
PDK2: An Underappreciated Regulator of Liver Metabolism


Abstract
:1. Introduction
2. The PDKs and PDH
3. Physiological Roles of PDKs
3.1. PDKs in Normal Physiology
3.2. Specific Roles for PDK2 in Normal Physiology:
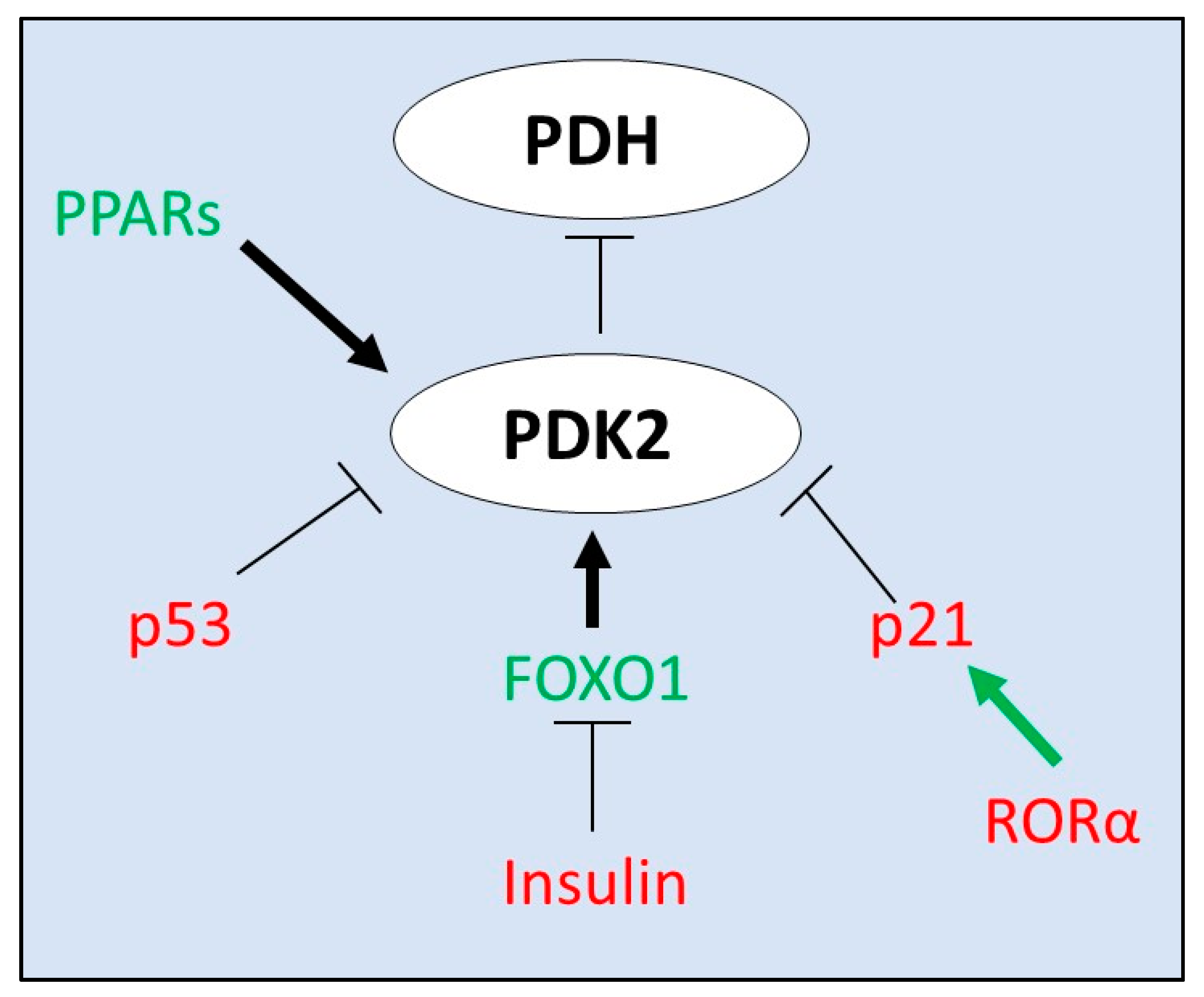
4. Transcriptional Regulators of PDK2 Expression
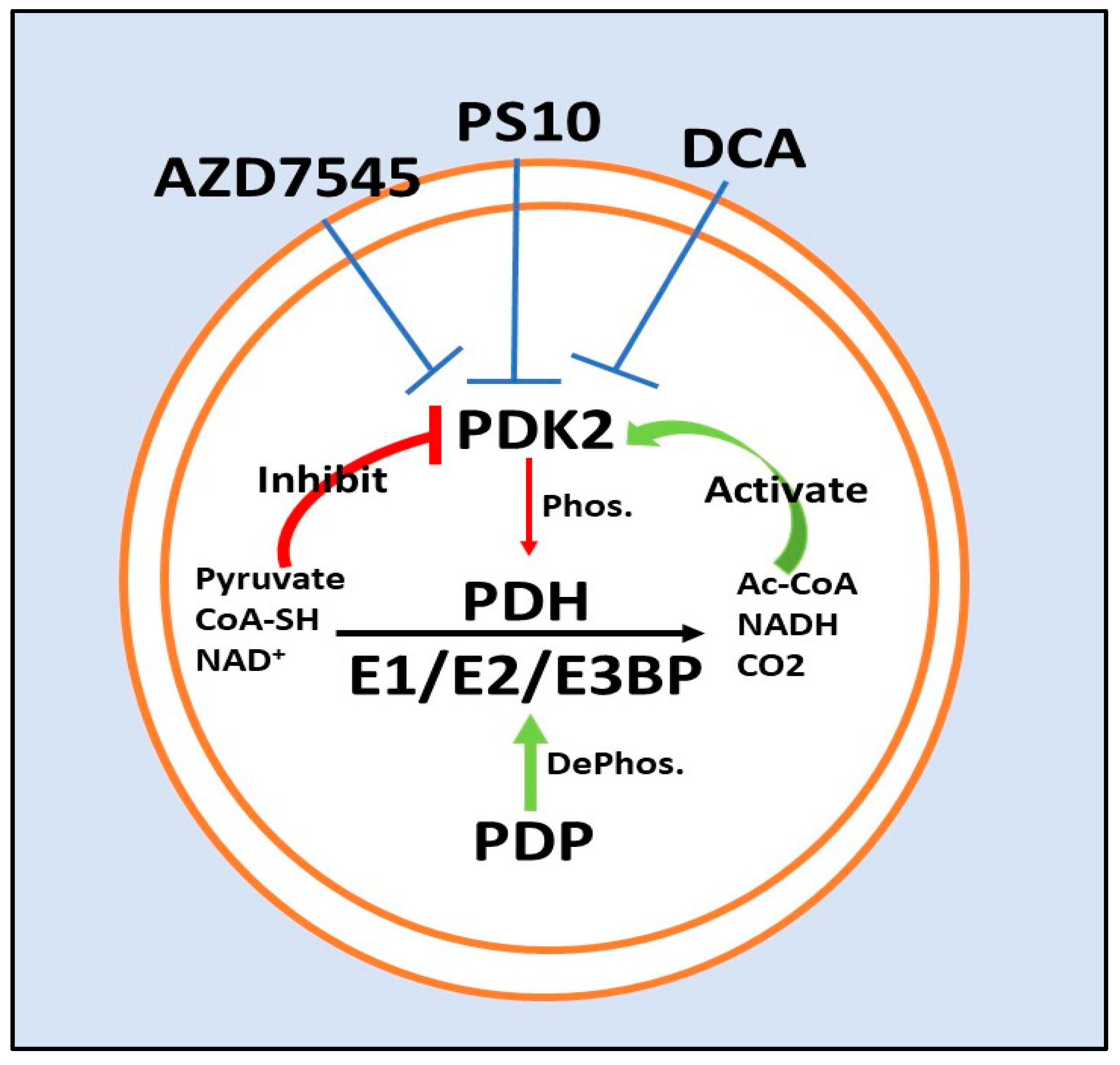
5. PDK2 Inhibitors
5.1. Dichloroacetate
5.2. ATP Binding Site/PS10
5.3. Lipoyl-Binding Domain/AZD7545
6. Pathophysiological Roles of PDKs in Disease
6.1. PDK2 and the Metabolic Syndrome
PDK2 and Nutrient Excess
6.2. PDK2 in Cancer
PDK2 in Hepatocellular Carcinoma
7. PDKs in Inflammation, a Growing Area of Interest
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bowker-Kinley, M.M.; Davis, I.W.; Wu, P.; Harris, A.R.; Popov, M.K. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 1998, 329, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolbright, B.L.; Rajendran, G.; Harris, R.A.; Taylor, J.A. Metabolic Flexibility in Cancer: Targeting the Pyruvate Dehydrogenase Kinase:Pyruvate Dehydrogenase Axis. Mol. Cancer Ther. 2019, 18, 1673–1681. [Google Scholar] [CrossRef] [Green Version]
- Klyuyeva, A.; Tuganova, A.; Kedishvili, N.Y.; Popov, K.M. Tissue-specific kinase expression and activity regulate flux through the pyruvate dehydrogenase complex. J. Biol. Chem. 2019, 294, 838–851. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Wu, P.; Bowker-Kinley, M.M.; Harris, R.A. Regulation of Pyruvate Dehydrogenase Kinase Expression by Peroxisome Proliferator-Activated Receptor- Ligands, Glucocorticoids, and Insulin. Diabetes 2002, 51, 276–283. [Google Scholar] [CrossRef] [Green Version]
- Sugden, M.C.; Holness, M.J. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate de-hydrogenase kinases. Arch Physiol. Biochem. 2006, 112, 139–149. [Google Scholar] [CrossRef]
- Jeoung, N.H.; Harris, R.A. Role of Pyruvate Dehydrogenase Kinase 4 in Regulation of Blood Glucose Levels. Korean Diabetes J. 2010, 34, 274–283. [Google Scholar] [CrossRef] [Green Version]
- Choiniere, J.; Wu, J.; Wang, L. Pyruvate Dehydrogenase Kinase 4 Deficiency Results in Expedited Cellular Proliferation through E2F1-Mediated Increase of Cyclins. Mol. Pharmacol. 2016, 91, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Choiniere, J.; Lin, M.J.; Wang, L.; Wu, J. Deficiency of pyruvate dehydrogenase kinase 4 sensitizes mouse liver to diethylnitrosamine and arsenic toxicity through inducing apoptosis. Liver Res. 2018, 2, 100–107. [Google Scholar] [CrossRef]
- Jeoung, N.H.; Rahimi, Y.; Wu, P.; Lee, W.N.P.; Harris, R.A. Fasting induces ketoacidosis and hypothermia in PDHK2/PDHK4-double-knockout mice. Biochem. J. 2012, 443, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Linn, T.C.; Pettit, F.H.; Reed, L.J. Alpha-keto acid dehydrogenase complexes. X. Regulation of the activity of the pyruvate dehy-drogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc. Natl. Acad. Sci. USA 1969, 62, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Roche, T.E.; Baker, J.C.; Yan, X.; Hiromasa, Y.; Gong, X.; Peng, T.; Dong, J.; Turkan, A.; Kasten, S.A. Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog. Nucleic Acid Res. Mol. Biol. 2001, 70, 33–75. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; McQuibban, G.A. The Mitochondrial Rhomboid Protease PARL Is Regulated by PDK2 to Integrate Mitochondrial Quality Control and Metabolism. Cell Rep. 2017, 18, 1458–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, J.; Thoudam, T.; Choi, E.J.; Kim, M.; Harris, R.A.; Lee, I. Loss of metabolic flexibility as a result of overexpression of pyruvate dehydrogenase kinases in muscle, liver and the immune system: Therapeutic targets in metabolic diseases. J. Diabetes Investig. 2021, 12, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Li, J.; Chuang, J.L.; Chuang, D.T. Distinct Structural Mechanisms for Inhibition of Pyruvate Dehydrogenase Kinase Isoforms by AZD7545, Dichloroacetate, and Radicicol. Structure 2007, 15, 992–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klyuyeva, A.; Tuganova, A.; Popov, K.M. Allosteric Coupling in Pyruvate Dehydrogenase Kinase 2. Biochemistry 2008, 47, 8358–8366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugden, M.C.; Holness, M.J. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydro-genase complex by PDKs. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E855–E862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Zhou, S.; Chang, S.S.; McFate, T.; Verma, A.; Califano, J.A. Mitochondrial Mutations Contribute to HIF1 Accumulation via Increased Reactive Oxygen Species and Up-regulated Pyruvate Dehydrogenease Kinase 2 in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2009, 15, 476–484. [Google Scholar] [CrossRef] [Green Version]
- Dunford, E.C.; Herbst, E.A.; Jeoung, N.H.; Gittings, W.; Inglis, J.G.; Vandenboom, R.; Leblanc, P.J.; Harris, R.A.; Peters, S.J. PDH activation during in vitro muscle contractions in PDH kinase 2 knockout mice: Effect of PDH kinase 1 compensation. Am. J. Physiol. Integr. Comp. Physiol. 2011, 300, R1487–R1493. [Google Scholar] [CrossRef] [Green Version]
- Fuller, S.J.; Randle, P.J. Reversible phosphorylation of pyruvate dehydrogenase in rat skeletal-muscle mitochondria. Effects of starvation and diabetes. Biochem. J. 1984, 219, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.Y.; Jeoung, N.H.; Park, K.-G.; Lee, I.-K. Transcriptional Regulation of Pyruvate Dehydrogenase Kinase. Diabetes Metab. J. 2012, 36, 328–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, Y.; Jeong, J.Y.; Jeoung, N.H.; Jeon, J.-H.; Park, B.-Y.; Kang, H.-J.; Ha, C.-M.; Choi, Y.-K.; Lee, S.J.; Ham, H.J.; et al. Inhibition of Pyruvate Dehydrogenase Kinase 2 Protects Against Hepatic Steatosis Through Modulation of Tricarboxylic Acid Cycle Anaplerosis and Ketogenesis. Diabetes 2016, 65, 2876–2887. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Sato, J.; Zhao, Y.; Jaskiewicz, J.; Popov, M.K.; Harris, A.R. Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. Biochem. J. 1998, 329, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Majer, M.; Popov, K.M.; Harris, R.A.; Bogardus, C.; Prochazka, M. Insulin Downregulates Pyruvate Dehydrogenase Kinase (PDK) mRNA: Potential Mechanism Contributing to Increased Lipid Oxidation in Insulin-Resistant Subjects. Mol. Genet. Metab. 1998, 65, 181–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.-S.; Huang, B.; Unterman, T.G.; Harris, R.A. Protein Kinase B- Inhibits Human Pyruvate Dehydrogenase Kinase-4 Gene Induction by Dexamethasone Through Inactivation of FOXO Transcription Factors. Diabetes 2004, 53, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuyama, T.; Kitayama, K.; Yamashita, H.; Mori, N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem. J. 2003, 375, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rena, G.; Guo, S.; Cichy, S.C.; Unterman, T.G.; Cohen, P. Phosphorylation of the Transcription Factor Forkhead Family Member FKHR by Protein Kinase B. J. Biol. Chem. 1999, 274, 17179–17183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Patil, S.; Chauhan, B.; Guo, S.; Powell, D.R.; Le, J.; Klotsas, A.; Matika, R.; Xiao, X.; Franks, R.; et al. FoxO1 regulates multiple metabolic pathways in the liver: Effects on gluconeogenic, glycolytic, and lipogenic gene expression. J. Biol. Chem. 2006, 281, 10105–10117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degenhardt, T.; Saramäki, A.; Malinen, M.; Rieck, M.; Väisänen, S.; Huotari, A.; Herzig, K.H.; Müller, R.; Carlberg, C. Three members of the human pyruvate dehydrogenase kinase gene family are direct targets of the peroxisome proliferator-activated receptor beta/delta. J. Mol. Biol. 2007, 372, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.; Choi, Y.; Na Kang, Y.; Jang, B.K.; Kang, K.J.; Jeon, Y.H.; Lee, H.; Jeon, J.; Koo, S.; Jeong, W.; et al. Retinoic acid-related orphan receptor alpha reprograms glucose metabolism in glutamine-deficient hepatoma cells. Hepatology 2015, 61, 953–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contractor, T.; Harris, C.R. p53 Negatively Regulates Transcription of the Pyruvate Dehydrogenase Kinase Pdk2. Cancer Res. 2012, 72, 560–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, T.E.; Hiromasa, Y. Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell. Mol. Life Sci. 2007, 64, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Steussy, C.N.; Popov, K.M.; Bowker-Kinley, M.M.; Sloan, R.B., Jr.; Harris, R.A.; Hamilton, J.A. Structure of pyruvate dehydrogenase kinase. Novel folding pattern for a serine protein kinase. J. Biol. Chem. 2001, 276, 37443–37450. [Google Scholar] [CrossRef] [Green Version]
- Brough, P.A.; Aherne, W.; Barril, X.; Borgognoni, J.; Boxall, K.; Cansfield, J.E.; Cheung, K.-M.J.; Collins, I.; Davies, N.G.M.; Drysdale, M.J.; et al. 4,5-Diarylisoxazole Hsp90 Chaperone Inhibitors: Potential Therapeutic Agents for the Treatment of Cancer. J. Med. Chem. 2008, 51, 196–218. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.D.; Staniszewska, A.; Shaw, T.; D’Alessandro, J.; Davis, B.; Surgenor, A.; Baker, L.; Matassova, N.; Murray, J.; Macias, A.; et al. VER-246608, a novel pan-isoform ATP competitive inhibitor of pyruvate dehydrogenase kinase, disrupts Warburg metabolism and induces context-dependent cytostasis in cancer cells. Oncotarget 2014, 5, 12862–12876. [Google Scholar] [CrossRef] [Green Version]
- Tuganova, A.; Yoder, M.D.; Popov, K.M. An Essential Role of Glu-243 and His-239 in the Phosphotransfer Reaction Catalyzed by Pyruvate Dehydrogenase Kinase. J. Biol. Chem. 2001, 276, 17994–17999. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Agatsuma, T.; Nakano, H. Targeting of the protein chaperone, HSP90, by the transformation suppressing agent, radicicol. Oncogene 1998, 16, 2639–2645. [Google Scholar] [CrossRef] [Green Version]
- Tso, S.-C.; Qi, X.; Gui, W.-J.; Wu, C.-Y.; Chuang, J.L.; Wernstedt-Asterholm, I.; Morlock, L.K.; Owens, K.R.; Scherer, P.E.; Williams, N.S.; et al. Structure-guided Development of Specific Pyruvate Dehydrogenase Kinase Inhibitors Targeting the ATP-binding Pocket. J. Biol. Chem. 2014, 289, 4432–4443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, M.L.; Roche, T.E. Mechanism of pyruvate inhibition of kidney pyruvate dehydrogenasea kinase and synergistic inhibition by pyruvate and ADP. J. Biol. Chem. 1979, 254, 7191–7196. [Google Scholar] [CrossRef]
- Knoechel, T.R.; Tucker, A.D.; Robinson, C.M.; Phillips, C.; Taylor, W.; Bungay, P.J.; Kasten, S.A.; Roche, T.E.; Brown, D.G. Regulatory roles of the N-terminal domain based on crystal structures of human pyruvate dehydrogenase kinase 2 containing physiological and synthetic ligands. Biochemistry 2006, 45, 402–415. [Google Scholar] [CrossRef]
- Holness, M.; Sugden, M. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans. 2003, 31, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kato, M.; Chuang, D.T. Pivotal Role of the C-terminal DW-motif in Mediating Inhibition of Pyruvate Dehydrogenase Kinase 2 by Dichloroacetate. J. Biol. Chem. 2009, 284, 34458–34467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehouse, S.; Cooper, R.H.; Randle, P.J. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem. J. 1974, 141, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Babu, E.; Ramachandran, S.; Coothankandaswamy, V.; Elangovan, S.; Prasad, P.D.; Ganapathy, V.; Thangaraju, M. Role of SLC5A8, a plasma membrane transporter and a tumor suppressor, in the antitumor activity of dichloroacetate. Oncogene 2011, 30, 4026–4037. [Google Scholar] [CrossRef] [Green Version]
- James, M.O.; Jahn, S.C.; Zhong, G.; Smeltz, M.G.; Hu, Z.; Stacpoole, P.W. Therapeutic applications of dichloroacetate and the role of glutathione transferase zeta-1. Pharmacol. Ther. 2017, 170, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Jahn, S.C.; Solayman, M.H.M.; Lorenzo, R.J.; Langaee, T.; Stacpoole, P.W.; James, M.O. GSTZ1 expression and chloride concentrations modulate sensitivity of cancer cells to dichloroacetate. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 1202–1210. [Google Scholar] [CrossRef] [Green Version]
- Stacpoole, P.W. The pharmacology of dichloroacetate. Metabolism 1989, 38, 1124–1144. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Nagaraja, N.V.; Hutson, A.D. Efficacy of dichloroacetate as a lactate-lowering drug. J. Clin. Pharmacol. 2003, 43, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Shroads, A.L.; Langaee, T.; Coats, B.S.; Kurtz, T.L.; Bullock, J.R.; Weithorn, D.; Gong, Y.; Wagner, D.A.; Ostrov, D.A.; Johnson, J.A.; et al. Human Polymorphisms in the Glutathione Transferase Zeta 1/Maleylacetoacetate Isomerase Gene Influence the Toxicokinetics of Dichloroacetate. J. Clin. Pharmacol. 2012, 52, 837–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunbar, E.M.; Coats, B.S.; Shroads, A.L.; Langaee, T.; Lew, A.; Forder, J.R.; Shuster, J.J.; Wagner, D.A.; Stacpoole, P.W. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Investig. New Drugs 2014, 32, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacpoole, P.W.; Martyniuk, C.J.; James, M.O.; Calcutt, N.A. Dichloroacetate-induced peripheral neuropathy. Int. Rev. Neurobiol. 2019, 145, 211–238. [Google Scholar] [CrossRef]
- Tataranni, T.; Agriesti, F.; Pacelli, C.; Ruggieri, V.; Laurenzana, I.; Mazzoccoli, C.; Della-Sala, G.; Panebianco, C.; Pazienza, V.; Capitanio, N.; et al. Dichloroacetate Affects Mitochondrial Function and Stemness-Associated Properties in Pancreatic Cancer Cell Lines. Cells 2019, 8, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tataranni, T.; Piccoli, C. Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications. Oxidative Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef]
- Chen, J.; Guccini, I.; di Mitri, D.; Brina, D.; Revandkar, A.; Sarti, M.; Pasquini, E.; Alajati, A.; Pinton, S.; Losa, M.; et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat. Genet. 2018, 50, 219–228. [Google Scholar] [CrossRef]
- Zachar, Z.; Marecek, J.; Maturo, C.; Gupta, S.; Stuart, S.D.; Howell, K.; Schauble, A.; Lem, J.; Piramzadian, A.; Karnik, S.; et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J. Mol. Med. 2011, 89, 1137–1148. [Google Scholar] [CrossRef]
- Newsholme, E.A.; Crabtree, B.; Ardawi, M.S.M. The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci. Rep. 1985, 5, 393–400. [Google Scholar] [CrossRef]
- Dutta, R.; Inouye, M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem. Sci. 2000, 25, 24–28. [Google Scholar] [CrossRef]
- Tso, S.-C.; Lou, M.; Wu, C.-Y.; Gui, W.-J.; Chuang, J.L.; Morlock, L.K.; Williams, N.S.; Wynn, R.M.; Qi, X.; Chuang, D.T. Development of Dihydroxyphenyl Sulfonylisoindoline Derivatives as Liver-Targeting Pyruvate Dehydrogenase Kinase Inhibitors. J. Med. Chem. 2017, 60, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Morrell, J.; Orme, J.; Butlin, R.; Roche, T.; Mayers, R.; Kilgour, E. AZD7545 is a selective inhibitor of pyruvate dehydrogenase kinase 2. Biochem. Soc. Trans. 2003, 31, 1168–1170. [Google Scholar] [CrossRef] [PubMed]
- Mayers, R.; Butlin, R.; Kilgour, E.; Leighton, B.; Martin, D.; Myatt, J.; Orme, J.; Holloway, B. AZD7545, a novel inhibitor of pyruvate dehydrogenase kinase 2 (PDHK2), activates pyruvate dehydrogenase in vivo and improves blood glucose control in obese (fa/fa) Zucker rats. Biochem. Soc. Trans. 2003, 31, 1165–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, R.M.; Kato, M.; Chuang, J.L.; Tso, S.-C.; Li, J.; Chuang, D.T. Pyruvate Dehydrogenase Kinase-4 Structures Reveal a Metastable Open Conformation Fostering Robust Core-free Basal Activity. J. Biol. Chem. 2008, 283, 25305–25315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate Immunity and Inflammation in NAFLD/NASH. Dig. Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, I.A.; Wong, V.W.-S.; Loomba, R. Treatment candidacy for pharmacologic therapies for NASH. Clin. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Tso, S.C.; Chuang, J.L.; Gui, W.J.; Lou, M.; Sharma, G.; Khemtong, C.; Qi, X.; Wynn, R.M.; Chuang, D.T. Targeting hepatic pyruvate dehydrogenase kinases restores insulin signaling and mitigates ChREBP-mediated lipo-genesis in diet-induced obese mice. Mol. Metab. 2018, 12, 12–24. [Google Scholar] [CrossRef]
- Lassailly, G.; Caiazzo, R.; Ntandja-Wandji, L.-C.; Gnemmi, V.; Baud, G.; Verkindt, H.; Ningarhari, M.; Louvet, A.; Leteurtre, E.; Raverdy, V.; et al. Bariatric Surgery Provides Long-term Resolution of Nonalcoholic Steatohepatitis and Regression of Fibrosis. Gastroenterology 2020, 159, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, H.; Zheng, J.; Tong, J. Integrated analysis of transcriptomic and metabolomic data demonstrates the significant role of pyruvate carboxylase in the progression of ovarian cancer. Aging 2020, 12, 21874–21889. [Google Scholar] [CrossRef]
- Lin, Q.; He, Y.; Wang, X.; Zhang, Y.; Hu, M.; Guo, W.; He, Y.; Zhang, T.; Lai, L.; Sun, Z.; et al. Targeting Pyruvate Carboxylase by a Small Molecule Suppresses Breast Cancer Progression. Adv. Sci. 2020, 7, 1903483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Sánchez, R.; Marín-Hernández, Á.; Gallardo-Pérez, J.C.; Pacheco-Velázquez, S.C.; Robledo-Cadena, D.X.; Padilla-Flores, J.A.; Saavedra, E.; Rodríguez-Enríquez, S. Physiological Role of Glutamate Dehydrogenase in Cancer Cells. Front. Oncol. 2020, 10, 429. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; de Berardinis, R.J.; Wen, X.; Corbin, I.R.; Sherry, A.D.; Malloy, C.R.; Jin, E.S. Active pyruvate dehydrogenase and impaired gluconeogenesis in orthotopic hepatomas of rats. Metabolism 2019, 101, 153993. [Google Scholar] [CrossRef]
- Fekir, K.; Dubois-Pot-Schneider, H.; Désert, R.; Daniel, Y.; Glaise, D.; Rauch, C.; Morel, F.; Fromenty, B.; Musso, O.; Cabillic, F.; et al. Retrodifferentiation of Human Tumor Hepatocytes to Stem Cells Leads to Metabolic Reprogramming and Chemo-resistance. Cancer Res. 2019, 79, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Xiong, X.; Huang, G.; Liu, J.; Sheng, S.; Wang, H.; Qin, W. Dichloroacetate Enhances Adriamycin-Induced Hepatoma Cell Toxicity In Vitro and In Vivo by Increasing Reactive Oxygen Species Levels. PLoS ONE 2014, 9, e92962. [Google Scholar] [CrossRef]
- Shen, Y.-C.; Ou, D.-L.; Hsu, C.; Lin, K.-L.; Chang, C.-Y.; Lin, C.-Y.; Liu, S.-H.; Cheng, A.-L. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br. J. Cancer 2012, 108, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Meng, G.; Li, B.; Chen, A.; Zheng, M.; Xu, T.; Zhang, H.; Dong, J.; Wu, J.; Yu, D.; Wei, J. Targeting aerobic glycolysis by dichloroacetate improves Newcastle disease virus-mediated viro-immunotherapy in hepatocellular carcinoma. Br. J. Cancer 2020, 122, 111–120. [Google Scholar] [CrossRef]
- Palmer, C.S.; Ostrowski, M.; Balderson, B.; Christian, N.; Crowe, S.M. Glucose Metabolism Regulates T Cell Activation, Differentiation, and Functions. Front. Immunol. 2015, 6, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menk, A.V.; Scharping, N.E.; Moreci, R.S.; Zeng, X.; Guy, C.; Salvatore, S.; Bae, H.; Xie, J.; Young, H.A.; Wendell, S.G.; et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep. 2018, 22, 1509–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, B.-K.; Park, S.; Kang, H.-J.; Kim, D.W.; Ham, H.J.; Ha, C.-M.; Choi, B.-J.; Lee, J.Y.; Oh, C.J.; Yoo, E.K.; et al. Pyruvate Dehydrogenase Kinase Is a Metabolic Checkpoint for Polarization of Macrophages to the M1 Phenotype. Front. Immunol. 2019, 10, 944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Z.; Xie, N.; Cui, H.; Moellering, D.R.; Abraham, E.; Thannickal, V.J.; Liu, G. Pyruvate Dehydrogenase Kinase 1 Participates in Macrophage Polarization via Regulating Glucose Metabolism. J. Immunol. 2015, 194, 6082–6089. [Google Scholar] [CrossRef]
- Galvan-Pena, S.; O’Neill, L.A. Metabolic reprograming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar]
- Na, Y.R.; Jung, D.; Song, J.; Park, J.-W.; Hong, J.J.; Seok, S.H. Pyruvate dehydrogenase kinase is a negative regulator of interleukin-10 production in macrophages. J. Mol. Cell Biol. 2020, 12, 543–555. [Google Scholar] [CrossRef] [Green Version]
- Woolbright, B.L.; Jaeschke, H. Alcoholic Hepatitis: Lost in Translation. J. Clin. Transl. Hepatol. 2017, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Inhibitor | Target | Efficacy | Notes | Refs |
|---|---|---|---|---|
| Dichloroacetate | pyruvate binding site | millimolar | common inhibitor | [4] |
| PS10 | ATP binding site | micromolar | high specificity for PDKs 2 and 4, and to a lesser degree PDK1 | [36] |
| azd7545 | PDH E2 lipoyl domain | micromolar | higher specificity for PDK1 and PDK2 | [37,38] |
| 2-chloropropionate | pyruvate binding site | millimolar | DCA analog | [4] |
| radicicol | ATP binding site | micromolar | also inhibits HSP90 | [39] |
| VER-246608 | ATP binding site | millimolar | pan-isoform inhibitor | [40] |
| Compound 17, PS10 derivative | ATP binding site inhibitor | micromolar | poorly defined thus far | [36,41] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woolbright, B.L.; Harris, R.A. PDK2: An Underappreciated Regulator of Liver Metabolism. Livers 2021, 1, 82-97. https://doi.org/10.3390/livers1020008
Woolbright BL, Harris RA. PDK2: An Underappreciated Regulator of Liver Metabolism. Livers. 2021; 1(2):82-97. https://doi.org/10.3390/livers1020008
Chicago/Turabian StyleWoolbright, Benjamin L., and Robert A. Harris. 2021. "PDK2: An Underappreciated Regulator of Liver Metabolism" Livers 1, no. 2: 82-97. https://doi.org/10.3390/livers1020008
APA StyleWoolbright, B. L., & Harris, R. A. (2021). PDK2: An Underappreciated Regulator of Liver Metabolism. Livers, 1(2), 82-97. https://doi.org/10.3390/livers1020008