
A Prediction of Drug Transport, Distribution, and Absorption Through a Multicompartmental Physiologically Based Pharmacokinetic Model †


 ,
,  , , , ,
, , , ,  and
and 
Abstract
1. Introduction
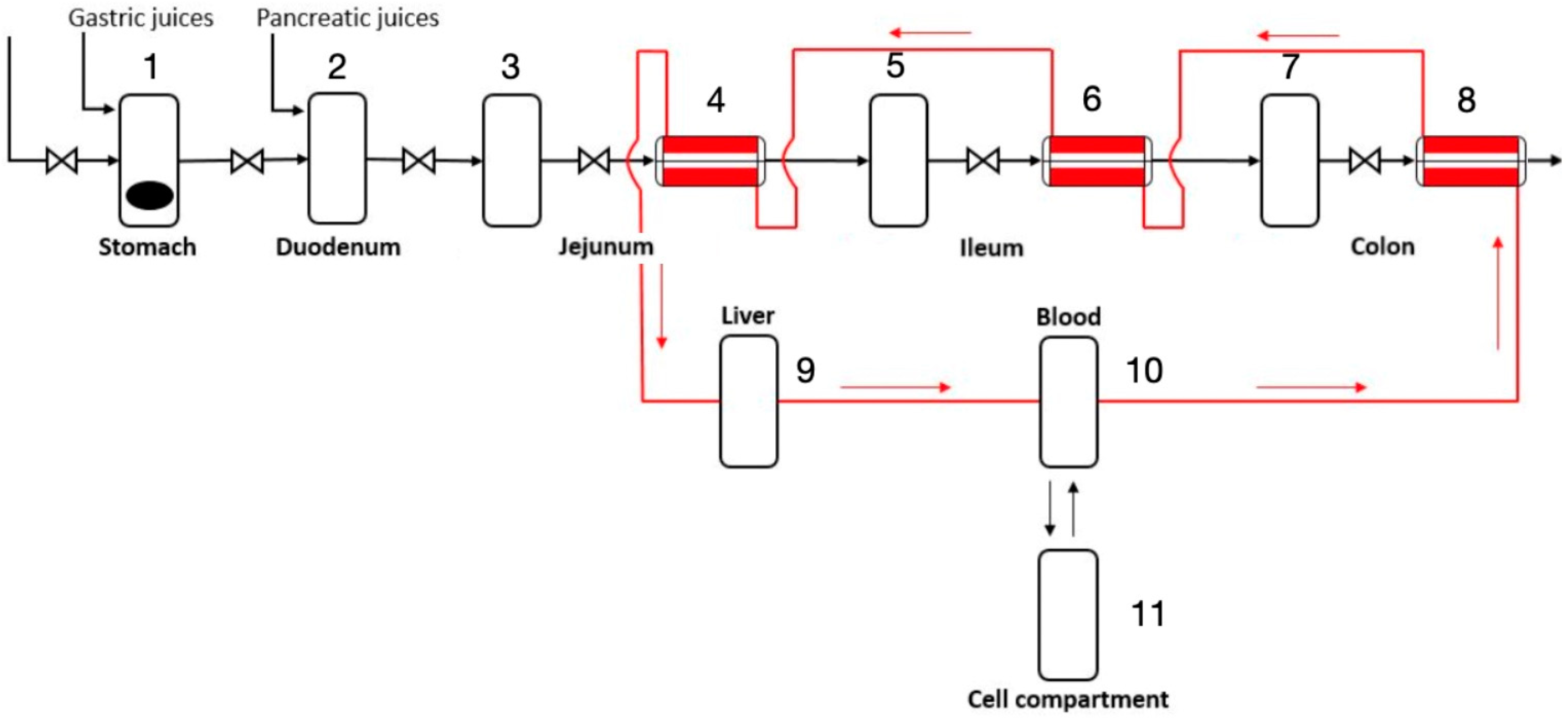
2. Materials and Methods
2.1. Stomach and Duodenum
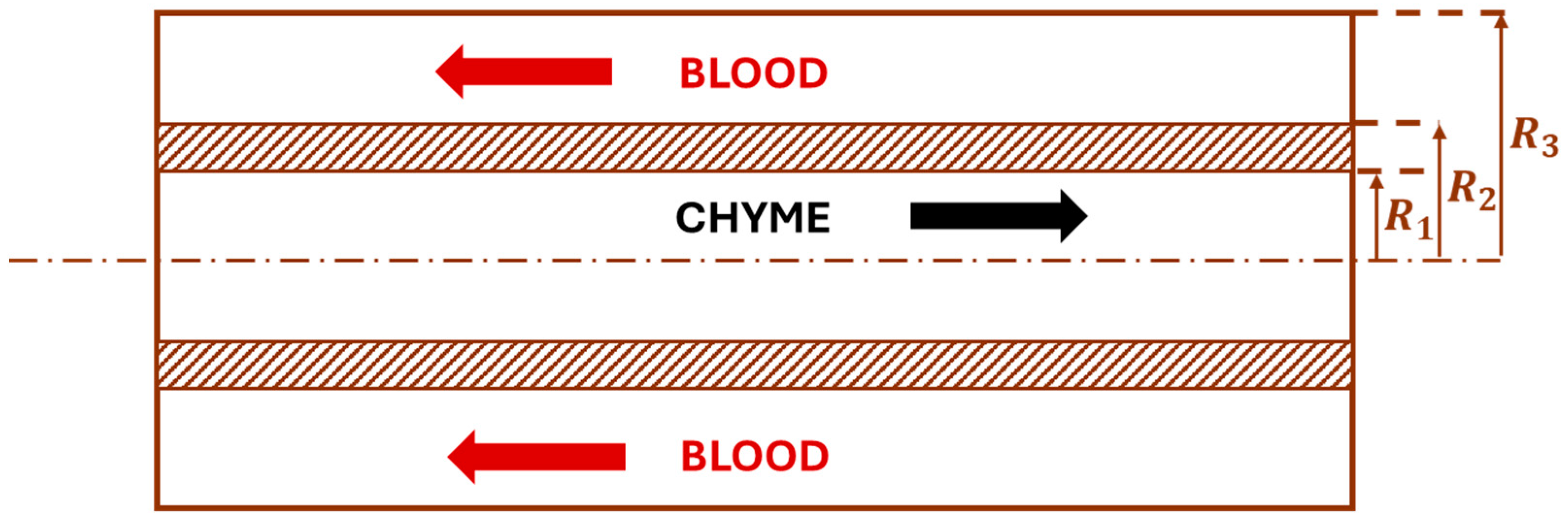
2.2. Jejunum, Ileum, and Colon
2.3. Cardiovascular System
2.4. Parameters Estimation
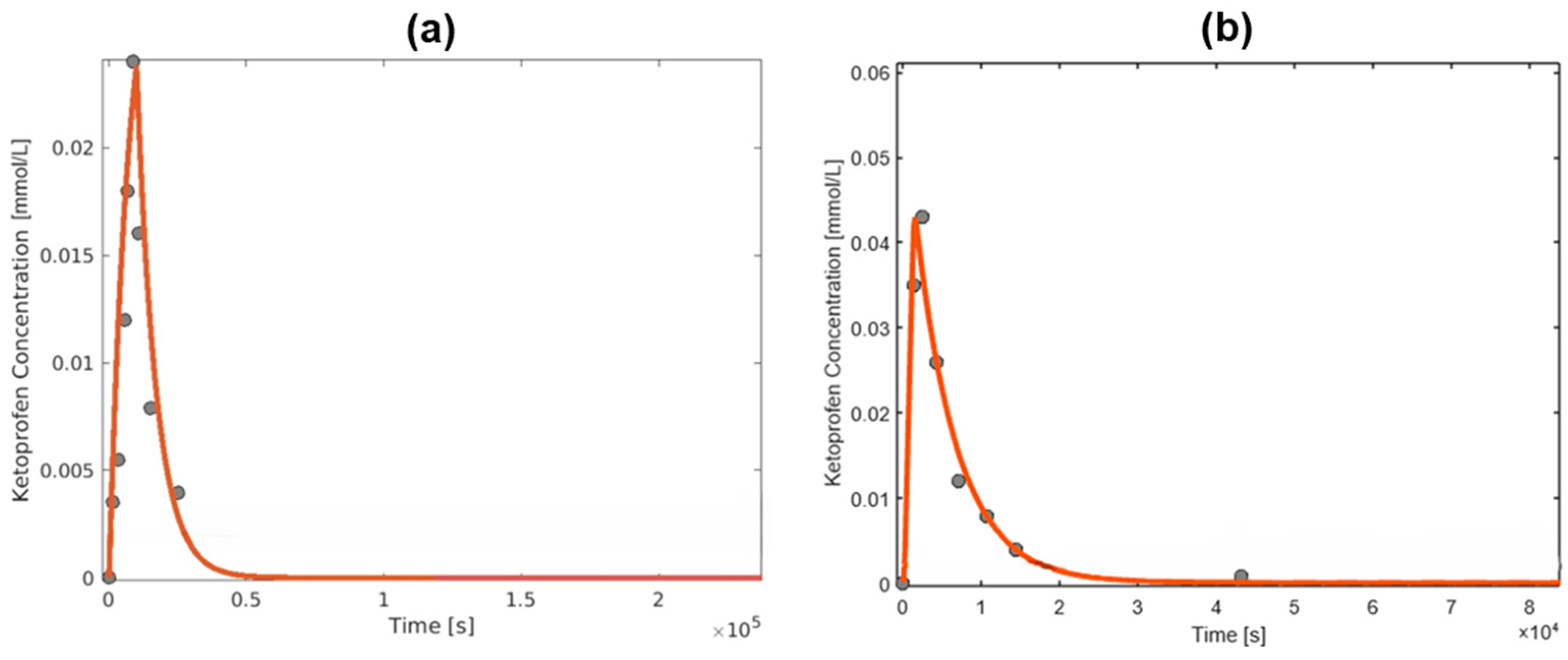
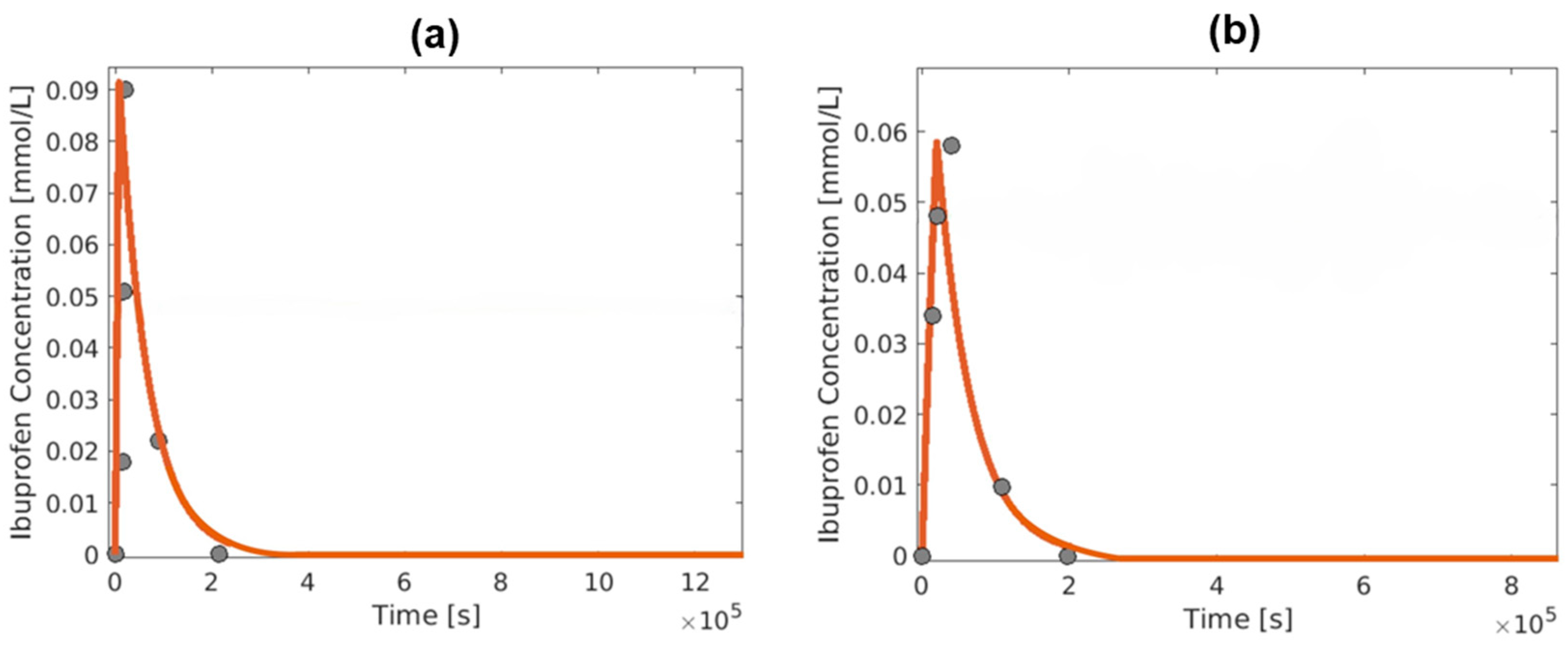
3. Results and Discussion
4. Conclusions
- Assessing and validating the model over a larger set of data, comprising a greater number of drugs.
- Comparing the model with previous works in terms of both computational burden and accuracy.
- Considering the possibility of incomplete solubilization of the drug and of enterohepatic circulation.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cabrera-Pérez, M.Á.; Pham-The, H. Computational Modeling of Human Oral Bioavailability: What Will Be Next? Expert. Opin. Drug Discov. 2018, 13, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Hua, S. Advances in Oral Drug Delivery for Regional Targeting in the Gastrointestinal Tract—Influence of Physiological, Pathophysiological and Pharmaceutical Factors. Front. Pharmacol. 2020, 11, 524. [Google Scholar] [CrossRef]
- Hirt, C.W.; Amsden, A.A.; Cook, J.L. An Arbitrary Lagrangian-Eulerian Computing Method for All Flow Speeds. J. Comput. Phys. 1974, 14, 227–253. [Google Scholar] [CrossRef]
- Kiela, P.R.; Ghishan, F.K. Physiology of Intestinal Absorption and Secretion. Best. Pract. Res. Clin. Gastroenterol. 2016, 30, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Peppas, N.A.; Narasimhan, B. Mathematical Models in Drug Delivery: How Modeling Has Shaped the Way We Design New Drug Delivery Systems. J. Control Release 2014, 190, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.M.; Rowland-Yeo, K. Basic Concepts in Physiologically Based Pharmacokinetic Modeling in Drug Discovery and Development. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gerlowski, L.E.; Jain, R.K. Physiologically Based Pharmacokinetic Modeling: Principles and Applications. J. Pharm. Sci. 1983, 72, 1103–1127. [Google Scholar] [CrossRef] [PubMed]
- Parrott, N.; Lave, T. Applications of Physiologically Based Absorption Models in Drug Discovery and Development. Mol. Pharm. 2008, 5, 760–775. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.; Peck, C.; Tucker, G. Physiologically-Based Pharmacokinetics in Drug Development and Regulatory Science. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 45–73. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, A.; Itaj, F.; Cacace, F.; Piemonte, V. Mathematical Modeling of the Gastrointestinal System for Preliminary Drug Absorption Assessment. Bioengineering 2024, 11, 813. [Google Scholar] [CrossRef] [PubMed]
- Pompa, M.; Capocelli, M.; Piemonte, V. A New Gastro-Intestinal Mathematical Model to Study Drug Bioavailability. Med. Eng. Phys. 2019, 74, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.D. Numerical Methods for Ordinary Differential Systems; Wiley: New York, NY, USA, 1991; Volume 146. [Google Scholar]
- Sugano, K.; Okazaki, A.; Sugimoto, S.; Tavornvipas, S.; Omura, A.; Mano, T. Solubility and Dissolution Profile Assessment in Drug Discovery. Drug Metab. Pharmacokinet. 2007, 22, 225–254. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Fujii, Y.; Tabata, F.; Ito, J.; Yukiko, M.; Kameda, A.; Akimoto, K.; Takahashi, M. Profiling and Trend Analysis of Food Effects on Oral Drug Absorption Considering Micelle Interaction and Solubilization by Bile Micelles. Drug Metab. Pharmacokinet. 2011, 26, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.B.; Bennett, P.N.; Rowland, M. Models of Hepatic Drug Clearance: Discrimination between the ‘Well Stirred’ and ‘Parallel-Tube’ Models. J. Pharm. Pharmacol. 1983, 35, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, B.; Lapicque, F.; Netter, P.; Monot, C.; Tamisier, J.N.; Thomas, P.; Royer, R.J. The Effect of Food on the Systemic Availability of Ketoprofen. Eur. J. Clin. Pharmacol. 1988, 33, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Pargal, A.; Kelkar, M.G.; Nayak, P.J. The Effect of Food on the Bioavailability of Ibuprofen and Flurbiprofen from Sustained Release Formulations. Biopharm. Drug Dispos. 1996, 17, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Gobeau, N.; Stringer, R.; De Buck, S.; Tuntland, T.; Faller, B. Evaluation of the GastroPlusTM Advanced Compartmental and Transit (ACAT) Model in Early Discovery. Pharm. Res. 2016, 33, 2126–2139. [Google Scholar] [CrossRef] [PubMed]
- Jamei, M.; Marciniak, S.; Feng, K.; Barnett, A.; Tucker, G.; Rostami-Hodjegan, A. The Simcyp Population-Based ADME Simulator. Expert. Opin. Drug Metab. Toxicol. 2009, 5, 211–223. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Parameter | Value |
|---|---|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiorboli, S.; D’Ambrosio, A.; Mazzeo, L.; Santori, F.; Bacco, L.; D’Antoni, F.; Palombo, G.; Merone, M.; Piemonte, V. A Prediction of Drug Transport, Distribution, and Absorption Through a Multicompartmental Physiologically Based Pharmacokinetic Model. Eng. Proc. 2024, 81, 13. https://doi.org/10.3390/engproc2024081013
Chiorboli S, D’Ambrosio A, Mazzeo L, Santori F, Bacco L, D’Antoni F, Palombo G, Merone M, Piemonte V. A Prediction of Drug Transport, Distribution, and Absorption Through a Multicompartmental Physiologically Based Pharmacokinetic Model. Engineering Proceedings. 2024; 81(1):13. https://doi.org/10.3390/engproc2024081013
Chicago/Turabian StyleChiorboli, Simone, Antonio D’Ambrosio, Leone Mazzeo, Francesca Santori, Luca Bacco, Federico D’Antoni, Giovanni Palombo, Mario Merone, and Vincenzo Piemonte. 2024. "A Prediction of Drug Transport, Distribution, and Absorption Through a Multicompartmental Physiologically Based Pharmacokinetic Model" Engineering Proceedings 81, no. 1: 13. https://doi.org/10.3390/engproc2024081013
APA StyleChiorboli, S., D’Ambrosio, A., Mazzeo, L., Santori, F., Bacco, L., D’Antoni, F., Palombo, G., Merone, M., & Piemonte, V. (2024). A Prediction of Drug Transport, Distribution, and Absorption Through a Multicompartmental Physiologically Based Pharmacokinetic Model. Engineering Proceedings, 81(1), 13. https://doi.org/10.3390/engproc2024081013