
Unveiling Mobilizable Multiresistance Clusters in Marine Bacteria †

Abstract
:1. Introduction
2. Materials and Methods
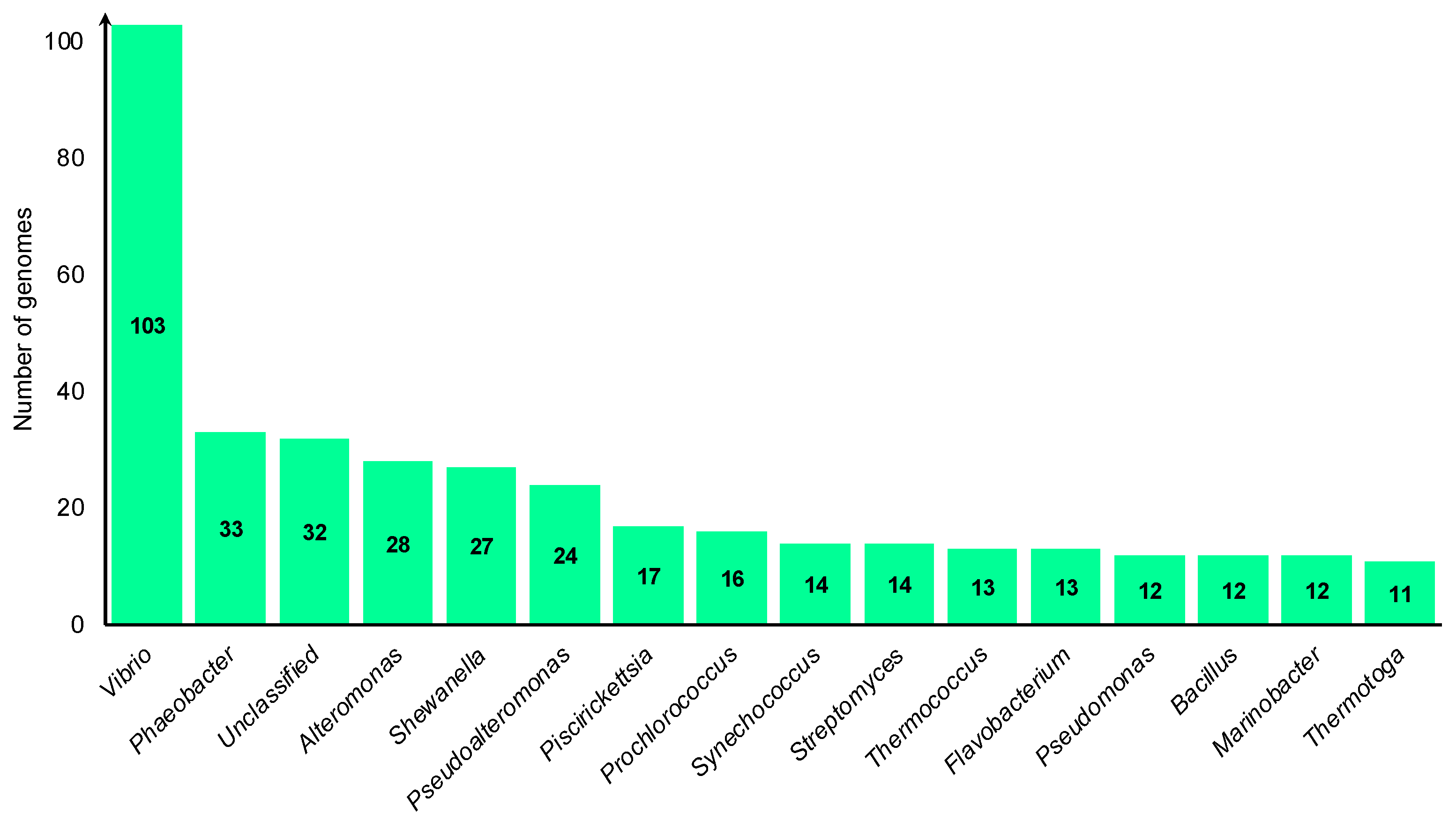
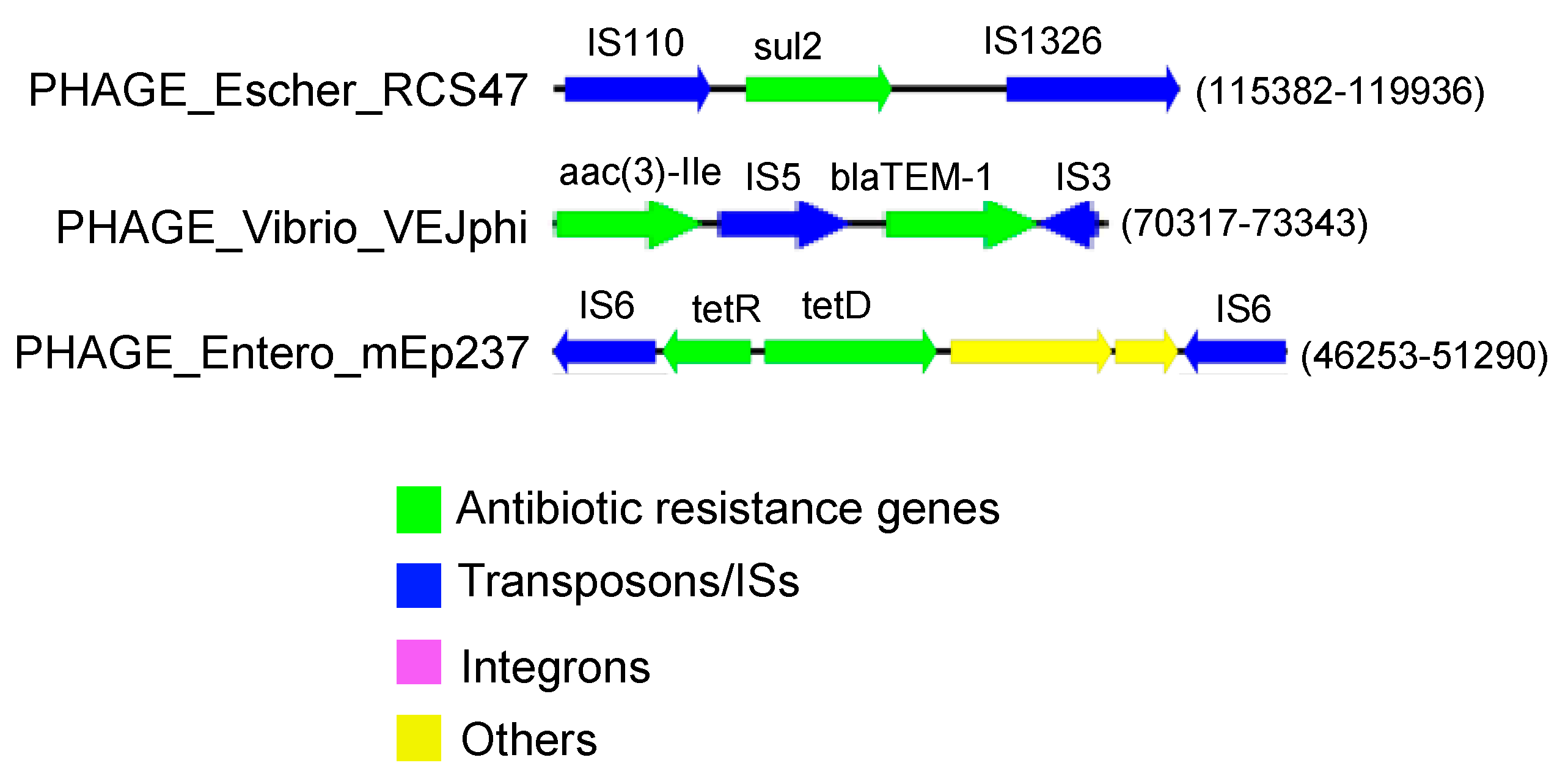
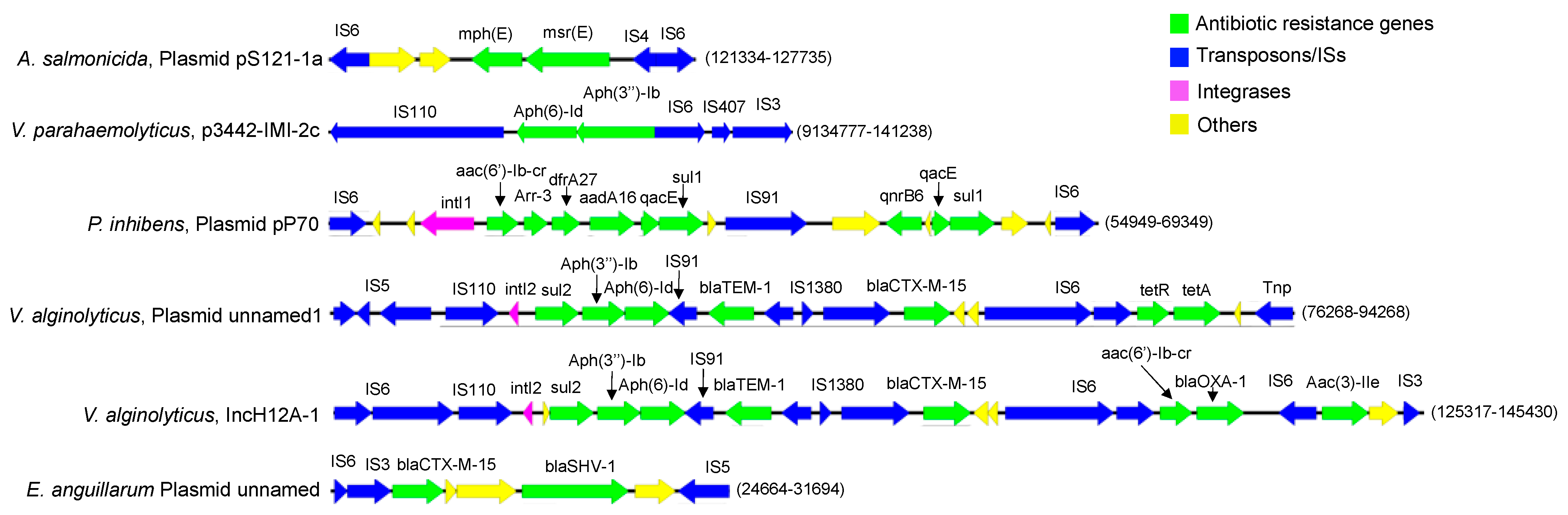
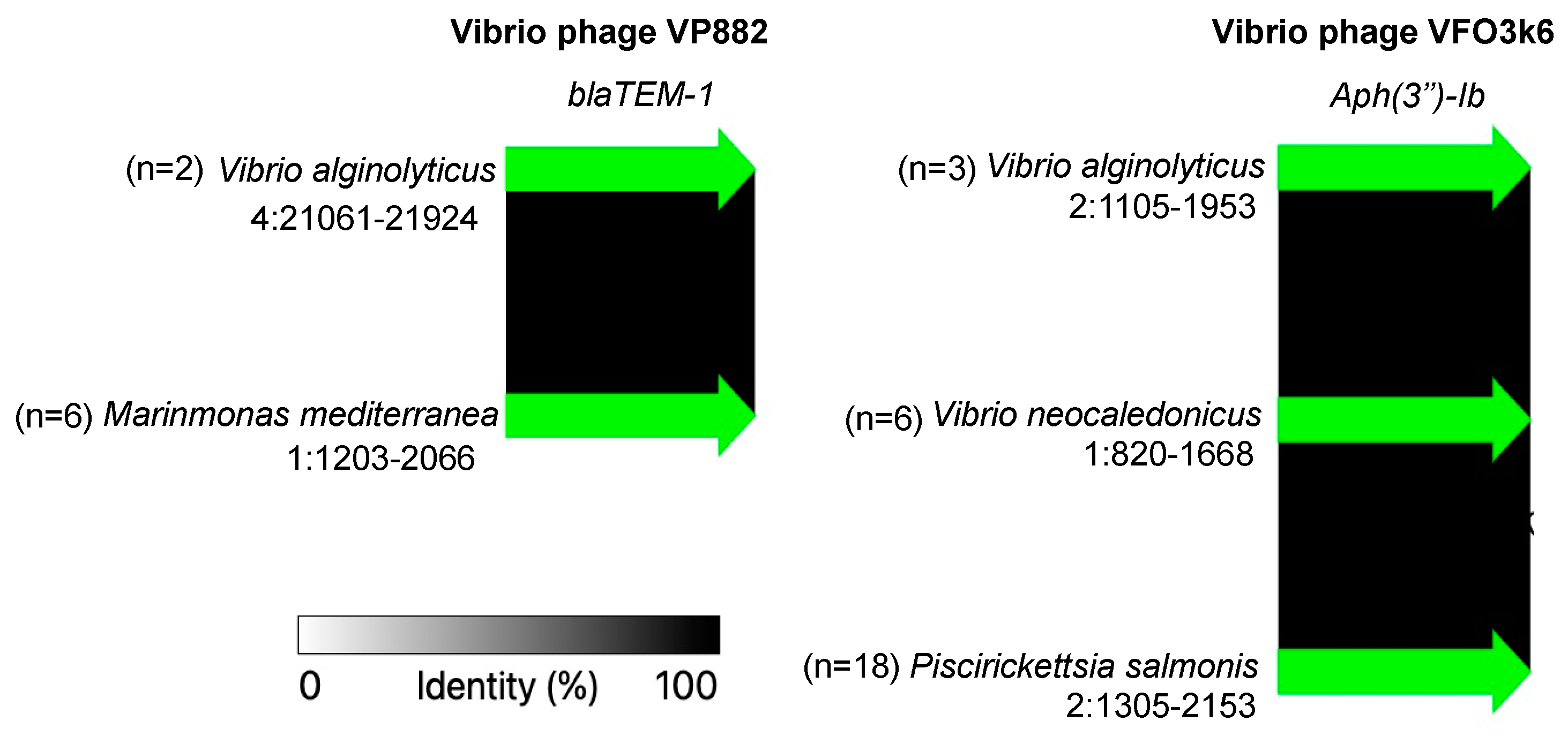
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marti, E.; Variatza, E.; Balcazar, J.L. The role of aquatic ecosystems as reservoirs of antibiotic resistance. Trends Microbiol. 2014, 22, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one fastq preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLOS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Wang, Q.; Cole, J.R.; Rosen, G.L. Using the rdp classifier to predict taxonomic novelty and reduce the search space for finding novel organisms. PLoS ONE 2012, 7, e32491. [Google Scholar] [CrossRef] [PubMed]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.H.; McDermott, P.F.; et al. Validating the amrfinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. Phaster: A better, faster version of the phast phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Xie, Z.; Tang, H. Isescan: Automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 2017, 33, 3340–3347. [Google Scholar] [CrossRef] [PubMed]
- Ghimpețeanu, O.M.; Pogurschi, E.N.; Popa, D.C.; Dragomir, N.; Drăgotoiu, T.; Mihai, O.D.; Petcu, C.D. Antibiotic use in livestock and residues in food-a public health threat: A review. Foods 2022, 11, 1430. [Google Scholar] [CrossRef]
- Huang, Z.; Anokyewaa, M.A.; Wang, J.; Jian, J.; Lu, Y. Pathogenicity and antibiotic resistance analysis of vibrio species found in coastal water at mainly beach of shenzhen, china. Front. Mar. Sci. 2022, 9, 980593. [Google Scholar] [CrossRef]
- Calero-Cáceres, W.; Ortuño-Gutiérrez, N.; Sunyoto, T.; Gomes-Dias, C.A.; Bastidas-Caldes, C.; Ramírez, M.S.; Harries, A.D. Whole-genome sequencing for surveillance of antimicrobial resistance in ecuador: Present and future implications. Rev. Panam. De Salud Pública 2023, 47, e8. [Google Scholar] [CrossRef] [PubMed]
- Khedkar, S.; Smyshlyaev, G.; Letunic, I.; Maistrenko, O.M.; Coelho, L.P.; Orakov, A.; Forslund, S.K.; Hildebrand, F.; Luetge, M.; Schmidt, T.S.; et al. Landscape of mobile genetic elements and their antibiotic resistance cargo in prokaryotic genomes. Nucleic Acids Res. 2022, 50, 3155–3168. [Google Scholar] [CrossRef] [PubMed]
- Haudiquet, M.; de Sousa, J.M.; Touchon, M.; Rocha, E.P.C. Selfish, promiscuous and sometimes useful: How mobile genetic elements drive horizontal gene transfer in microbial populations. Philos. Trans. R. Soc. B 2022, 377, 20210234. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.L. Bacteriophage-mediated horizontal gene transfer: Transduction. In Bacteriophages: Biology, Technology, Therapy; Harper, D.R., Abedon, S.T., Burrowes, B.H., McConville, M.L., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 151–192. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Taxonomy | Antibiotic Class | AST | ARG | Plasmids | Prophages |
|---|---|---|---|---|---|
| Vibrio cholerae | Betalactam | Ampicillin Imipenem Meropenem Ceftazidime | blaTEM-1 blaS1 YEM-1 | IncFII_1 (2) IncH12A-1 (2) Unnamed plasmids (3) | PHAGE_Vibrio_K139 PHAGE_Vibrio_VP882 PHAGE_Klebsi_ST16 PHAGE_Escher_520873 (2) |
| Aminoglycoside | Kanamycin | cpxA | |||
| Tetracycline | Oxytetracycline | tetA | |||
| Vibrio alginolyticus | Betalactam | Ampicillin Imipenem Aztreonam | blaOXA50 MexR | p3442-IMI-2c Unnamed plasmids (3) | PHAGE_Vibrio_K139 PHAGE_Vibrio_VfO3K6 (2) PHAGE_Vibrio_VCY (2) PHAGE_Erwini_vB_EhrS (2) |
| Aminoglycoside | Kanamycin | AAC(6’)-34 | |||
| Tetracycline | Oxytetracycline | tetA, tetB | |||
| Fluoroquinolones | Ciprofloxacin | oqxA, oqxB | |||
| Phenicol | Chloramphenicol | Cat | |||
| Vibrio parahaemolyticus | Betalactam | Ampicillin Imipenem Aztreonam | blaXCARB2 MexR | Unnamed plasmids (3) | PHAGE_Vibrio_K139 PHAGE_Klebsi_ST16 PHAGE_Klebsi_ST17 PHAGE_Staphy_Spbeta |
| Aminoglycoside | Kanamycin | AAC(6’)-34 | |||
| Tetracycline | Oxytetracycline | tetB | |||
| Polymyxin B | Colistin | PmrC | |||
| Phaeobacter inhibens | Betalactam | Ampicillin | blaTEM-1 | IncFII_1 Unnamed plasmids (4) | PHAGE_Entero_mEp237 PHAGE_Entero_mEp235 |
| Fluoroquinolones | Levofloxacin | gyrA, gyrB | |||
| Phenociol | Florfenicol | floR | |||
| Aeromonas salmonicida | Aminoglycoside | Kanamycin | cpxA | IncFII_1 Unnamed plasmids (2) | PHAGE_Entero_mEp237 (2) PHAGE_Klebsi_ST17 PHAGE_Salmon_118970_sal3 (2) |
| Tetracycline | Oxytetracycline | tetB | |||
| Edwardsiella anguillarum | Betalactam | Ampicillin | blaTEM-1 | Unnamed plasmids (2) | PHAGE_Entero_mEp235 |
| Polymyxin B | Colistin | eptA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farooq, A.; Rafique, A. Unveiling Mobilizable Multiresistance Clusters in Marine Bacteria. Eng. Proc. 2023, 56, 189. https://doi.org/10.3390/ASEC2023-16306
Farooq A, Rafique A. Unveiling Mobilizable Multiresistance Clusters in Marine Bacteria. Engineering Proceedings. 2023; 56(1):189. https://doi.org/10.3390/ASEC2023-16306
Chicago/Turabian StyleFarooq, Adeel, and Asma Rafique. 2023. "Unveiling Mobilizable Multiresistance Clusters in Marine Bacteria" Engineering Proceedings 56, no. 1: 189. https://doi.org/10.3390/ASEC2023-16306