
Characterization of Structure and Morphology of Cellulose Lyocell Microfibers Extracted from PAN Matrix

 ,
,  , , , , , , , and
, , , , , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Production of Test Specimens
2.2. Characterization of Cellulose Microfibers
Fourier Transforms Infrared Spectroscopy (FT-IR)
2.3. Scanning Electron Microscope (SEM)
2.4. X-Ray Diffraction (XRD)
2.5. Atomic Force Microscopy
3. Results
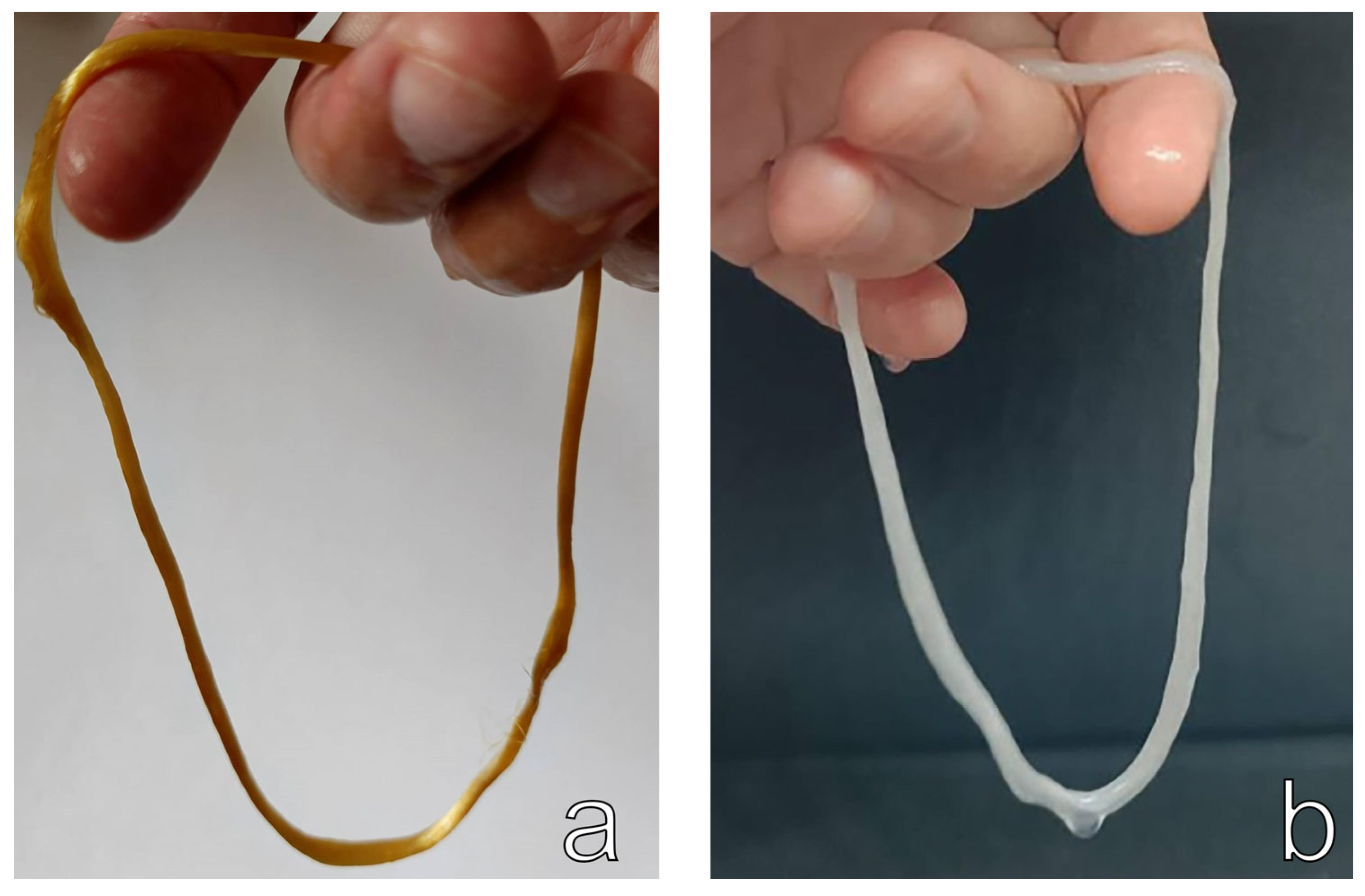
3.1. Production of Cellulose Microfibers
3.2. Morphological Studies
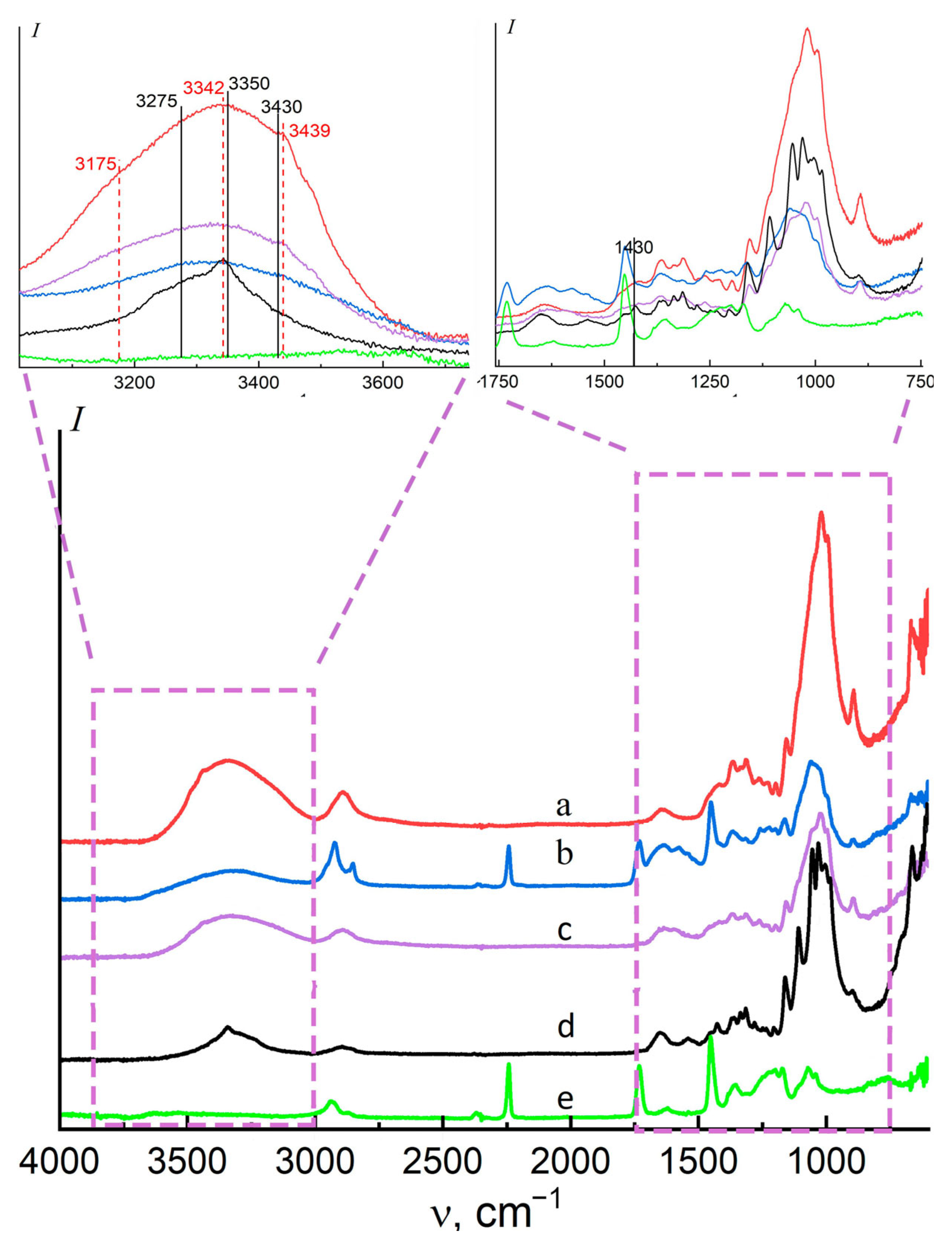
3.3. Fourier Transform Infrared Spectroscopy (FT-IR)
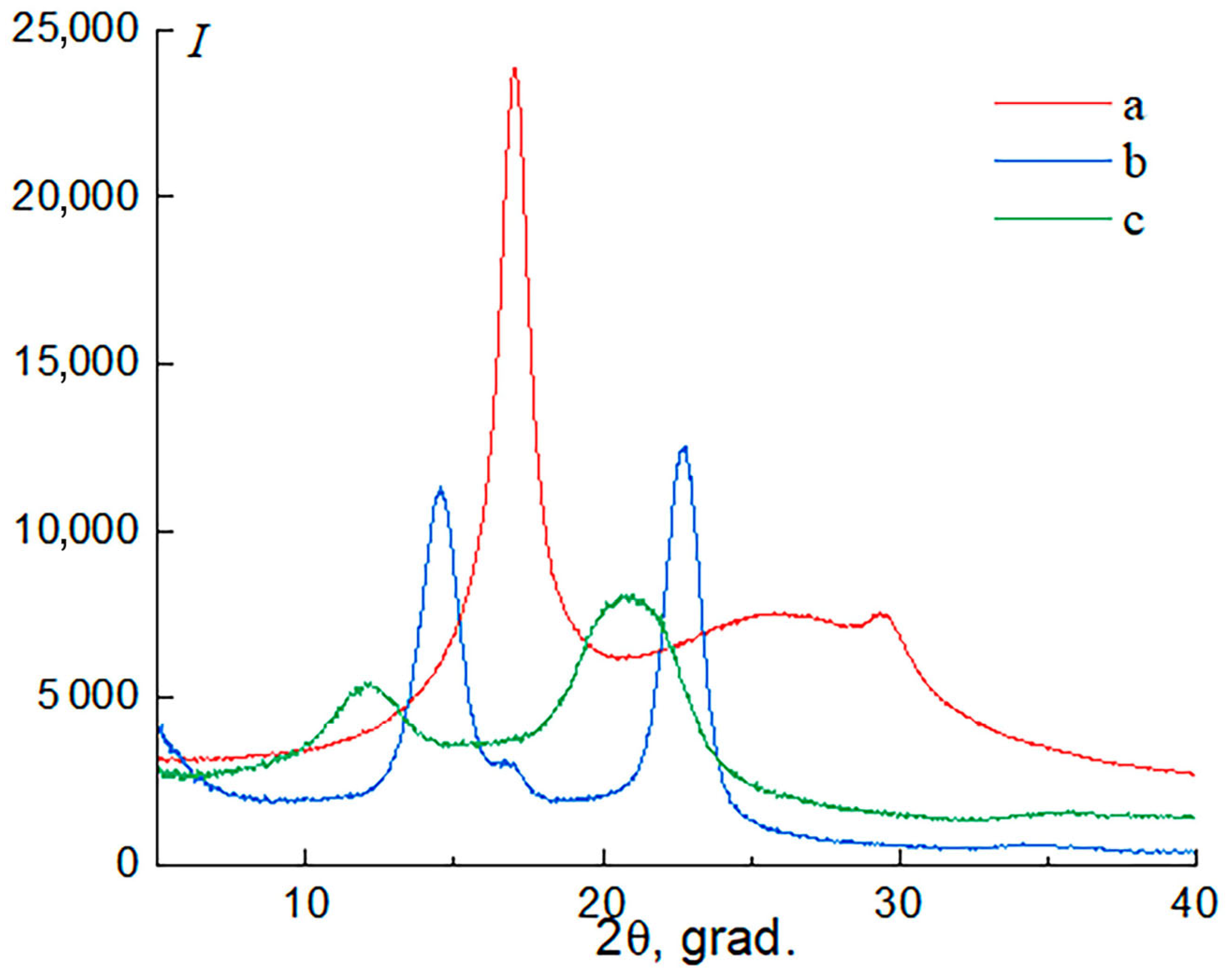
3.4. X-Ray Diffraction (XRD)
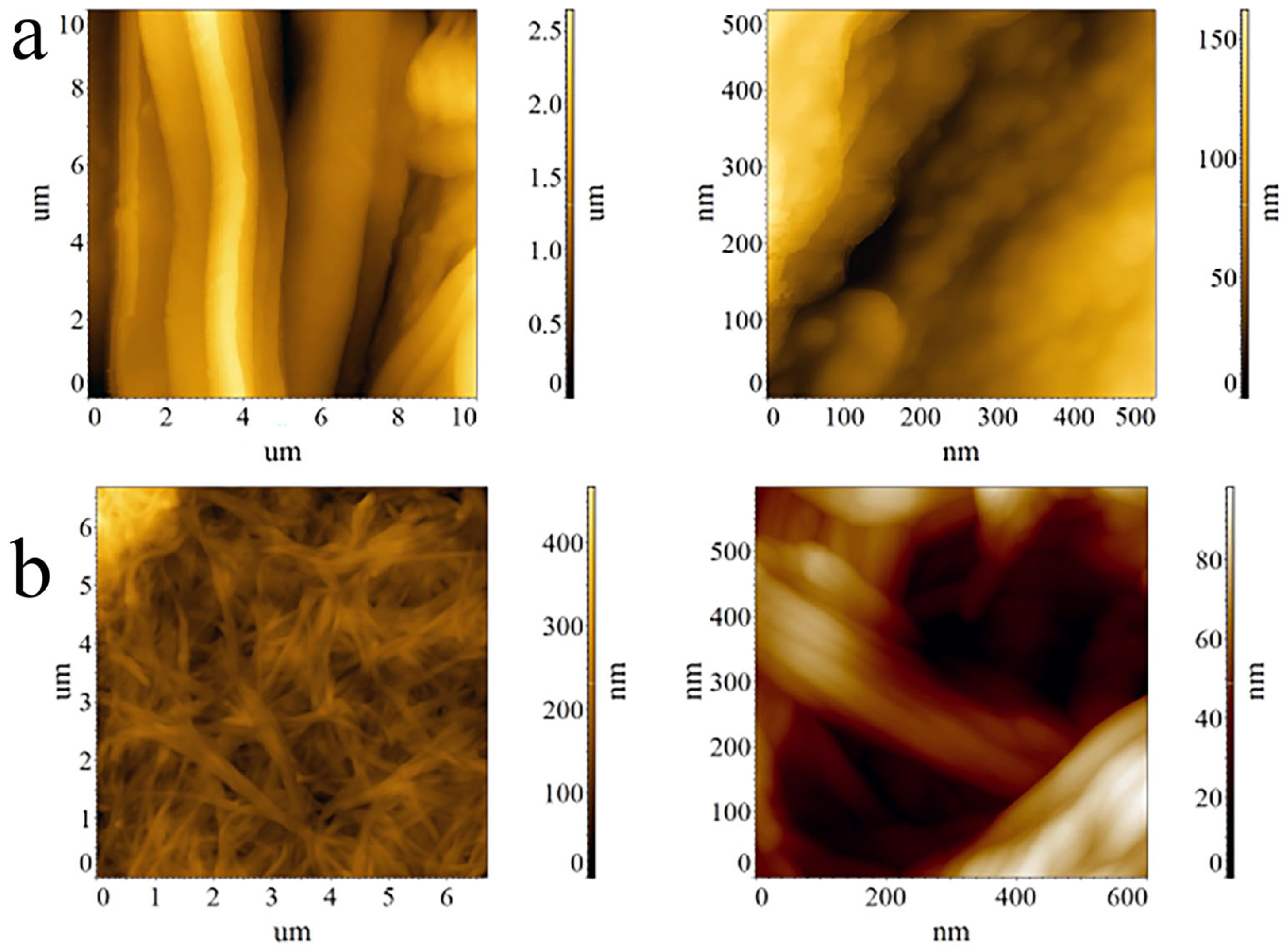
3.5. AFM Investigations of Cellulose Microfibers and Bacterial Cellulose
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Aleshina, L.A.; Mikhailina, A.A.; Lugovskaya, L.A. Structure of the coniferous bleached cellulose in the different states. For. Bull. 2015, 2, 107–114. Available online: https://rucont.ru/efd/416762 (accessed on 10 December 2024).
- Gibb, B.C. The centenary (maybe) of the hydrogen bond. Nat. Chem. 2020, 12, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L. The Nature of the Chemical Bond; Cornell University Press: Ithaca, NY, USA, 1960; p. 644. [Google Scholar]
- Wohlert, M.; Benselfelt, T.; Wågberg, L.; Furó, I.; Berglund, L.A.; Wohlert, J. Cellulose and the role of hydrogen bonds: Not in charge of everything. Cellulose 2022, 29, 1–23. [Google Scholar] [CrossRef]
- Liyanage, S.; Acharya, S.; Parajuli, P.; Shamshina, J.L.; Abidi, N. Production and Surface Modification of Cellulose Bioproducts. Polymers 2021, 13, 3433. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin/Heidelberg, Germany, 1994; p. 569. [Google Scholar] [CrossRef]
- Baskakov, S.A.; Baskakova, Y.V.; Kabachkov, E.N.; Kichigina, G.A.; Kushch, P.P.; Kiryukhin, D.P.; Krasnikova, S.S.; Badamshina, E.R.; Vasil’ev, S.G.; Soldatenkov, T.A.; et al. Cellulose from Annual Plants and Its Use for the Production of the Films Hydrophobized with Tetrafluoroethylene Telomers. Molecules 2022, 27, 6002. [Google Scholar] [CrossRef]
- Makarov, I.S.; Golova, L.K.; Vinogradov, M.I.; Egorov, Y.E.; Kulichikhin, V.G.; Mikhailov, Y.M. New Hydrated Cellulose Fiber Based on Flax Cellulose. Russ. J. Gen. Chem. 2021, 91, 1807–1815. [Google Scholar] [CrossRef]
- Tarchevsky, J.A.; Marchenko, G.N. Cellulose: Biosynthesis and Structure; Springer: Berlin/Heidelberg, Germany, 1991; p. 322. [Google Scholar] [CrossRef]
- Korchagina, A.A.; Gismatulina, Y.A.; Budaeva, V.V.; Zolotukhin, V.N.; Bychin, N.V.; Sakovich, G.V. Miscanthus × Giganteus var. KAMIS as a new feedstock for cellulose nitrates. J. Sib. Fed. Univ. Chem. 2020, 13, 565–577. [Google Scholar] [CrossRef]
- Makarov, I.S.; Golova, L.K.; Vinogradov, M.I.; Levin, I.S.; Gromovykh, T.I.; Arkharova, N.A.; Kulichikhin, V.G. Cellulose Fibers from Solutions of Bacterial Cellulose in N-Methylmorpholine N-Oxide. Fibre Chem. 2019, 51, 175–181. [Google Scholar] [CrossRef]
- Miri, M.; Ghasemian, A.; Resalati, H.; Zeinaly, F. Total chlorine-free bleaching of Populus deltoides kraft pulp by oxone. Int. J. Carbohydr. Chem. 2015, 2015, 381242. [Google Scholar] [CrossRef]
- Gromovykh, T.I.; Feldman, N.B.; Tikhonova, O.A.; Lutsenko, S.V.; Timashev, P.S.; Bardakova, K.N.; Churbanov, S.N.; Kiselyova, O.I.; Kraeva, M.N.; Grinevich, A.S. Elaboration of a bacterial cellulose matrix for the immobilisation of Escherichia coli cell. Int. J. Nanotechnol. 2018, 15, 288–300. [Google Scholar] [CrossRef]
- Colvin, J.R. The size of the cellulose microfibril. JCB 1963, 17, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Zhao, S.; Shen, W.; Collings, C.; Ding, S.-Y. Direct Measurement of Plant Cellulose Microfibril and Bundles in Native Cell Walls. Front. Plant Sci. 2020, 11, 479. [Google Scholar] [CrossRef] [PubMed]
- Newman, R.H.; Hill, S.J.; Harris, P.J. Wide-angle x-ray scattering and solid-state nuclear magnetic resonance data combined to test models for cellulose microfibrils in mung bean cell walls. Plant Physiol. 2013, 163, 1558–1567. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Hong, M. Solid-state NMR investigations of cellulose structure and interactions with matrix polysaccharides in plant primary cell walls. J. Exp. Bot. 2016, 67, 503–514. [Google Scholar] [CrossRef]
- Ioelovich, M. Nanostructured cellulose: Review. BioRes 2008, 3, 1403–1418. [Google Scholar] [CrossRef]
- Fink, H.P.; Purz, H.; Bohn, A.; Kunze, J. Structural investigations of bacterial cellulose. Macromol. Symp. 1997, 120, 207–217. [Google Scholar] [CrossRef]
- Nicolas, W.J.; Ghosal, D.; Tocheva, E.I.; Meyerowitz, E.M.; Jensen, G.J. Structure of the Bacterial Cellulose Ribbon and Its Assembly-Guiding Cytoskeleton by Electron Cryotomography. J. Bacteriol. 2021, 203, e00371-20. [Google Scholar] [CrossRef]
- Park, S.; Baker, J.-O.; Himmel, M.-E.; Parilla, P.-A.; Johnson, D.-K. Cellulose crystallinity index: Measurement techniques and their impact on interpreting cellulose performance. Biotechnol. Biofuels 2010, 3, 10. [Google Scholar] [CrossRef]
- Frone, A.N.; Chiulan, I.; Panaitescu, D.M.; Nicolae, C.A.; Ghiurea, M.; Galan, A.-M. Isolation of cellulose nanocrystals from plum seed shells, structural and morphological characterization. Mater. Lett. 2017, 194, 160–163. [Google Scholar] [CrossRef]
- Trache, D.; Tarchoun, A.F.; Derradji, M.; Hamidon, T.S.; Masruchin, N.; Brosse, N.; Hussin, M.H. Nanocellulose: From Fundamentals to Advanced Applications. Front. Chem. 2020, 8, 392. [Google Scholar] [CrossRef]
- Mao, J.; Abushammala, H.; Brown, N.; Laborie, M.-P. Comparative Assessment of Methods for Producing Cellulose I Nanocrystals from Cellulosic Sources. ACS Symp. Ser. 2017, 1251, 19–53. [Google Scholar] [CrossRef]
- Lavoine, N.; Desloges, I.; Dufresne, A.; Bras, J. Microfibrillated cellulose—Its barrier properties and applications in cellulosic materials: A review. Carbohydr. Polym. 2012, 90, 735–764. [Google Scholar] [CrossRef] [PubMed]
- da Silva Maradini, G.; Oliveira, M.P.; da Silva Guanaes, G.M.; Passamani, G.Z.; Carreira, L.G.; Boschetti, W.T.; Monteiro, S.N.; Pereira, A.C.; de Oliveira, B.F. Characterization of Polyester Nanocomposites Reinforced with Conifer Fiber Cellulose Nanocrystals. Polymers 2020, 12, 2838. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Dong, C.; Gao, Y.; Jin, Z. A Green and Iridescent Composite of Cellulose Nanocrystals with Wide Solvent Resistance and Strong Mechanical Properties. ACS Sustain. Chem. Eng. 2021, 9, 6764–6775. [Google Scholar] [CrossRef]
- Taheri, H.; Mastali, M.; Falah, M.; Abdollahnejad, Z.; Ghiassi, B.; Perrot, A.; Kawashima, S. Microfibrillated cellulose as a new approach to develop lightweight cementitious composites: Rheological, Mechanical, and microstructure perspectives. CBM 2022, 342, 128008. [Google Scholar] [CrossRef]
- Ul-Islam, M.; Khan, S.; Ullah, M.W.; Park, J.K. Bacterial cellulose composites: Synthetic strategies and multiple applications in bio-medical and electro-conductive fields. Biotechnol. J. 2015, 10, 1847–1861. [Google Scholar] [CrossRef]
- Cheng, Q.; Wang, S.; Han, Q. Novel process for isolating fibrils from cellulose fibers by high-intensity ultrasonication. II. Fibril characterization. J. Appl. Polym. Sci. 2010, 115, 2756–2762. [Google Scholar] [CrossRef]
- Lahiji, R.R.; Xu, X.; Reifenberger, R.; Raman, A.; Rudie, A.; Moon, R.J. Atomic Force Microscopy Characterization of Cellulose Nanocrystals. Langmuir 2010, 26, 4480–4488. [Google Scholar] [CrossRef]
- Parvej, M.S.; Wang, X.; Jiang, L. AFM based nanomechanical characterization of cellulose nanofibril. J. Compos. Mater. 2020, 54, 1–7. [Google Scholar] [CrossRef]
- Vinogradov, M.I.; Golova, L.K.; Makarov, I.S.; Bondarenko, G.N.; Levin, I.S.; Arkharova, N.A.; Kulichikhin, V.G. Transformation of Specific Dispersion Interactions between Cellulose and Polyacrylonitrile in Solutions into Covalent Interactions in Fibers. Materials 2023, 16, 5843. [Google Scholar] [CrossRef]
- Golova, L.K.; Borodina, O.E.; Kuznetsova, L.K.; Lyubova, T.A.; Krylova, T.B. The Solid-Phase MMO Process. Fibre Chem. 2000, 32, 243–251. [Google Scholar] [CrossRef]
- Meyer, K.H.; Lotmar, W. Sur l’élasticité de la cellulose. (Sur la constitution de la partie cristallisée de la cellulose IV). Helv. Chim. Acta 1936, 19, 68–86. [Google Scholar] [CrossRef]
- Diddens, I.; Murphy, B.; Krisch, M.; Muller, M. Anisotropic Elastic Properties of Cellulose Measured Using Inelastic X-ray Scattering. Macromolecules 2008, 41, 9755. [Google Scholar] [CrossRef]
- Hellwig, J.; Durán, V.L.; Pettersson, T. Measuring elasticity of wet cellulose fibres with AFM using indentation and a linearized Hertz model. Anal. Methods 2018, 10, 3820–3823. [Google Scholar] [CrossRef]
- Eichhorn, S.; Young, R. The Young’s modulus of a microcrystalline cellulose. Cellulose 2001, 8, 197–207. [Google Scholar] [CrossRef]
- Cheng, Q.; Wang, S. A method for testing the elastic modulus of single cellulose fibrils via atomic force microscopy. Compos. A Appl. Sci. Manuf. 2008, 39, 1838–1843. [Google Scholar] [CrossRef]
- Kroon-Batenburg, L.M.J.; Kroon, J.; Northolt, M.G. Chain modulus and intramolecular hydrogen bonding in native and regenerated cellulose fibers. Polym. Commun. 1986, 27, 290–292. [Google Scholar]
- Moud, A.A. Cellulose Nanocrystals Examined by Atomic Force Microscopy: Applications and Fundamentals. ACS Food Sci. Technol. 2022, 2, 1789–1818. [Google Scholar] [CrossRef]
- Makarov, I.; Vinogradov, M.; Golubev, Y.; Palchikova, E.; Kulanchikov, Y.; Grishin, T. Development of Cellulose Microfibers from Mixed Solutions of PAN-Cellulose in N-Methylmorpholine-N-Oxide. Polymers 2024, 16, 1869. [Google Scholar] [CrossRef]
- Makarov, I.S.; Golova, L.K.; Kuznetsova, L.K.; Shlyakhtin, A.V.; Nifantiev, I.E.; Kulichikhin, V.G. Method for Preparing a Solution of Acrylonitrile-Based Copolymer in n-Methylmorpholine-n-Oxide. RF Patent 2541473, 13 June 2013. [Google Scholar]
- Golova, L.K.; Romanov, V.V.; Lunina, O.B.; Platonov, V.A.; Papkov, S.P.; Khorozova, O.D.; Yakshin, V.V.; Belasheva, T.P.; Sokira, A.N. Method for Producing Solution for Spinning Fibers. RF Patent 1645308, 30 April 1991. [Google Scholar]
- Fink, H.P.; Hofmann, D.; Philipp, B. Some aspects of lateral chain order in cellulosics from X-ray scattering. Cellulose 1995, 2, 51–70. [Google Scholar] [CrossRef]
- Segal, L.; Creely, J.J.; Martin, A.E.; Conrad, C.M. An Empirical Method for Estimating the Degree of Crystallinity of Native Cellulose Using the X-Ray Diffractometer. Text. Res. J. 1959, 29, 786–794. [Google Scholar] [CrossRef]
- Scherrer, P. Bestimmung der Größe und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. Nachr. Ges. Wiss. Gottingen Math.-Phys. Kl. 1918, 1918, 98–100. Available online: http://eudml.org/doc/59018 (accessed on 10 December 2024).
- Pogorelova, N.A.; Chernigova, S.V.; Rogachev, E.A. Morphological features of the structure of bacterial cellulose and nanocomposites based on it for the manufacture of modern wound dressings. Vestnik Omskogo GAU 2019, 4, 131–141. [Google Scholar]
- Pavlov, I.N.; Kuznetsov, P.S.; Shilov, A.I. Study of the process of freeze drying of bacterial nanocellulose. Polzunovsky Vestn. 2020, 4, 88–94. [Google Scholar] [CrossRef]
- Bolgova, A.L.; Shevtsov, A.V.; Arkharova, N.A.; Karimov, D.N.; Makarov, I.S.; Gromovykh, T.I.; Klechkovskaya, V.V. Microstructure of Gel Films of Bacterial Cellulose Synthesized under Static Conditions of Cultivation of the Gluconacetobacter hansenii GH-1/2008 Strain on Nutrient Media with Different Carbon Sources. Crystallogr. Rep. 2023, 68, 607–614. [Google Scholar] [CrossRef]
- Choi, S.M.; Shin, E.J. The Nanofication and Functionalization of Bacterial Cellulose and Its Applications. Nanomaterials 2020, 10, 406. [Google Scholar] [CrossRef]
- Skvortsova, Z.N.; Gromovykh, T.I.; Grachev, V.S.; Traskin, V.Y. Physicochemical Mechanics of Bacterial Cellulose. Colloid. J. 2019, 81, 366–376. [Google Scholar] [CrossRef]
- Watanabe, K.; Tabuchi, M.; Morinaga, Y.; Yoshinaga, F. Structural Features and Properties of Bacterial Cellulose Produced in Agitated Culture. Cellulose 1998, 5, 187. [Google Scholar] [CrossRef]
- Czaja, W.; Romanovicz, D.; Brown, R.M. Structural investigations of microbial cellulose produced in stationary and agitated culture. Cellulose 2004, 11, 3. [Google Scholar] [CrossRef]
- Nelson, M.L.; O’Connor, R.T. Relation of Certain Infrared Bands to Cellulose Crystallinity and Crystal Lattice Type. Part II. A New Infrared Ratio for Estimation of Crystallinity in Celluloses I and II. J. Appl. Polym. Sci. 1964, 8, 1325–1341. [Google Scholar] [CrossRef]
- Bazarnova, N.G.; Karpova, E.V.; Katrakov, I.B.; Markin, V.I.; Mikushina, I.V.; Ol’khov, Y.A.; Khudenko, S.V. Methods for the study of wood and its derivatives. Benefit. Baranul. Alt. State Univ. 2002, 160. [Google Scholar]
- Kadimaliev, D.; Kezina, E.; Telyatnik, V.; Revin, V.; Parchaykina, O.; Syusin, I. Residual Brewer’s yeast biomass and bacterial cellulose as an alternative to toxic phenol-formaldehyde binders in production of pressed materials from waste wood. BioRes 2015, 10, 1644–1656. [Google Scholar] [CrossRef]
- Ivanova, N.V. Mathematical processing of the IR spectrum of cellulose. J. Appl. Spectrosc. 1989, 51, 301–306. [Google Scholar] [CrossRef]
- Carrillo, F.; Colom, X.; Sunol, J.J.; Saurina, J. Structural FTIR analysis and thermal characterization of lyocell and viscose-type fibres. Eur. Polym. J. 2004, 40, 2229–2234. [Google Scholar] [CrossRef]
- Nelson, M.L.; O’Connor, R.T. Relation of certain infrared bands to cellulose crystallinity and crystal lattice type. Part I. Spectra of lattice types I, II, III and amorphous cellulose. J. Appl. Polym. Sci. 1964, 8, 1311–1324. [Google Scholar] [CrossRef]
- Oh, S.Y.; Yoo, D.I.; Shin, Y.; Seo, G. FTIR analysis of cellulose treated with sodium hydroxide and carbon dioxide. Carbohydr. Res. 2005, 340, 417–428. [Google Scholar] [CrossRef]
- Nada, A.-A.M.A.; Kamel, S.; El-Sakhawy, M. Thermal behaviour and infrared spectroscopy of cellulose carbamates. Polym. Degrad. Stab. 2000, 70, 347–355. [Google Scholar] [CrossRef]
- Kulichikhin, V.; Golova, L.; Makarov, I.; Bondarenko, G.; Makarova, V.; Ilyin, S.; Skvortsov, I.; Berkovich, A. Solutions of acrylonitrile copolymers in N-methylmorpholine-Noxide: Structure, properties, fiber spinning. Eur. Polym. J. 2017, 92, 326–337. [Google Scholar] [CrossRef]
- Makarov, I.S.; Smyslov, A.G.; Palchikova, E.E.; Vinogradov, M.I.; Shandryuk, G.A.; Levin, I.S.; Arkharova, N.A.; Kulichikhin, V.G. Nonwoven materials based on natural and artificial fibers. Cellulose 2024, 31, 1927–1940. [Google Scholar] [CrossRef]
- Lee, C.M.; Gu, J.; Kafle, K.; Catchmark, J.; Kim, S.H. Cellulose produced by Gluconacetobacter xylinus strains ATCC 53524 and ATCC 23768: Pellicle formation, post-synthesis aggregation and fiber density. Carbohydr. Polym. 2015, 133, 270–276. [Google Scholar] [CrossRef]
- Wada, M.; Okano, T.; Sugiyama, J. Allomorphs of native crystalline cellulose I evaluated by two equatoriald-spacings. J. Wood Sci. 2001, 47, 124–128. [Google Scholar] [CrossRef]
- Kaplan, D.L. Biopolymers from Renewable Resources; Springer: Berlin/Heidelberg, Germany, 1998; p. 420. [Google Scholar] [CrossRef]
- Azubuike, C.P.; Rodríguez, H.; Okhamafe, A.O.; Rogers, R.D. Physicochemical properties of maize cob cellulose powders reconstituted from ionic liquid solution. Cellulose 2012, 19, 425–433. [Google Scholar] [CrossRef]
- Salem, K.S.; Kasera, N.K.; Rahman, M.A.; Jameel, H.; Habibi, Y.; Eichhorn, S.J.; French, A.D.; Pal, L.; Lucia, L.A. Comparison and assessment of methods for cellulose crystallinity determination. Chem. Soc. Rev. 2023, 52, 6417–6446. [Google Scholar] [CrossRef] [PubMed]
- Kreze, T.; Strnad, S.; Stana-Kleinschek, K.; Ribitsch, V. Influence of aqueous medium on mechanical properties of conventional and new environmentally friendly regenerated cellulose fibers. Mater. Res. Innov. 2001, 4, 107–114. [Google Scholar] [CrossRef]
- Sakurada, I.; Nukushina, Y.; Ito, T. Experimental determination of the elastic modulus of crystalline regions of oriented polymers. J. Polym. Sci. 1962, 57, 651–660. [Google Scholar] [CrossRef]
- Young, R.J.; Lovell, P.A. Introduction to Polymers, 2nd ed.; Chapman & Hall: London, UK, 1991; p. 443. [Google Scholar] [CrossRef]
- Chen, P.; Ogawa, Y.; Nishiyama, Y.; Ismail, A.E.; Mazeau, K. Linear, Non-Linear and Plastic Bending Deformation of Cellulose Nanocrystals. Phys. Chem. Chem. Phys. 2016, 18, 19880–19887. [Google Scholar] [CrossRef]
- Ganster, J.; Fink, H.-P.; Fraatz, J.; Nywlt, M. Relation between structure and elastic constants of man-made cellulosic fibres: I. a two phase anisotropic model with contiguity parameter. Acta Polymer. 1994, 45, 312–318. [Google Scholar] [CrossRef]
- Jiang, G.; Huang, W.; Li, L.; Wang, X.; Pang, F.; Zhang, Y.; Wang, H. Structure and properties of regenerated cellulose fibers from different technology processes. Carbohydr. Polym. 2012, 87, 2012–2018. [Google Scholar] [CrossRef]
- Tsouko, E.; Kourmentza, C.; Ladakis, D.; Kopsahelis, N.; Mandala, I.; Papanikolaou, S.; Paloukis, F.; Alves, V.; Koutinas, A. Bacterial Cellulose Production from Industrial Waste and by-Product Streams. Int. J. Mol. Sci. 2015, 16, 14832–14849. [Google Scholar] [CrossRef]
- French, A.D.; Cintrón, M.S. Cellulose polymorphy, crystallite size, and the segal crystallinity index. Cellulose 2013, 20, 583–588. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makarov, I.; Palchikova, E.; Vinogradov, M.; Golubev, Y.; Legkov, S.; Gromovykh, P.; Makarov, G.; Arkharova, N.; Karimov, D.; Gainutdinov, R. Characterization of Structure and Morphology of Cellulose Lyocell Microfibers Extracted from PAN Matrix. Polysaccharides 2025, 6, 10. https://doi.org/10.3390/polysaccharides6010010
Makarov I, Palchikova E, Vinogradov M, Golubev Y, Legkov S, Gromovykh P, Makarov G, Arkharova N, Karimov D, Gainutdinov R. Characterization of Structure and Morphology of Cellulose Lyocell Microfibers Extracted from PAN Matrix. Polysaccharides. 2025; 6(1):10. https://doi.org/10.3390/polysaccharides6010010
Chicago/Turabian StyleMakarov, Igor, Ekaterina Palchikova, Markel Vinogradov, Yaroslav Golubev, Sergey Legkov, Petr Gromovykh, Georgy Makarov, Natalia Arkharova, Denis Karimov, and Radmir Gainutdinov. 2025. "Characterization of Structure and Morphology of Cellulose Lyocell Microfibers Extracted from PAN Matrix" Polysaccharides 6, no. 1: 10. https://doi.org/10.3390/polysaccharides6010010
APA StyleMakarov, I., Palchikova, E., Vinogradov, M., Golubev, Y., Legkov, S., Gromovykh, P., Makarov, G., Arkharova, N., Karimov, D., & Gainutdinov, R. (2025). Characterization of Structure and Morphology of Cellulose Lyocell Microfibers Extracted from PAN Matrix. Polysaccharides, 6(1), 10. https://doi.org/10.3390/polysaccharides6010010