
Clinical and Molecular Features of Anti-CENP-B Autoantibodies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical Significance of ACAs
3. ACAs and Relationships with Cancer
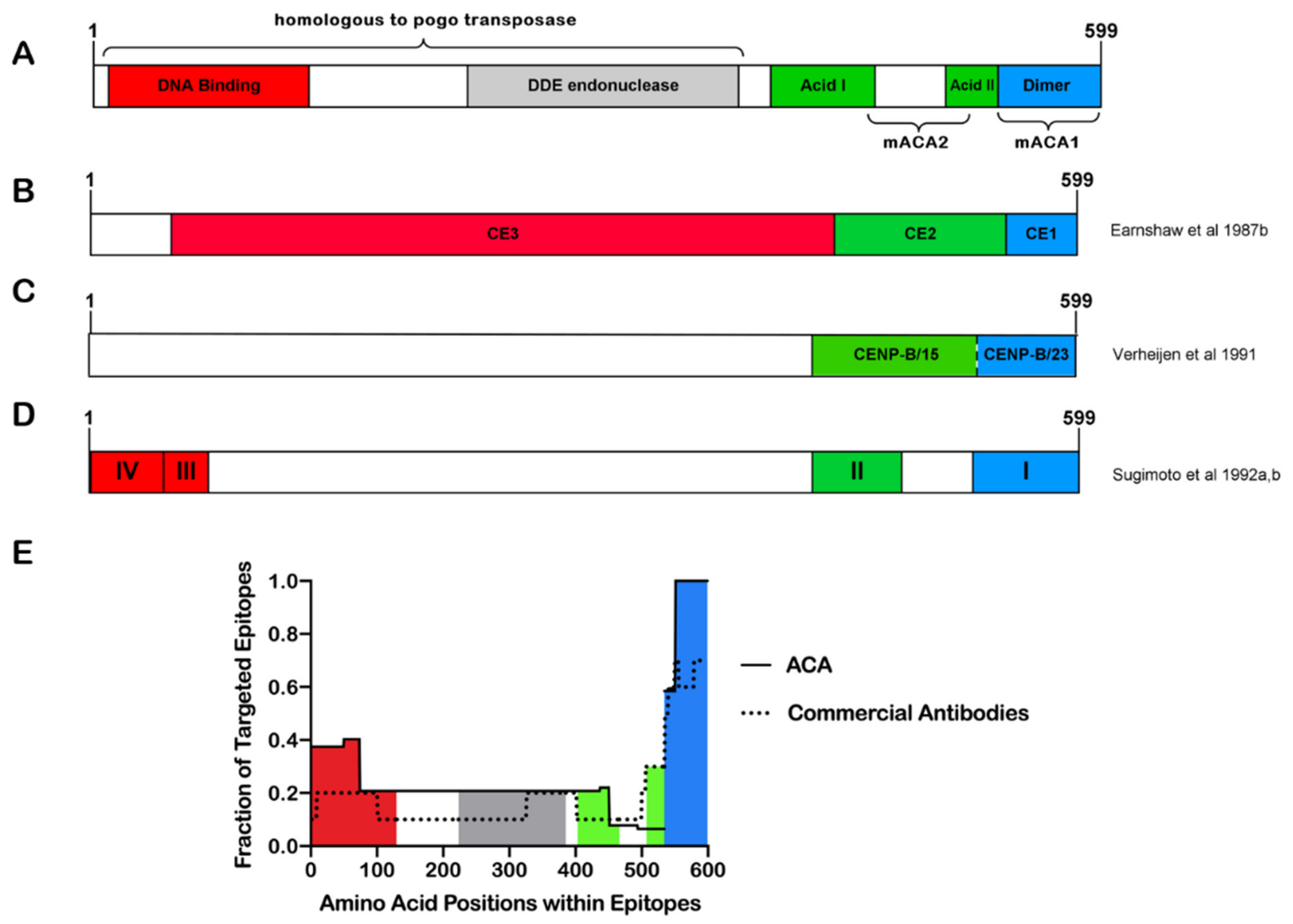
4. Autoantibodies Targeting CENP-B Define ACAs
5. Structure and Origin of CENP-B
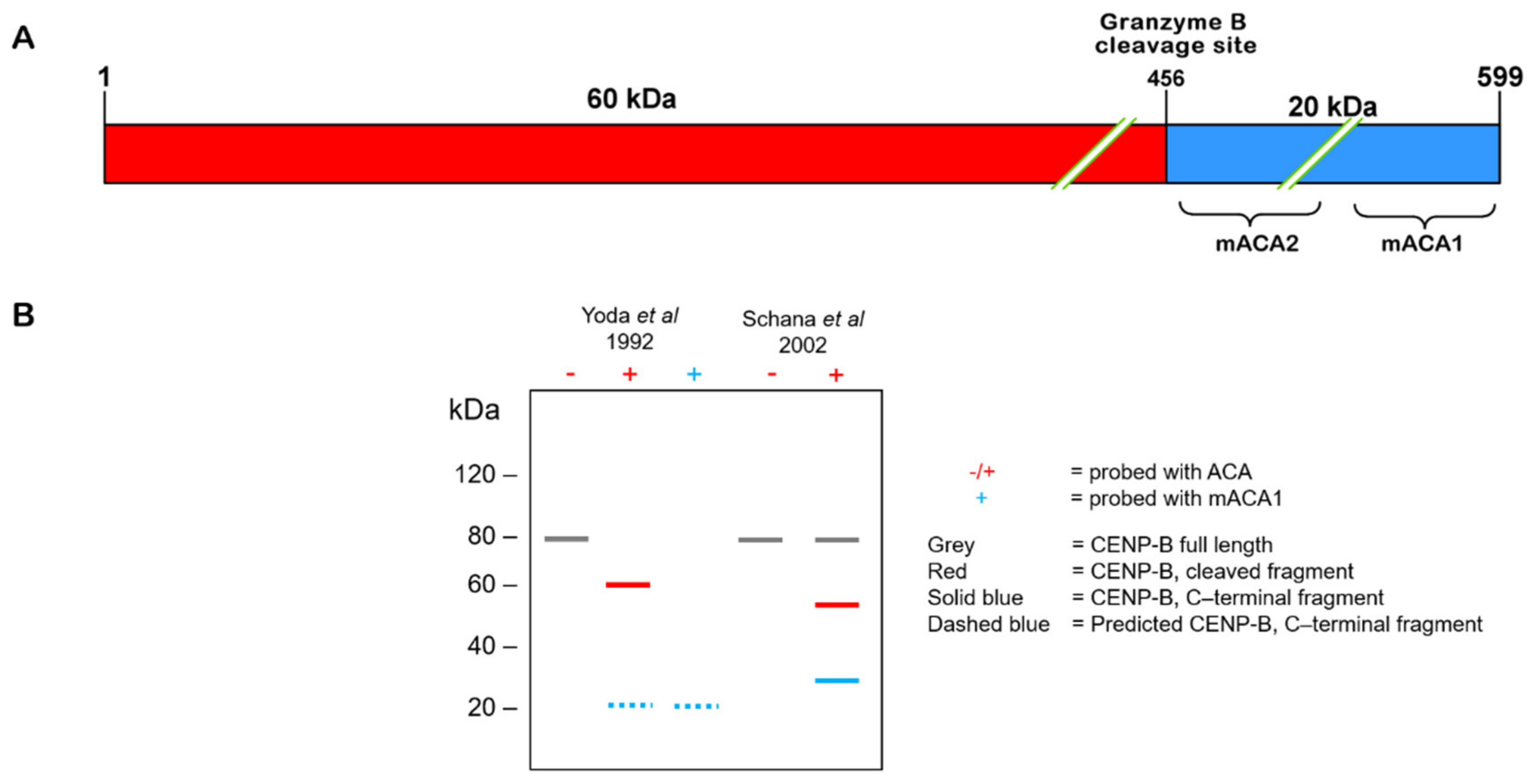
6. Enzymatic Cleavage of CENP-B
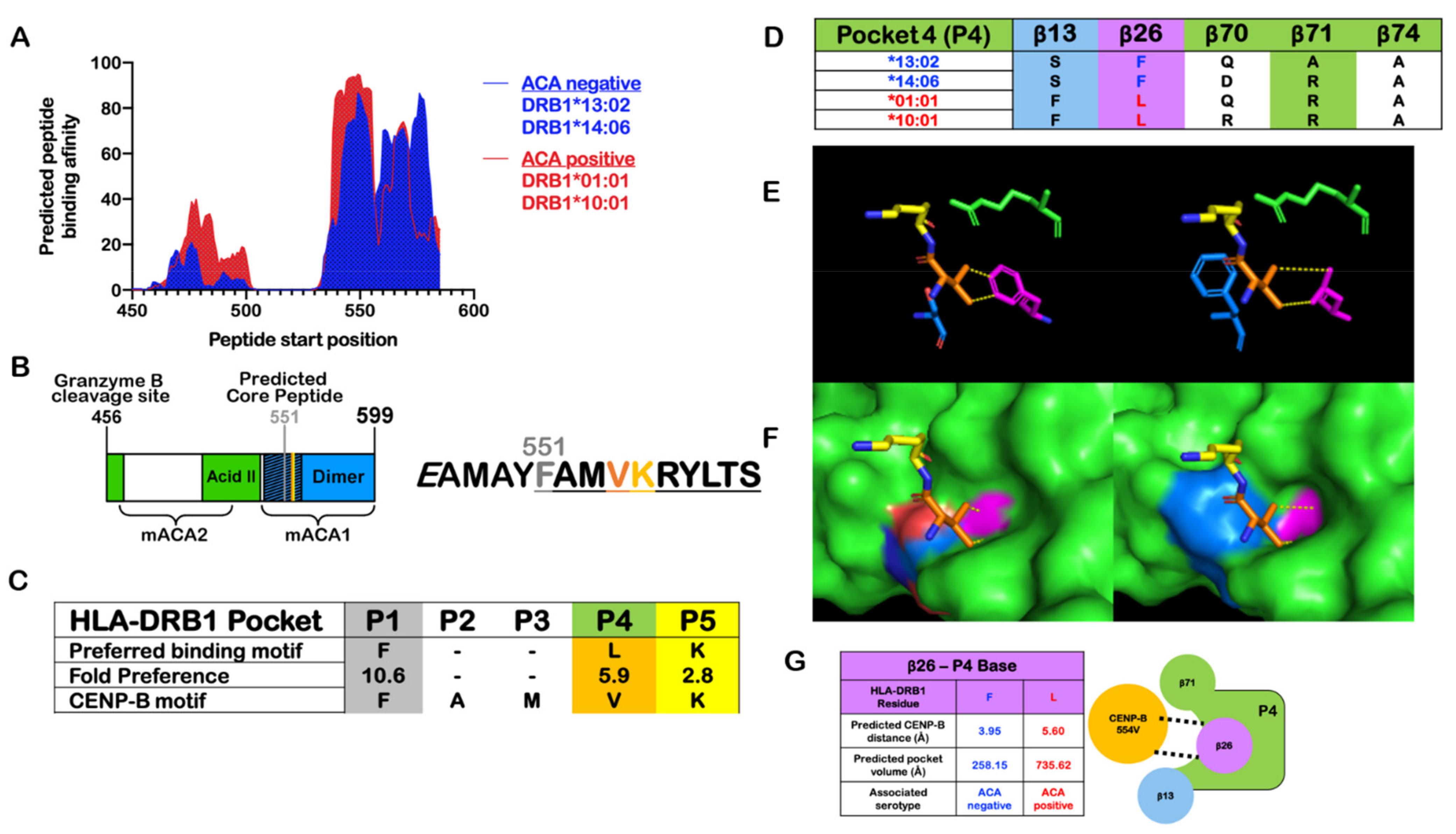
7. CENP-B Immunogenicity
8. Why Are CENP-B Autoantibodies Formed?
9. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abu-Shakra, M.; Buskila, D.; Ehrenfeld, M.; Conrad, K.; Shoenfeld, Y. Cancer and autoimmunity: Autoimmune and rheumatic features in patients with malignancies. Ann. Rheum Dis. 2001, 60, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Casiano, C.A.; Mediavilla-Varela, M.; Tan, E.M. Tumor-associated antigen arrays for the serological diagnosis of cancer. Mol. Cell Proteom. 2006, 5, 1745–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, E.M.; Zhang, J. Autoantibodies to tumor-associated antigens: Reporters from the immune system. Immunol. Rev. 2008, 222, 328–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.; Qu, Y.; Li, J.; Wang, X.; Wang, K.; Wang, P.; Jiang, B.H.; Zhang, J. Serological proteome analysis approach-based identification of ENO1 as a tumor-associated antigen and its autoantibody could enhance the sensitivity of CEA and CYFRA 21-1 in the detection of non-small cell lung cancer. Oncotarget 2017, 8, 36664–36673. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Ren, P.; Liu, M.; Imai, H.; Tan, E.M.; Zhang, J.Y. Using immunomic approach to enhance tumor-associated autoantibody detection in diagnosis of hepatocellular carcinoma. Clin. Immunol. 2014, 152, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacqueline, C.; Finn, O.J. Antibodies specific for disease-associated antigens (DAA) expressed in non-malignant diseases reveal potential new tumor-associated antigens (TAA) for immunotherapy or immunoprevention. Semin. Immunol. 2020, 47, 101394. [Google Scholar] [CrossRef]
- Reuschenbach, M.; von Knebel Doeberitz, M.; Wentzensen, N. A systematic review of humoral immune responses against tumor antigens. Cancer Immunol. Immunother. 2009, 58, 1535–1544. [Google Scholar] [CrossRef] [Green Version]
- Betancur, J.F.; Londono, A.; Estrada, V.E.; Puerta, S.L.; Osorno, S.M.; Loaiza, A.; Carmona, J.A.; Gomez-Puerta, J.A. Uncommon patterns of antinuclear antibodies recognizing mitotic spindle apparatus antigens and clinical associations. Medicine 2018, 97, e11727. [Google Scholar] [CrossRef]
- Earnshaw, W.C. Discovering centromere proteins: From cold white hands to the A, B, C of CENPs. Nat. Rev. Mol. Cell Biol. 2015, 16, 443–449. [Google Scholar] [CrossRef]
- Fritzler, M.J.; Rattner, J.B.; Luft, L.M.; Edworthy, S.M.; Casiano, C.A.; Peebles, C.; Mahler, M. Historical perspectives on the discovery and elucidation of autoantibodies to centromere proteins (CENP) and the emerging importance of antibodies to CENP-F. Autoimmun. Rev. 2011, 10, 194–200. [Google Scholar] [CrossRef]
- Li, S.; Li, X.; Xu, A.; Zhang, B.; He, X.; Chen, H.; Huang, J. Screening and clinical evaluation of dominant peptides of centromere protein F antigen for early diagnosis of hepatocellular carcinoma. Mol. Med. Rep. 2018, 17, 4720–4728. [Google Scholar] [CrossRef] [PubMed]
- Welner, S.; Trier, N.H.; Frisch, M.; Locht, H.; Hansen, P.R.; Houen, G. Correlation between centromere protein-F autoantibodies and cancer analyzed by enzyme-linked immunosorbent assay. Mol. Cancer 2013, 12, 95. [Google Scholar] [CrossRef] [Green Version]
- Beck, J.S. Variations in the morphological patterns of “autoimmune” nuclear fluorescence. Lancet 1961, 1, 1203–1205. [Google Scholar] [CrossRef]
- Van den Hoogen, F.; Khanna, D.; Fransen, J.; Johnson, S.R.; Baron, M.; Tyndall, A.; Matucci-Cerinic, M.; Naden, R.P.; Medsger, T.A., Jr.; Carreira, P.E.; et al. 2013 classification criteria for systemic sclerosis: An American college of rheumatology/European league against rheumatism collaborative initiative. Ann. Rheum. Dis. 2013, 72, 1747–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foltz, D.R.; Jansen, L.E.; Black, B.E.; Bailey, A.O.; Yates, J.R., 3rd; Cleveland, D.W. The human CENP-A centromeric nucleosome-associated complex. Nat. Cell Biol. 2006, 8, 458–469. [Google Scholar] [CrossRef]
- Hori, T.; Amano, M.; Suzuki, A.; Backer, C.B.; Welburn, J.P.; Dong, Y.; McEwen, B.F.; Shang, W.H.; Suzuki, E.; Okawa, K.; et al. CCAN makes multiple contacts with centromeric DNA to provide distinct pathways to the outer kinetochore. Cell 2008, 135, 1039–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, S.; Bukowski-Wills, J.C.; Sanchez-Pulido, L.; Alves Fde, L.; Wood, L.; Chen, Z.A.; Platani, M.; Fischer, L.; Hudson, D.F.; Ponting, C.P.; et al. The protein composition of mitotic chromosomes determined using multiclassifier combinatorial proteomics. Cell 2010, 142, 810–821. [Google Scholar] [CrossRef] [Green Version]
- Damoiseaux, J.; Andrade, L.E.C.; Carballo, O.G.; Conrad, K.; Francescantonio, P.L.C.; Fritzler, M.J.; Garcia de la Torre, I.; Herold, M.; Klotz, W.; Cruvinel, W.M.; et al. Clinical relevance of HEp-2 indirect immunofluorescent patterns: The International Consensus on ANA patterns (ICAP) perspective. Ann. Rheum. Dis. 2019, 78, 879–889. [Google Scholar] [CrossRef] [Green Version]
- Tramposch, H.D.; Smith, C.D.; Senecal, J.L.; Rothfield, N. A long-term longitudinal study of anticentromere antibodies. Arthritis Rheum. 1984, 27, 121–124. [Google Scholar] [CrossRef]
- Hildebrandt, S.; Weiner, E.S.; Earnshaw, W.C.; Zanetti, M.; Rothfield, N.F. Idiotypic analysis of human anticentromere autoantibodies. Autoimmunity 1991, 9, 131–140. [Google Scholar] [CrossRef]
- Vazquez-Abad, D.; Russell, C.A.; Cusick, S.M.; Earnshaw, W.C.; Rothfield, N.F. Longitudinal study of anticentromere and antitopoisomerase-I isotypes. Clin. Immunol. Immunopathol. 1995, 74, 257–270. [Google Scholar] [CrossRef]
- Baer, A.N.; Medrano, L.; McAdams-DeMarco, M.; Gniadek, T.J. Association of Anticentromere Antibodies with More Severe Exocrine Glandular Dysfunction in Sjogren’s Syndrome: Analysis of the Sjogren’s International Collaborative Clinical Alliance Cohort. Arthritis Care Res. 2016, 68, 1554–1559. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.L.; Tsay, G.J.; Tsai, R.T. Anticentromere antibodies in subjects with no apparent connective tissue disease. Ann. Rheum. Dis. 1993, 52, 586–589. [Google Scholar] [CrossRef] [Green Version]
- Atalay, C.; Atalay, G.; Yilmaz, K.B.; Altinok, M. The role of anti-CENP-B and anti-SS-B antibodies in breast cancer. Neoplasma 2005, 52, 32–35. [Google Scholar]
- Atalay, C.; Dogan, L.; Atalay, G. Anti-CENP-B antibodies are associated with prolonged survival in breast cancer. Future Oncol. 2010, 6, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Madrid, F.F.; Maroun, M.C.; Olivero, O.A.; Long, M.; Stark, A.; Grossman, L.I.; Binder, W.; Dong, J.; Burke, M.; Nathanson, S.D.; et al. Autoantibodies in breast cancer sera are not epiphenomena and may participate in carcinogenesis. BMC Cancer 2015, 15, 407. [Google Scholar] [CrossRef] [Green Version]
- Briasoulis, E.; Kamposioras, K.; Tzovaras, V.; Pafitanis, G.; Kostoula, A.; Mavridis, A.; Pavlidis, N. CENP-B specific anti-centromere autoantibodies heralding small-cell lung cancer. A case study and review of the literature. Lung Cancer 2008, 60, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Zhang, Y.; Jiang, Y.; Li, H.; Chen, J.; Ming, F.; Wang, W.; Yu, J.; Zeng, T.; Tian, Y.; et al. The clinical significance of anti-mitotic spindle apparatus antibody (MSA) and anti-centromere antibody (ACA) detected in patients with small cell lung cancer (SCLC). Am. J. Clin. Exp. Immunol. 2017, 6, 21–26. [Google Scholar]
- He, Y.; Rozeboom, L.; Rivard, C.J.; Ellison, K.; Dziadziuszko, R.; Yu, H.; Zhou, C.; Hirsch, F.R. MHC class II expression in lung cancer. Lung Cancer 2017, 112, 75–80. [Google Scholar] [CrossRef]
- Igusa, T.; Hummers, L.K.; Visvanathan, K.; Richardson, C.; Wigley, F.M.; Casciola-Rosen, L.; Rosen, A.; Shah, A.A. Autoantibodies and scleroderma phenotype define subgroups at high-risk and low-risk for cancer. Ann. Rheum. Dis. 2018, 77, 1179–1186. [Google Scholar] [CrossRef]
- Joseph, C.G.; Darrah, E.; Shah, A.A.; Skora, A.D.; Casciola-Rosen, L.A.; Wigley, F.M.; Boin, F.; Fava, A.; Thoburn, C.; Kinde, I.; et al. Association of the autoimmune disease scleroderma with an immunologic response to cancer. Science 2014, 343, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.A.; Rosen, A. Cancer and systemic sclerosis: Novel insights into pathogenesis and clinical implications. Curr. Opin. Rheumatol. 2011, 23, 530–535. [Google Scholar] [CrossRef]
- Shah, A.A.; Rosen, A.; Hummers, L.; Wigley, F.; Casciola-Rosen, L. Close temporal relationship between onset of cancer and scleroderma in patients with RNA polymerase I/III antibodies. Arthritis Rheum. 2010, 62, 2787–2795. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.Y.; Hill, C.L.; Pontifex, E.K.; Roberts-Thomson, P.J. Breast cancer and systemic sclerosis: A clinical description of 21 patients in a population-based cohort study. Rheumatol. Int. 2008, 28, 895–899. [Google Scholar] [CrossRef]
- Gauderon, A.; Roux-Lombard, P.; Spoerl, D. Antinuclear Antibodies With a Homogeneous and Speckled Immunofluorescence Pattern Are Associated with Lack of Cancer While Those with a Nucleolar Pattern with the Presence of Cancer. Front. Med. 2020, 7, 165. [Google Scholar] [CrossRef]
- Earnshaw, W.; Bordwell, B.; Marino, C.; Rothfield, N. Three human chromosomal autoantigens are recognized by sera from patients with anti-centromere antibodies. J. Clin. Invest. 1986, 77, 426–430. [Google Scholar] [CrossRef] [Green Version]
- Earnshaw, W.C.; Machlin, P.S.; Bordwell, B.J.; Rothfield, N.F.; Cleveland, D.W. Analysis of anticentromere autoantibodies using cloned autoantigen CENP-B. Proc. Natl. Acad. Sci. USA 1987, 84, 4979–4983. [Google Scholar] [CrossRef] [Green Version]
- Rothfield, N.; Whitaker, D.; Bordwell, B.; Weiner, E.; Senecal, J.L.; Earnshaw, W. Detection of anticentromere antibodies using cloned autoantigen CENP-B. Arthritis Rheum. 1987, 30, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Migita, H.; Hagishita, Y.; Yata, H.; Himeno, M. An antigenic determinant on human centromere protein B (CENP-B) available for production of human-specific anticentromere antibodies in mouse. Cell Struct Funct. 1992, 17, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheijen, R.; de Jong, B.A.; Oberye, E.H.; van Venrooij, W.J. Molecular cloning of a major CENP-B epitope and its use for the detection of anticentromere autoantibodies. Mol. Biol. Rep. 1992, 16, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Earnshaw, W.C.; Sullivan, K.F.; Machlin, P.S.; Cooke, C.A.; Kaiser, D.A.; Pollard, T.D.; Rothfield, N.F.; Cleveland, D.W. Molecular cloning of cDNA for CENP-B, the major human centromere autoantigen. J. Cell Biol. 1987, 104, 817–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, K.; Muro, Y.; Himeno, M. Anti-helix-loop-helix domain antibodies: Discovery of autoantibodies that inhibit DNA binding activity of human centromere protein B (CENP-B). J. Biochem. 1992, 111, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Whyte, J.; Soriano, E.; Earnshaw, W.C.; McHugh, N.J. Frequency of autoantibodies to a major epitope on the carboxyl terminal fragment of CENP-B in patients with autoimmune disease. Br. J. Rheumatol. 1995, 34, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Zian, Z.; Bennani Mechita, M.; Hamdouch, K.; Maamar, M.; Barakat, A.; Ghailani Nourouti, N.; El Aouad, R.; Valdivia, M.M.; Arji, N. Proteomics characterization of CENP-B epitope in Moroccan scleroderma patients with anti-centromere autoantibodies. Immunol. Lett. 2020, 221, 1–5. [Google Scholar] [CrossRef]
- Eisenberg, R.A.; Earnshaw, W.C.; Bordwell, B.J.; Craven, S.Y.; Cheek, R.; Rothfield, N.F. Isotype analysis of the anti-CENP-B anticentromere autoantibody: Evidence for restricted clonality. Arthritis Rheum. 1989, 32, 1315–1318. [Google Scholar] [CrossRef]
- Iwahara, J.; Kigawa, T.; Kitagawa, K.; Masumoto, H.; Okazaki, T.; Yokoyama, S. A helix-turn-helix structure unit in human centromere protein B (CENP-B). EMBO J. 1998, 17, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Masumoto, H.; Masukata, H.; Muro, Y.; Nozaki, N.; Okazaki, T. A human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J. Cell Biol. 1989, 109, 1963–1973. [Google Scholar] [CrossRef]
- Muro, Y.; Masumoto, H.; Yoda, K.; Nozaki, N.; Ohashi, M.; Okazaki, T. Centromere protein B assembles human centromeric alpha-satellite DNA at the 17-bp sequence, CENP-B box. J. Cell Biol. 1992, 116, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Pluta, A.F.; Saitoh, N.; Goldberg, I.; Earnshaw, W.C. Identification of a subdomain of CENP-B that is necessary and sufficient for localization to the human centromere. J. Cell Biol. 1992, 116, 1081–1093. [Google Scholar] [CrossRef] [Green Version]
- Yoda, K.; Kitagawa, K.; Masumoto, H.; Muro, Y.; Okazaki, T. A human centromere protein, CENP-B, has a DNA binding domain containing four potential alpha helices at the NH2 terminus, which is separable from dimerizing activity. J. Cell Biol. 1992, 119, 1413–1427. [Google Scholar] [CrossRef] [Green Version]
- Kasinathan, S.; Henikoff, S. Non-B-Form DNA Is Enriched at Centromeres. Mol. Biol. Evol. 2018, 35, 949–962. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, K.F.; Glass, C.A. CENP-B is a highly conserved mammalian centromere protein with homology to the helix-loop-helix family of proteins. Chromosoma 1991, 100, 360–370. [Google Scholar] [CrossRef]
- Yoda, K.; Nakamura, T.; Masumoto, H.; Suzuki, N.; Kitagawa, K.; Nakano, M.; Shinjo, A.; Okazaki, T. Centromere protein B of African green monkey cells: Gene structure, cellular expression, and centromeric localization. Mol. Cell Biol. 1996, 16, 5169–5177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa-Cisneros, O.; Fraire-Velazquez, S.; Moreno, J.; Herrera-Esparza, R. CENP-B autoantigen is a conserved protein from humans to higher plants: Identification of the aminoterminal domain in Phaseolus vulgaris. Rev. Rhum. Engl. Ed. 1997, 64, 368–374. [Google Scholar] [PubMed]
- Thongchum, R.; Nishihara, H.; Srikulnath, K.; Hirai, H.; Koga, A. The CENP-B box, a nucleotide motif involved in centromere formation, has multiple origins in New World monkeys. Genes Genet. Syst. 2020, 94, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Ohzeki, J.; Nakano, M.; Okada, T.; Masumoto, H. CENP-B box is required for de novo centromere chromatin assembly on human alphoid DNA. J. Cell Biol. 2002, 159, 765–775. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Ohzeki, J.; Nakano, M.; Yoda, K.; Brinkley, W.R.; Larionov, V.; Masumoto, H. CENP-B controls centromere formation depending on the chromatin context. Cell 2007, 131, 1287–1300. [Google Scholar] [CrossRef] [Green Version]
- Logsdon, G.A.; Gambogi, C.W.; Liskovykh, M.A.; Barrey, E.J.; Larionov, V.; Miga, K.H.; Heun, P.; Black, B.E. Human Artificial Chromosomes that Bypass Centromeric DNA. Cell 2019, 178, 624–639.e19. [Google Scholar] [CrossRef] [Green Version]
- Fowler, K.J.; Hudson, D.F.; Salamonsen, L.A.; Edmondson, S.R.; Earle, E.; Sibson, M.C.; Choo, K.H. Uterine dysfunction and genetic modifiers in centromere protein B-deficient mice. Genome Res. 2000, 10, 30–41. [Google Scholar]
- Hudson, D.F.; Fowler, K.J.; Earle, E.; Saffery, R.; Kalitsis, P.; Trowell, H.; Hill, J.; Wreford, N.G.; de Kretser, D.M.; Cancilla, M.R.; et al. Centromere protein B null mice are mitotically and meiotically normal but have lower body and testis weights. J. Cell Biol. 1998, 141, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Perez-Castro, A.V.; Shamanski, F.L.; Meneses, J.J.; Lovato, T.L.; Vogel, K.G.; Moyzis, R.K.; Pedersen, R. Centromeric protein B null mice are viable with no apparent abnormalities. Dev. Biol. 1998, 201, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Lampson, M.; Efimov, A.; Yen, T.J. Chromosome instability in tumor cells due to defects in Aurora B mediated error correction at kinetochores. Cell Cycle 2018, 17, 2622–2636. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Vleugel, M.; Backer, C.B.; Hori, T.; Fukagawa, T.; Cheeseman, I.M.; Lampson, M.A. Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J. Cell Biol. 2010, 188, 809–820. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Ballister, E.R.; Lampson, M.A. Aurora B dynamics at centromeres create a diffusion-based phosphorylation gradient. J. Cell Biol. 2011, 194, 539–549. [Google Scholar] [CrossRef]
- Armas-Portela, R.; Kremer, L.; Avila, J. The centromere protein CENP-B behaves as a microtubule-associated protein. Acta Histochem. Suppl. 1991, 41, 37–43. [Google Scholar]
- Balczon, R.D.; Brinkley, B.R. Tubulin interaction with kinetochore proteins: Analysis by in vitro assembly and chemical cross-linking. J. Cell Biol. 1987, 105, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Tudor, M.; Lobocka, M.; Goodell, M.; Pettitt, J.; O’Hare, K. The pogo transposable element family of Drosophila melanogaster. Mol. Gen. Genet. 1992, 232, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Kipling, D.; Warburton, P.E. Centromeres, CENP-B and Tigger too. Trends Genet. 1997, 13, 141–145. [Google Scholar] [CrossRef]
- Upadhyay, U.; Srivastava, S.; Khatri, I.; Nanda, J.S.; Subramanian, S.; Arora, A.; Singh, J. Ablation of RNA interference and retrotransposons accompany acquisition and evolution of transposases to heterochromatin protein CENPB. Mol. Biol. Cell 2017, 28, 1132–1146. [Google Scholar] [CrossRef]
- Capy, P.; Vitalis, R.; Langin, T.; Higuet, D.; Bazin, C. Relationships between transposable elements based upon the integrase-transposase domains: Is there a common ancestor? J. Mol. Evol. 1996, 42, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Casola, C.; Hucks, D.; Feschotte, C. Convergent domestication of pogo-like transposases into centromere-binding proteins in fission yeast and mammals. Mol. Biol. Evol. 2008, 25, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Dupeyron, M.; Baril, T.; Bass, C.; Hayward, A. Phylogenetic analysis of the Tc1/mariner superfamily reveals the unexplored diversity of pogo-like elements. Mob. DNA 2020, 11, 21. [Google Scholar] [CrossRef]
- Gao, B.; Wang, Y.; Diaby, M.; Zong, W.; Shen, D.; Wang, S.; Chen, C.; Wang, X.; Song, C. Evolution of pogo, a separate superfamily of IS630-Tc1-mariner transposons, revealing recurrent domestication events in vertebrates. Mob. DNA 2020, 11, 25. [Google Scholar] [CrossRef]
- Marshall, O.J.; Choo, K.H. Putative CENP-B paralogues are not present at mammalian centromeres. Chromosoma 2012, 121, 169–179. [Google Scholar] [CrossRef]
- Kitagawa, K.; Masumoto, H.; Ikeda, M.; Okazaki, T. Analysis of protein-DNA and protein-protein interactions of centromere protein B (CENP-B) and properties of the DNA-CENP-B complex in the cell cycle. Mol. Cell Biol. 1995, 15, 1602–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawaramoto, M.S.; Park, S.Y.; Tanaka, Y.; Nureki, O.; Kurumizaka, H.; Yokoyama, S. Crystal structure of the human centromere protein B (CENP-B) dimerization domain at 1.65—A resolution. J. Biol. Chem. 2003, 278, 51454–51461. [Google Scholar] [CrossRef] [Green Version]
- Otake, K.; Ohzeki, J.I.; Shono, N.; Kugou, K.; Okazaki, K.; Nagase, T.; Yamakawa, H.; Kouprina, N.; Larionov, V.; Kimura, H.; et al. CENP-B creates alternative epigenetic chromatin states permissive for CENP-A or heterochromatin assembly. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Schachna, L.; Wigley, F.M.; Morris, S.; Gelber, A.C.; Rosen, A.; Casciola-Rosen, L. Recognition of Granzyme B-generated autoantigen fragments in scleroderma patients with ischemic digital loss. Arthritis Rheum. 2002, 46, 1873–1884. [Google Scholar] [CrossRef]
- Casciola-Rosen, L.; Andrade, F.; Ulanet, D.; Wong, W.B.; Rosen, A. Cleavage by granzyme B is strongly predictive of autoantigen status: Implications for initiation of autoimmunity. J. Exp. Med. 1999, 190, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Darrah, E.; Rosen, A. Granzyme B cleavage of autoantigens in autoimmunity. Cell Death Differ. 2010, 17, 624–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, S.J.; Rajotte, R.V.; Korbutt, G.S.; Bleackley, R.C. Granzyme B: A natural born killer. Immunol. Rev. 2003, 193, 31–38. [Google Scholar] [CrossRef]
- Hendel, A.; Hiebert, P.R.; Boivin, W.A.; Williams, S.J.; Granville, D.J. Granzymes in age-related cardiovascular and pulmonary diseases. Cell Death Differ. 2010, 17, 596–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, S.S.; Reed, T.J.; Berthier, C.C.; Tsou, P.S.; Liu, J.; Gudjonsson, J.E.; Khanna, D.; Kahlenberg, J.M. Scleroderma keratinocytes promote fibroblast activation independent of transforming growth factor beta. Rheumatology 2017, 56, 1970–1981. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Asano, Y.; Sugawara, K.; Yamashita, T.; Nakamura, K.; Saigusa, R.; Ichimura, Y.; Toyama, T.; Taniguchi, T.; Akamata, K.; et al. Epithelial Fli1 deficiency drives systemic autoimmunity and fibrosis: Possible roles in scleroderma. J. Exp. Med. 2017, 214, 1129–1151. [Google Scholar] [CrossRef]
- Genth, E.; Mierau, R.; Genetzky, P.; von Muhlen, C.A.; Kaufmann, S.; von Wilmowsky, H.; Meurer, M.; Krieg, T.; Pollmann, H.J.; Hartl, P.W. Immunogenetic associations of scleroderma-related antinuclear antibodies. Arthritis Rheum. 1990, 33, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Reveille, J.D.; Owerbach, D.; Goldstein, R.; Moreda, R.; Isern, R.A.; Arnett, F.C. Association of polar amino acids at position 26 of the HLA-DQB1 first domain with the anticentromere autoantibody response in systemic sclerosis (scleroderma). J. Clin. Invest. 1992, 89, 1208–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, N.J.; Whyte, J.; Artlett, C.; Briggs, D.C.; Stephens, C.O.; Olsen, N.J.; Gusseva, N.G.; Maddison, P.J.; Black, C.M.; Welsh, K. Anti-centromere antibodies (ACA) in systemic sclerosis patients and their relatives: A serological and HLA study. Clin. Exp. Immunol. 1994, 96, 267–274. [Google Scholar] [CrossRef]
- Parveen, S.; Morshed, S.A.; Arima, K.; Nishioka, M.; Czaja, A.J.; Chow, W.C.; Ng, H.S. Antibodies to Ro/La, Cenp-B, and snRNPs antigens in autoimmune hepatitis of North America versus Asia: Patterns of immunofluorescence, ELISA reactivities, and HLA association. Dig. Dis. Sci. 1998, 43, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Oka, S.; Kawasaki, A.; Shimada, K.; Sugii, S.; Matsushita, T.; Hashimoto, A.; Komiya, A.; Fukui, N.; Kobayashi, K.; et al. Human Leukocyte Antigen and Systemic Sclerosis in Japanese: The Sign of the Four Independent Protective Alleles, DRB1*13:02, DRB1*14:06, DQB1*03:01, and DPB1*02:01. PLoS ONE 2016, 11, e0154255. [Google Scholar] [CrossRef] [Green Version]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef]
- Alvarez, I.; Collado, J.; Daura, X.; Colome, N.; Rodriguez-Garcia, M.; Gallart, T.; Canals, F.; Jaraquemada, D. The rheumatoid arthritis-associated allele HLA-DR10 (DRB1*1001) shares part of its repertoire with HLA-DR1 (DRB1*0101) and HLA-DR4 (DRB*0401). Arthritis Rheum. 2008, 58, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Kameda, H.; Pandey, J.P.; Kaburaki, J.; Inoko, H.; Kuwana, M. Immunoglobulin allotype gene polymorphisms in systemic sclerosis: Interactive effect of MHC class II and KM genes on anticentromere antibody production. Ann. Rheum. Dis. 1998, 57, 366–370. [Google Scholar] [CrossRef]
- Casciola-Rosen, L.; Nagaraju, K.; Plotz, P.; Wang, K.; Levine, S.; Gabrielson, E.; Corse, A.; Rosen, A. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J. Exp. Med. 2005, 201, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Conrad, K.; Stahnke, G.; Liedvogel, B.; Mehlhorn, J.; Barth, J.; Blasum, C.; Altmeyer, P.; Sonnichsen, N.; Frank, K.H. Anti-CENP-B response in sera of uranium miners exposed to quartz dust and patients with possible development of systemic sclerosis (scleroderma). J. Rheumatol. 1995, 22, 1286–1294. [Google Scholar] [PubMed]
- Lee, S.; Hayashi, H.; Kumagai-Takei, N.; Matsuzaki, H.; Yoshitome, K.; Nishimura, Y.; Uragami, K.; Kusaka, M.; Yamamoto, S.; Ikeda, M.; et al. Clinical evaluation of CENP-B and Scl-70 autoantibodies in silicosis patients. Exp. Ther. Med. 2017, 13, 2616–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdivia, M.M.; Figueroa, J.; Iglesias, C.; Ortiz, M. A novel centromere monospecific serum to a human autoepitope on the histone H3-like protein CENP-A. FEBS Lett. 1998, 422, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Hamaguchi, Y.; Yanaba, K.; Bouaziz, J.D.; Uchida, J.; Fujimoto, M.; Matsushita, T.; Matsushita, Y.; Horikawa, M.; Komura, K.; et al. B-lymphocyte depletion reduces skin fibrosis and autoimmunity in the tight-skin mouse model for systemic sclerosis. Am. J. Pathol. 2006, 169, 954–966. [Google Scholar] [CrossRef] [Green Version]
- Yoshizaki, A. Pathogenic roles of B lymphocytes in systemic sclerosis. Immunol. Lett. 2018, 195, 76–82. [Google Scholar] [CrossRef]
- Maehara, T.; Kaneko, N.; Perugino, C.A.; Mattoo, H.; Kers, J.; Allard-Chamard, H.; Mahajan, V.S.; Liu, H.; Murphy, S.J.; Ghebremichael, M.; et al. Cytotoxic CD4+ T lymphocytes may induce endothelial cell apoptosis in systemic sclerosis. J. Clin. Invest. 2020, 130, 2451–2464. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prasad, R.M.; Bellacosa, A.; Yen, T.J. Clinical and Molecular Features of Anti-CENP-B Autoantibodies. J. Mol. Pathol. 2021, 2, 281-295. https://doi.org/10.3390/jmp2040024
Prasad RM, Bellacosa A, Yen TJ. Clinical and Molecular Features of Anti-CENP-B Autoantibodies. Journal of Molecular Pathology. 2021; 2(4):281-295. https://doi.org/10.3390/jmp2040024
Chicago/Turabian StylePrasad, Rahul M., Alfonso Bellacosa, and Tim J. Yen. 2021. "Clinical and Molecular Features of Anti-CENP-B Autoantibodies" Journal of Molecular Pathology 2, no. 4: 281-295. https://doi.org/10.3390/jmp2040024
APA StylePrasad, R. M., Bellacosa, A., & Yen, T. J. (2021). Clinical and Molecular Features of Anti-CENP-B Autoantibodies. Journal of Molecular Pathology, 2(4), 281-295. https://doi.org/10.3390/jmp2040024