
TAM Receptors in the Pathophysiology of Liver Disease



Abstract
:1. Biology of TAM Receptors
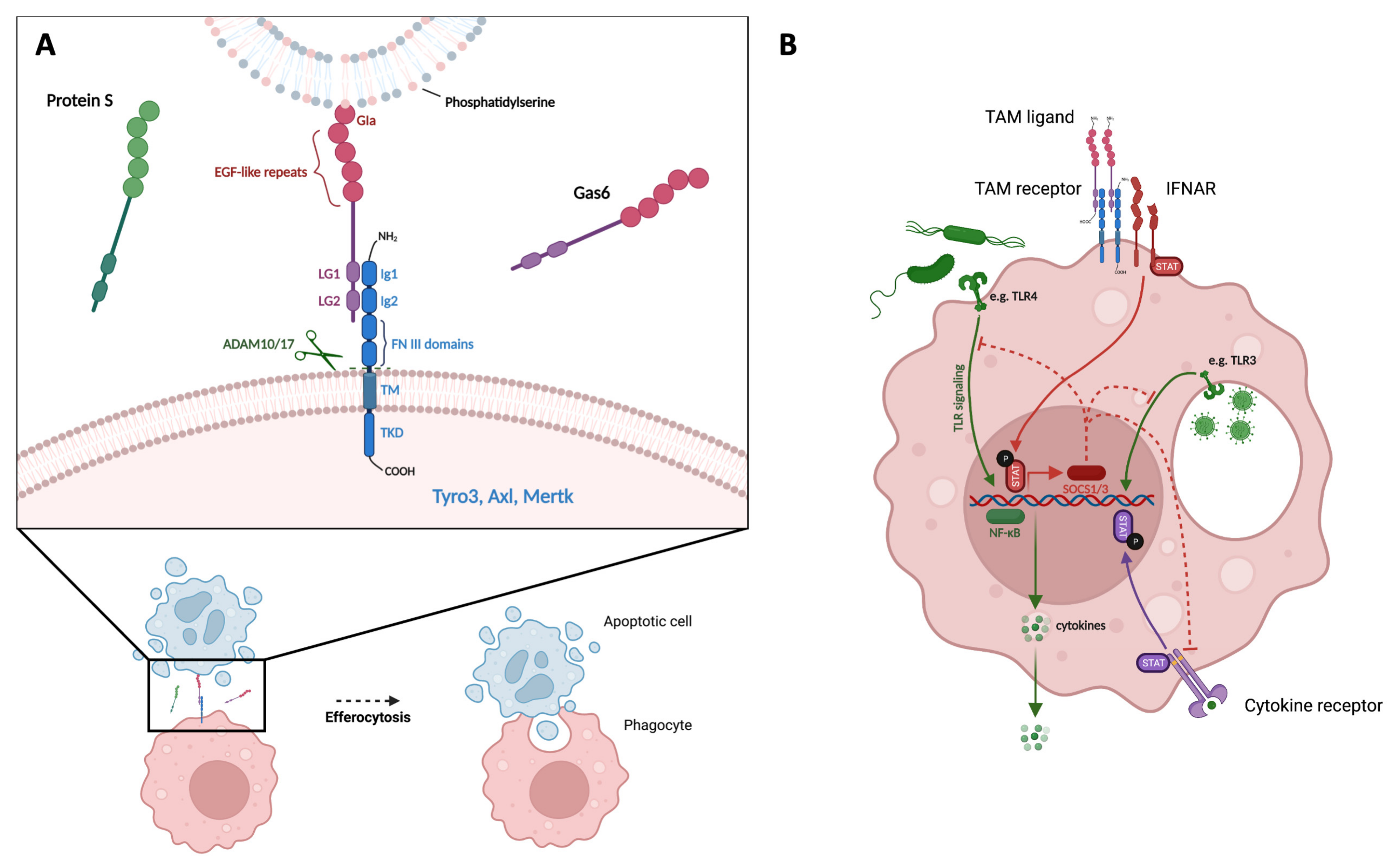
1.1. TAM Receptor and Ligand Structure
1.2. Function of TAM Receptors
1.3. Regulation of TAM Receptor Expression
2. TAM Receptors in Liver Disease
2.1. Liver Homeostasis
2.2. Acute Liver Injury
2.3. Liver Steatosis
2.4. Liver Fibrosis
2.5. Cirrhosis-Associated Immune Dysfunction
2.6. Hepatocellular Carcinoma
2.7. Cholestatic Diseases and Cholangiocarcinoma
3. Conclusions and Future Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM Receptor Tyrosine Kinases: Biologic Functions, Signaling, and Potential Therapeutic Targeting in Human Cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelillo-Scherrer, A.; Burnier, L.; Flores, N.; Savi, P.; DeMol, M.; Schaeffer, P.; Herbert, J.; Lemke, G.; Goff, S.P.; Matsushima, G.K.; et al. Role of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. J. Clin. Investig. 2005, 115, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, S.J.; Powell, M.J.; Franci, C.; Chan, E.W.; Friera, A.M.; Atchison, R.E.; McLaughlin, J.; Swift, S.E.; Pali, E.S.; Yam, G.; et al. Multiple roles for the receptor tyrosine kinase Axl in tumor formation. Cancer Res. 2005, 65, 9294–9303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto, A.L.; Weber, J.L.; Lai, C. Expression of the Receptor Protein-Tyrosine Kinases Tyro-3, Axl, and Mer in the Developing Rat Central Nervous System. J. Comp. Neurol. 2000, 425, 295–314. [Google Scholar] [CrossRef]
- Prieto, A.L.; Weber, J.L.; Tracy, S.; Heeb, M.J.; Lai, C. Gas6, a ligand for the receptor protein-tyrosine kinase Tyro-3, is widelyexpressed in the central nervous system. Brain Res. 1999, 816, 646–661. [Google Scholar] [CrossRef]
- Pierce, A.M.; Keating, A.K. TAM receptor tyrosine kinases: Expression, disease and oncogenesis in the central nervous system. Brain Res. 2014, 1542, 206–220. [Google Scholar] [CrossRef] [Green Version]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider’, C. The Protein Encoded by a Growth Arrest-Specific Gene (gas6) Is a New Member of the Vitamin K-Dependent Proteins Related to Protein S, a Negative Coregulator in the Blood Coagulation Cascade. Mol. Cell. Biol. 1993, 13, 4976–4985. [Google Scholar]
- Dahlbäck, B.; Villoutreix, B.O. Regulation of blood coagulation by the protein C anticoagulant pathway: Novel insights into structure-function relationships and molecular recognition. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1311–1320. [Google Scholar] [CrossRef]
- Nomura, K.; Vilalta, A.; Allendorf, D.H.; Hornik, T.C.; Brown, G.C. Activated Microglia Desialylate and Phagocytose Cells via Neuraminidase, Galectin-3, and Mer Tyrosine Kinase. J. Immunol. 2017, 198, 4792–4801. [Google Scholar] [CrossRef] [Green Version]
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Knyazev, P.G.; Clout, N.J.; Cheburkin, Y.; Göhring, W.; Ullrich, A.; Timpl, R.; Hohenester, E. Structural basis for Gas6-Axl signalling. EMBO J. 2006, 25, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the Product of Growth Arrest-specific Gene 6 as a Common Ligand for Axl, Sky, and Mer Receptor Tyrosine Kinases. Cell Biol. Metab. 1996, 271, 30022–30027. [Google Scholar] [CrossRef] [Green Version]
- Heiring, C.; Dahlbäck, B.; Muller, Y.A. Ligand recognition and homophilic interactions in Tyro3: Structural insights into the Axl/Tyro3 receptor tyrosine kinase family. J. Biol. Chem. 2004, 279, 6952–6958. [Google Scholar] [CrossRef] [Green Version]
- Tsou, W.-I.; Nguyen, K.-Q.N.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor Tyrosine Kinases, TYRO3, AXL, and MER, Demonstrate Distinct Patterns and Complex Regulation of Ligand-induced Activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef] [Green Version]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Ge, Y.; Ma, P.; Ma, Q.; Ma, J.; Wang, H.; Xue, S.; Han, D. Immunoexpression of Tyro 3 Family Receptors—Tyro 3, Axl, and Mer—and Their Ligand Gas6 in Postnatal Developing Mouse Testis. J. Histochem. Cytochem. 2005, 53, 1355–1364. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM Receptors Are Pleiotropic Inhibitors of the Innate Immune Response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-Y.; Wang, P.-G.; An, J. The Multifaceted Roles of TAM Receptors during Viral Infection. Virol. Sin. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Tutusaus, A.; Marí, M.; Ortiz-Pérez, J.T.; Nicolaes, G.A.F.; Morales, A.; De Frutos, P.G. Role of Vitamin K-dependent Factors Protein S and GAS6 and TAM Receptors in SARS-CoV-2 Infection and COVID-19-Associated Immunothrombosis. Cells 2020, 9, 2186. [Google Scholar] [CrossRef]
- Waterborg, C.E.J.; A Broeren, M.G.; Davidson, E.N.B.; I Koenders, M.; Lent, P.L.E.M.V.; Berg, W.B.V.D.; Van Der Kraan, P.M.; Loo, F.A.J.V.D. The level of synovial AXL expression determines the outcome of inflammatory arthritis, possibly depending on the upstream role of TGF-β1. Rheumatology 2018, 58, 536–546. [Google Scholar] [CrossRef]
- Brenig, R.; Pop, O.T.; Triantafyllou, E.; Geng, A.; Singanayagam, A.; Perez-Shibayama, C.; Besse, L.; Cupovic, J.; Künzler, P.; Boldanova, T.; et al. Expression of AXL receptor tyrosine kinase relates to monocyte dysfunction and severity of cirrhosis. Life Sci. Alliance 2019, 3, e201900465. [Google Scholar] [CrossRef]
- Zizzo, G.; Cohen, P.L. IL-17 Stimulates Differentiation of Human Anti-Inflammatory Macrophages and Phagocytosis of Apoptotic Neutrophils in Response to IL-10 and Glucocorticoids. J. Immunol. 2013, 190, 5237–5246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McColl, A.; Bournazos, S.; Franz, S.; Perretti, M.; Morgan, B.P.; Haslett, C.; Dransfield, I. Glucocorticoids Induce Protein S-Dependent Phagocytosis of Apoptotic Neutrophils by Human Macrophages. J. Immunol. 2009, 183, 2167–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, N.A.; Bensinger, S.J.; Hong, C.; Beceiro, S.; Bradley, M.N.; Zelcer, N.; Deniz, J.; Ramírez, C.; Díaz, M.; Gallardo, G.; et al. Apoptotic Cells Promote Their Own Clearance and Immune Tolerance through Activation of the Nuclear Receptor LXR. Immunity 2009, 31, 245–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahuczky, G.; Kristóf, E.; Majai, G.; Fésüs, L. Differentiation and Glucocorticoid Regulated Apopto-Phagocytic Gene Expression Patterns in Human Macrophages. Role of Mertk in Enhanced Phagocytosis. PLoS ONE 2011, 6, e21349. [Google Scholar] [CrossRef] [Green Version]
- Malawista, A.; Wang, X.; Trentalange, M.; Allore, H.G.; Montgomery, R.R. Coordinated expression of tyro3, axl, and mer receptors in macrophage ontogeny. Macrophage 2016, 3, e1261. [Google Scholar] [CrossRef] [Green Version]
- Sanjurjo, L.; Aran, G.; Téllez, E.; Amézaga, N.; Armengol, C.; López, D.; Prats, C.; Sarrias, M.-R. CD5L Promotes M2 Macrophage Polarization through Autophagy-Mediated Upregulation of ID3. Front. Immunol. 2018, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Grabiec, A.M.; Goenka, A.; E Fife, M.; Fujimori, T.; Hussell, T. Axl and MerTK receptor tyrosine kinases maintain human macrophage efferocytic capacity in the presence of viral triggers. Eur. J. Immunol. 2018, 48, 855–860. [Google Scholar] [CrossRef]
- Zagórska, A.; Través, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 2014, 15, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Shibata, T.; Habiel, D.M.; Coelho, A.L.; Kunkel, S.L.; Lukacs, N.W.; Hogaboam, C.M. Axl receptor blockade ameliorates pulmonary pathology resulting from primary viral infection and viral exacerbation of asthma. J. Immunol. 2014, 192, 3569–3581. [Google Scholar] [CrossRef] [Green Version]
- Orme, J.; DU, Y.; Vanarsa, K.; Mayeux, J.; Li, L.; Mutwally, A.; Arriens, C.; Min, S.; Hutcheson, J.; Davis, L.S.; et al. Heightened cleavage of Axl receptor tyrosine kinase by ADAM metalloproteases may contribute to disease pathogenesis in SLE. Clin. Immunol. 2016, 169, 58–68. [Google Scholar] [CrossRef] [Green Version]
- O’Bryan, J.P.; Fridell, Y.; Koski, R.; Varnum, B.; Liu, E.T. The Transforming Receptor Tyrosine Kinase, Axl, Is Post-translationally Regulated by Proteolytic Cleavage. J. Biol. Chem. 1995, 270, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Ekman, C.; Stenhoff, J.; Dahlbäck, B. Gas6 is complexed to the soluble tyrosine kinase receptor Axl in human blood. J. Thromb. Haemost. 2010, 8, 838–844. [Google Scholar] [CrossRef]
- Lu, Y.; Wan, J.; Yang, Z.; Lei, X.; Niu, Q.; Jiang, L.; Passtoors, W.M.; Zang, A.; Fraering, P.C.; Wu, F. Regulated intramembrane proteolysis of the AXL receptor kinase generates an intracellular domain that localizes in the nucleus of cancer cells. FASEB J. 2016, 31, 1382–1397. [Google Scholar] [CrossRef] [Green Version]
- Qi, N.; Liu, P.; Zhang, Y.; Wu, H.; Chen, Y.; Han, D. Development of a Spontaneous Liver Disease Resembling Autoimmune Hepatitis in Mice Lacking Tyro3, Axl and Mer Receptor Tyrosine Kinases. PLoS ONE 2013, 8, e66604. [Google Scholar] [CrossRef]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [Green Version]
- Zagórska, A.; Través, P.G.; Jiménez-García, L.; Strickland, J.D.; Oh, J.; Tapia, F.J.; Mayoral, R.; Burrola, P.; Copple, B.L.; Lemke, G. Differential regulation of hepatic physiology and injury by the TAM receptors Axl and Mer. Life Sci. Alliance 2020, 3, e202000694. [Google Scholar] [CrossRef]
- Triantafyllou, E.; Pop, O.T.; Possamai, L.A.; Wilhelm, A.; Liaskou, E.; Singanayagam, A.; Bernsmeier, C.; Khamri, W.; Petts, G.; Dargue, R.; et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut 2018, 67, 333–347. [Google Scholar] [CrossRef]
- Llacuna, L.; Bárcena, C.; Bellido-Martín, L.; Fernández, L.; Stefanovic, M.; Marí, M.; García-Ruiz, C.; Fernández-Checa, J.C.; de Frutos, P.G.; Morales, A. Growth arrest-specific protein 6 is hepatoprotective against murine ischemia/reperfusion injury. Hepatology 2010, 52, 1371–1379. [Google Scholar] [CrossRef] [Green Version]
- Lafdil, F.; Chobert, M.-N.; Deveaux, V.; Zafrani, E.-S.; Mavier, P.; Nakano, T.; Laperche, Y.; Brouillet, A. Growth arrest-specific protein 6 deficiency impairs liver tissue repair after acute toxic hepatitis in mice. J. Hepatol. 2009, 51, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Tutusaus, A.; de Gregorio, E.; Cucarull, B.; Cristóbal, H.; Aresté, C.; Graupera, I.; Coll, M.; Colell, A.; Gausdal, G.; Lorens, J.B.; et al. A Functional Role of GAS6/TAM in Nonalcoholic Steatohepatitis Progression Implicates AXL as Therapeutic Target. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 349–368. [Google Scholar] [CrossRef]
- Petta, S.; Valenti, L.; Marra, F.; Grimaudo, S.; Tripodo, C.; Bugianesi, E.; Cammà, C.; Cappon, A.; Di Marco, V.; Di Maira, G.; et al. MERTK rs4374383 polymorphism affects the severity of fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 682–690. [Google Scholar] [CrossRef]
- Fourcot, A.; Couchie, D.; Chobert, M.; Zafrani, E.; Mavier, P.; Laperche, Y.; Brouillet, A. Gas6 deficiency prevents liver inflammation, steatohepatitis, and fibrosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, 1043–1053. [Google Scholar] [CrossRef] [Green Version]
- Cai, B.; Dongiovanni, P.; Corey, K.E.; Wang, X.; Shmarakov, I.O.; Zheng, Z.; Kasikara, C.; Davra, V.; Meroni, M.; Chung, R.T.; et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2019, 31, 406–421.e7. [Google Scholar] [CrossRef]
- Bárcena, C.; Stefanovic, M.; Tutusaus, A.; Joannas, L.; Menéndez, A.; García-Ruiz, C.; Sancho-Bru, P.; Caballería, J.; Rothlin, C.V. Gas6/Axl pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J. Hepatol. 2015, 63, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Lafdil, F.; Chobert, M.N.; Couchie, D.; Brouillet, A.; Zafrani, E.S.; Mavier, P.; Laperche, Y. Induction of Gas6 protein in CCl4-induced rat liver injury and anti-apoptotic effect on hepatic stellate cells. Hepatology 2006, 44, 228–239. [Google Scholar] [CrossRef]
- Cavalli, M.; Pan, G.; Nord, H.; Arzt, E.W.; Wallerman, O.; Wadelius, C. Genetic prevention of hepatitis C virus-induced liver fibrosis by allele-specific downregulation of MERTK. Hepatol. Res. 2016, 47, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, J.H.M.; van der Poll, T.; van ’t Veer, C.V. TAM receptors, Gas6, and protein S: Roles in inflammation and hemostasis. Blood 2014, 123, 2460–2469. [Google Scholar] [CrossRef]
- Zermatten, M.G.; Fraga, M.; Moradpour, D.; Calderara, D.B.; Aliotta, A.; Stirnimann, G.; De Gottardi, A.; Alberio, L. Hemostatic Alterations in Patients with Cirrhosis: From Primary Hemostasis to Fibrinolysis. Hepatology 2020, 71, 2135–2148. [Google Scholar] [CrossRef] [PubMed]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albillos, A.; de la Hera, A.; Reyes, E.; Monserrat, J.; Muñoz, L.; Nieto, M.; Prieto, A.; Sanz, E.; Alvarez-Mon, M. Tumour necrosis factor-alpha expression by activated monocytes and altered T-cell homeostasis in ascitic alcoholic cirrhosis: Amelioration with norfloxacin. J. Hepatol. 2004, 40, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, L.; Albillos, A.; Nieto, M.; Reyes, E.; Lledó, L.; Monserrat, J.; Sanz, E.; de la Hera, A.; Alvarez-Mon, M. Mesenteric Th1 polarization and monocyte TNF-α production: First steps to systemic inflammation in rats with cirrhosis. Hepatology 2005, 42, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Fiuza, C.; Salcedo, M.; Clemente, G.; Tellado, J.M. Granulocyte Colony-Stimulating Factor Improves Deficient In Vitro Neutrophil Transendothelial Migration in Patients with Advanced Liver Disease. Clin. Vaccine Immunol. 2002, 9, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Devière, J.; Content, J.; Denys, C.; Vandenbussche, P.; Schandene, L.; Wybran, J.; Dupont, E. Excessive In Vitro Bacterial Lipopolysaccharide-induced Production of Monokines in Cirrhosis. Hepatology 1990, 11, 628–634. [Google Scholar] [CrossRef]
- Singanayagam, A.; Triantafyllou, E. Macrophages in Chronic Liver Failure: Diversity, Plasticity and Therapeutic Targeting. Front. Immunol. 2021, 12, 4. [Google Scholar] [CrossRef]
- Runyon, B.A.; Morrissey, R.L.; Hoefs, J.C.; Wyle, F.A. Opsonic activity of human ascitic fluid: A potentially important protective mechanism against spontaneous bacterial peritonitis. Hepatology 1985, 5, 634–637. [Google Scholar] [CrossRef]
- Helmy, K.Y.; Katschke, K.J., Jr.; Gorgani, N.N.; Kljavin, N.M.; Elliott, J.M.; Diehl, L.; Scales, S.J.; Ghilardi, N.; van Lookeren Campagne, M. CRIg: A Macrophage Complement Receptor Required for Phagocytosis of Circulating Pathogens. Cell 2006, 124, 915–927. [Google Scholar] [CrossRef] [Green Version]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Bernsmeier, C.; Pop, O.T.; Singanayagam, A.; Triantafyllou, E.; Patel, V.; Weston, C.J.; Curbishley, S.; Sadiq, F.; Vergis, N.; Khamri, W.; et al. Patients With Acute-on-Chronic Liver Failure Have Increased Numbers of Regulatory Immune Cells Expressing the Receptor Tyrosine Kinase MERTK. Gastroenterology 2015, 148, 603–615.e14. [Google Scholar] [CrossRef]
- Moreau, R.; Jalan, R.; Gines, P.; Pavesi, M.; Angeli, P.; Cordoba, J.; Durand, F.; Gustot, T.; Saliba, F.; Domenicali, M.; et al. Acute-on-Chronic Liver Failure Is a Distinct Syndrome That Develops in Patients with Acute Decompensation of Cirrhosis. Gastroenterology 2013, 144, 1426–1437.e9. [Google Scholar] [CrossRef]
- Arvaniti, V.; D’Amico, G.; Fede, G.; Manousou, P.; Tsochatzis, E.; Pleguezuelo, M.; Burroughs, A.K. Infections in Patients with Cirrhosis Increase Mortality Four-Fold and Should Be Used in Determining Prognosis. Gastroenterology 2010, 139, 1246–1256.e5. [Google Scholar] [CrossRef]
- Triantafyllou, E.; Woollard, K.J.; McPhail, M.; Antoniades, C.G.; Possamai, L.A. The Role of Monocytes and Macrophages in Acute and Acute-on-Chronic Liver Failure. Front. Immunol. 2018, 9, 2948. [Google Scholar] [CrossRef]
- Han, J.; Bae, J.; Choi, C.-Y.; Choi, S.-P.; Kang, H.-S.; Jo, E.-K.; Park, J.; Lee, Y.S.; Moon, H.-S.; Park, C.-G.; et al. Autophagy induced by AXL receptor tyrosine kinase alleviates acute liver injury via inhibition of NLRP3 inflammasome activation in mice. Autophagy 2016, 12, 2326–2343. [Google Scholar] [CrossRef] [Green Version]
- Bellan, M.; Sainaghi, P.P.; Minh, M.T.; Minisini, R.; Molinari, L.; Baldrighi, M.; Salmi, L.; Barbaglia, M.N.; Castello, L.M.; Ravanini, P.; et al. Gas6 as a predictor of esophageal varices in patients affected by hepatitis C virus related-chronic liver disease. Biomark. Med. 2018, 12, 27–34. [Google Scholar] [CrossRef]
- Staufer, K.; Dengler, M.; Huber, H.; Marculescu, R.; Stauber, R.; Lackner, C.; Dienes, H.-P.; Kivaranovic, D.; Schachner, C.; Zeitlinger, M.; et al. The non-invasive serum biomarker soluble Axl accurately detects advanced liver fibrosis and cirrhosis. Cell Death Dis. 2017, 8, e3135. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Holstein, E.; Binder, M.; Mikulits, W. Dynamics of Axl Receptor Shedding in Hepatocellular Carcinoma and Its Implication for Theranostics. Int. J. Mol. Sci. 2018, 19, 4111. [Google Scholar] [CrossRef] [Green Version]
- Reichl, P.; Dengler, M.; Van Zijl, F.; Huber, H.; Führlinger, G.; Reichel, C.; Sieghart, W.; Peck-Radosavljevic, M.; Grubinger, M.; Mikulits, W. Axl activates autocrine transforming growth factor-β signaling in hepatocellular carcinoma. Hepatology 2015, 61, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM receptors): Implications for macrophages in the tumor microenvironment. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Haider, C.; Hnat, J.; Wagner, R.; Huber, H.; Timelthaler, G.; Grubinger, M.; Coulouarn, C.; Schreiner, W.; Schlangen, K.; Sieghart, W.; et al. Transforming Growth Factor-β and Axl Induce CXCL5 and Neutrophil Recruitment in Hepatocellular Carcinoma. Hepatology 2018, 69, 222–236. [Google Scholar] [CrossRef] [Green Version]
- Chai, Z.-T.; Zhang, X.-P.; Ao, J.-Y.; Zhu, X.-D.; Wu, M.-C.; Lau, W.Y.; Sun, H.-C.; Cheng, S.-Q. AXL Overexpression in Tumor-Derived Endothelial Cells Promotes Vessel Metastasis in Patients with Hepatocellular Carcinoma. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Shen, L.; Lei, S.; Zhang, B.; Li, S.; Huang, L.; Czachor, A.; Breitzig, M.; Gao, Y.; Huang, M.; Mo, X.; et al. Skipping of exon 10 in Axl pre-mRNA regulated by PTBP1 mediates invasion and metastasis process of liver cancer cells. Theranostics 2020, 10, 5719–5735. [Google Scholar] [CrossRef]
- Li, X.-Y.; Wen, J.-Y.; Jia, C.-C.; Wang, T.-T.; Li, X.; Dong, M.; Lin, Q.; Chen, Z.-H.; Ma, X.-K.; Wei, L.; et al. MicroRNA-34a-5p enhances sensitivity to chemotherapy by targeting AXL in hepatocellular carcinoma MHCC-97L cells. Oncol. Lett. 2015, 10, 2691–2698. [Google Scholar] [CrossRef] [Green Version]
- Sauzay, C.; Petit, A.; Bourgeois, A.-M.; Barbare, J.-C.; Chauffert, B.; Galmiche, A.; Houessinon, A. Alpha-foetoprotein (AFP): A multi-purpose marker in hepatocellular carcinoma. Clin. Chim. Acta 2016, 463, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wu, A.; Ding, Z.; Liang, S.; Zhang, C. Soluble Axl Is a Novel Diagnostic Biomarker of Hepatocellular Carcinoma in Chinese Patients with Chronic Hepatitis B Virus Infection. Cancer Res. Treat. 2020, 52, 789–797. [Google Scholar] [CrossRef] [Green Version]
- Dengler, M.; Staufer, K.; Huber, H.; Stauber, R.; Bantel, H.; Weiss, K.H.; Starlinger, P.; Pock, H.; Plachky, P.K.; Gotthardt, D.N.; et al. Soluble Axl is an accurate biomarker of cirrhosis and hepatocellular carcinoma development: Results from a large scale multicenter analysis. Oncotarget 2017, 8, 46234–46248. [Google Scholar] [CrossRef]
- Trojan, J. Cabozantinib for the Treatment of Advanced Hepatocellular Carcinoma: Current Data and Future Perspectives. Drugs 2020, 80, 1203–1210. [Google Scholar] [CrossRef]
- Hayashi, M.; Abe, K.; Fujita, M.; Takahashi, A.; Hashimoto, Y.; Ohira, H. Serum Gas6 and Axl as non-invasive biomarkers of advanced histological stage in primary biliary cholangitis. Hepatol. Res. 2020, 50, 1337–1346. [Google Scholar] [CrossRef]
- Khamko, R.; Daduang, J.; Settasatian, C.; Limpaiboon, T. OPCML Exerts Antitumor Effects in Cholangiocarcinoma via AXL/STAT3 Inactivation and Rho GTPase Down-regulation. Cancer Genom.-Proteom. 2021, 18, 771–780. [Google Scholar] [CrossRef]
- Jung, D.E.; Park, S.B.; Kim, K.; Kim, C.; Song, S.Y. CG200745, an HDAC inhibitor, induces anti-tumour effects in cholangiocarcinoma cell lines via miRNAs targeting the Hippo pathway. Sci. Rep. 2017, 7, 10921. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, T.; Kato, K.; Fujihara, S.; Iwama, H.; Yamashita, T.; Kobayashi, K.; Kamada, H.; Morishita, A.; Kobara, H.; Mori, H.; et al. Antitumor effect of metformin on cholangiocarcinoma: In vitro and in vivo studies. Oncol. Rep. 2015, 34, 2987–2996. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Axl | MerTK | Tyro-3 | |
|---|---|---|---|
| Bone Marrow |

|

|

|
| Blood |


|


|

|
| Liver |
 
|
 
|

|
 = murine data;
= murine data;  = human data. (Health vs. Disease).
= human data. (Health vs. Disease).| Liver Pathophysiology | Axl Signaling | MerTK Signaling |
|---|---|---|
| Homeostasis |
 | |
| Acute liver injury |

|
 |
| Steatosis |

|

|
| Fibrosis |
 
| |
| Cirrhosis-associated immune dysfunction |

|

|
| Hepatocellular carcinoma |

| ø |
 = murine data;
= murine data;  = human data; ø = lack of findings.
= human data; ø = lack of findings.Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flint, E.; Triantafyllou, E.; Bernsmeier, C. TAM Receptors in the Pathophysiology of Liver Disease. Livers 2022, 2, 15-29. https://doi.org/10.3390/livers2010002
Flint E, Triantafyllou E, Bernsmeier C. TAM Receptors in the Pathophysiology of Liver Disease. Livers. 2022; 2(1):15-29. https://doi.org/10.3390/livers2010002
Chicago/Turabian StyleFlint, Emilio, Evangelos Triantafyllou, and Christine Bernsmeier. 2022. "TAM Receptors in the Pathophysiology of Liver Disease" Livers 2, no. 1: 15-29. https://doi.org/10.3390/livers2010002
APA StyleFlint, E., Triantafyllou, E., & Bernsmeier, C. (2022). TAM Receptors in the Pathophysiology of Liver Disease. Livers, 2(1), 15-29. https://doi.org/10.3390/livers2010002