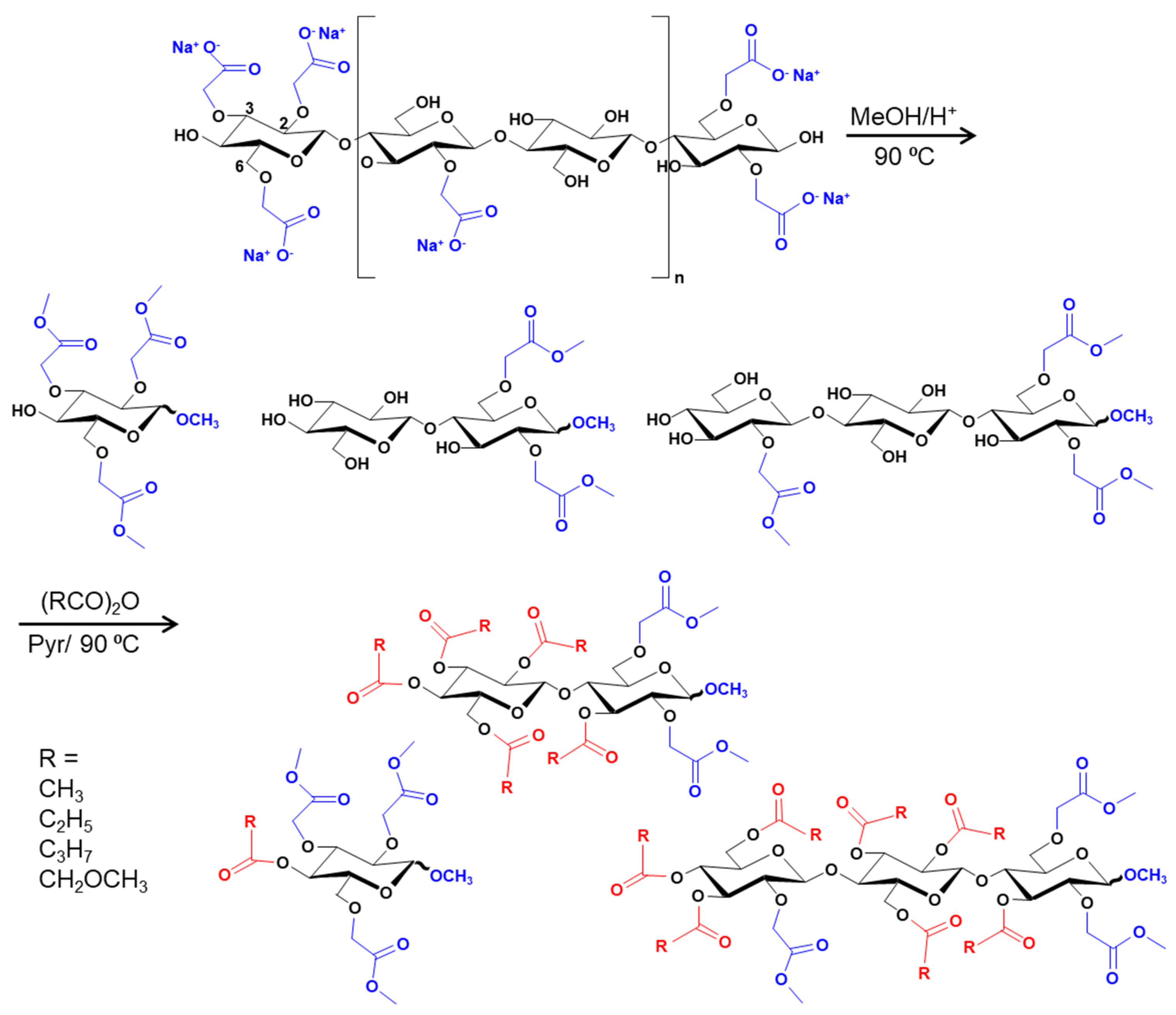
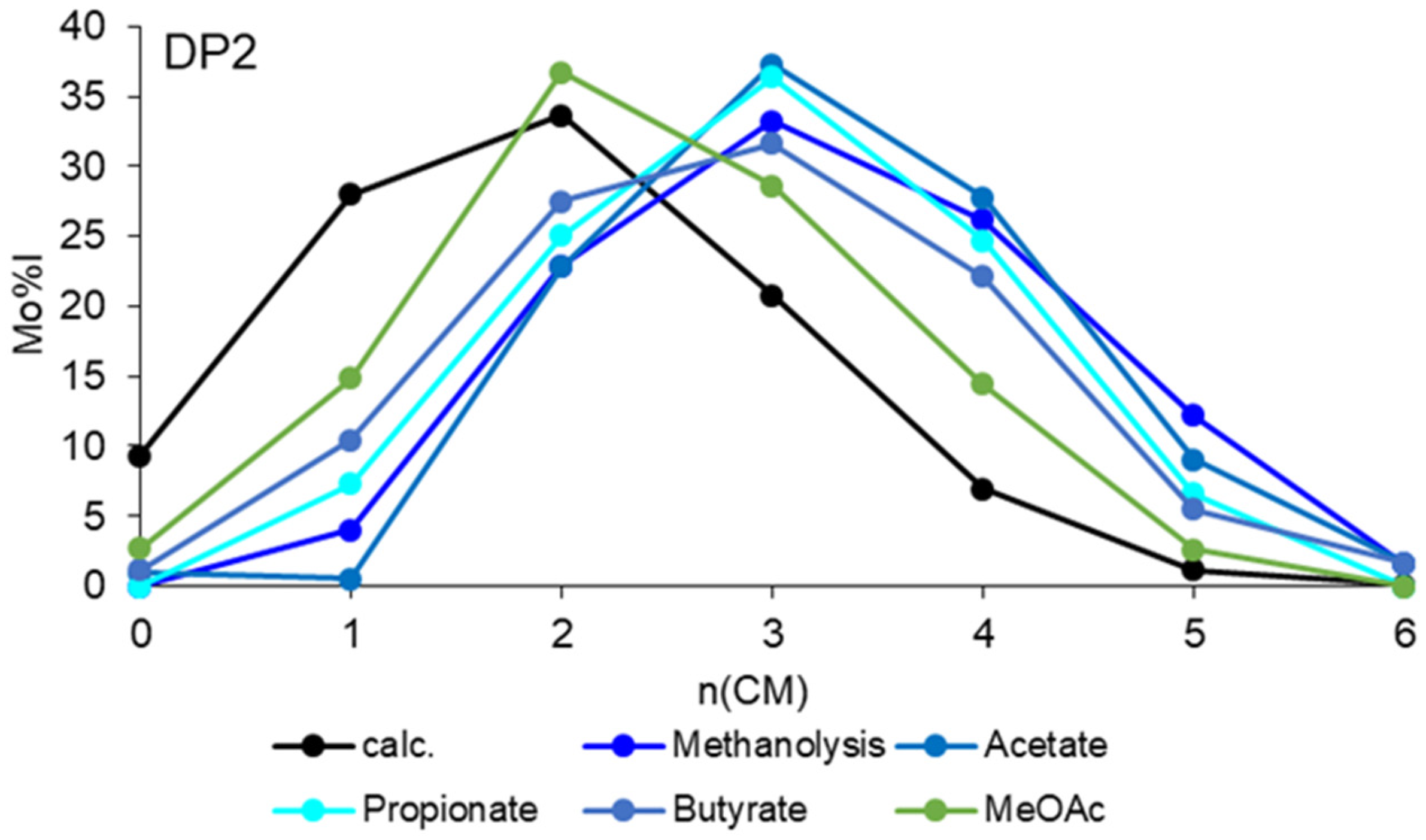
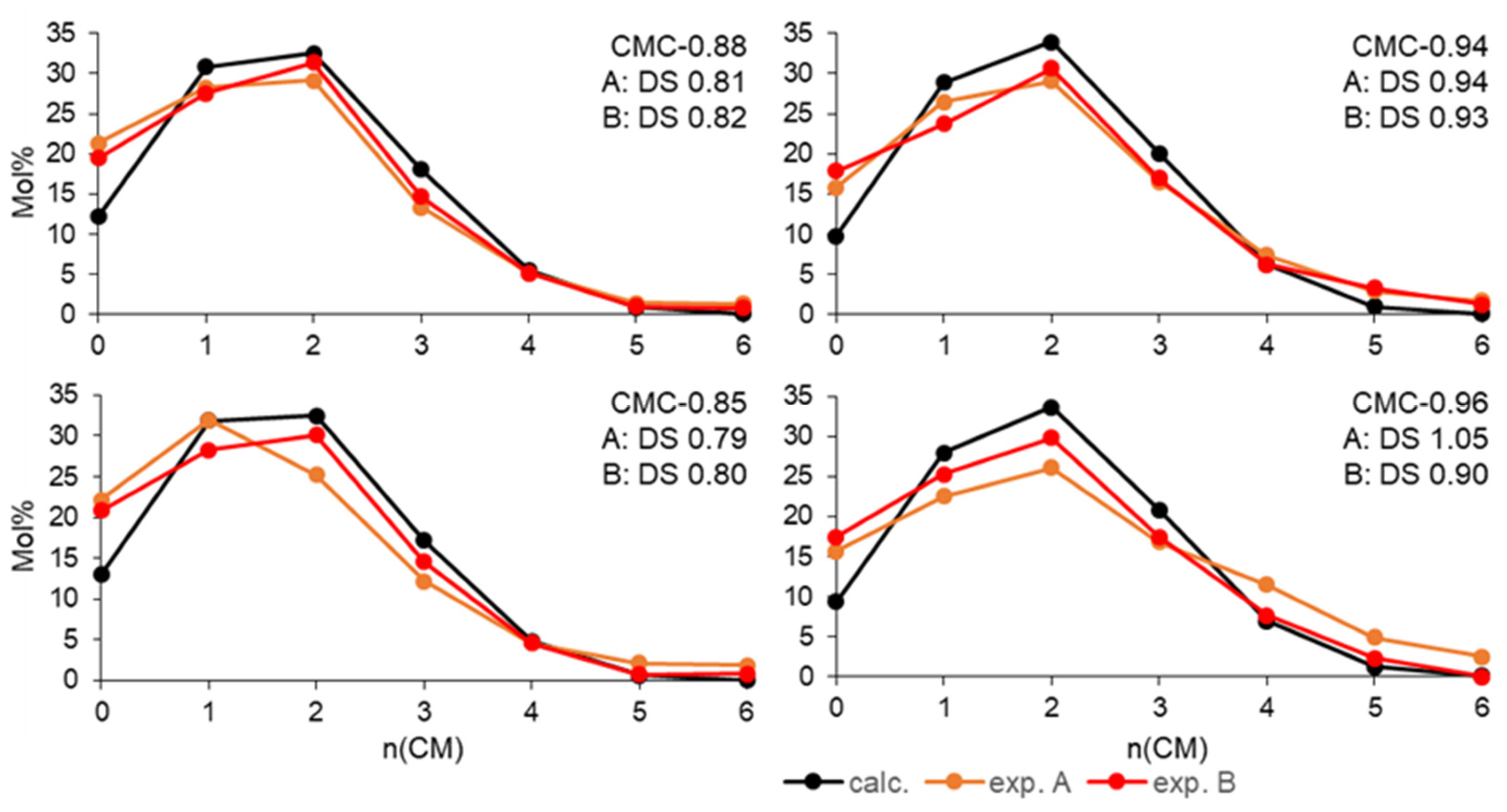



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steingaß, F.; Adden, A.; Mischnick, P. Analysis of the Substituent Distribution in Carboxymethyl-1,4-glucans on Different Structural Levels—An Ongoing Challenge. Polysaccharides 2024, 5, 332-357. https://doi.org/10.3390/polysaccharides5030022
Steingaß F, Adden A, Mischnick P. Analysis of the Substituent Distribution in Carboxymethyl-1,4-glucans on Different Structural Levels—An Ongoing Challenge. Polysaccharides. 2024; 5(3):332-357. https://doi.org/10.3390/polysaccharides5030022
Chicago/Turabian StyleSteingaß, Franziska, Anne Adden, and Petra Mischnick. 2024. "Analysis of the Substituent Distribution in Carboxymethyl-1,4-glucans on Different Structural Levels—An Ongoing Challenge" Polysaccharides 5, no. 3: 332-357. https://doi.org/10.3390/polysaccharides5030022
APA StyleSteingaß, F., Adden, A., & Mischnick, P. (2024). Analysis of the Substituent Distribution in Carboxymethyl-1,4-glucans on Different Structural Levels—An Ongoing Challenge. Polysaccharides, 5(3), 332-357. https://doi.org/10.3390/polysaccharides5030022