
Mechanisms of Inflammasome Activation and Involvement in Liver Disease


College of Pharmacy, Yeungnam University, Gyeongsan 38541, Republic of Korea
J. Mol. Pathol. 2024, 5(2), 171-186; https://doi.org/10.3390/jmp5020011
Submission received: 28 September 2023
/
Revised: 13 November 2023
/
Accepted: 10 April 2024
/
Published: 13 April 2024
(This article belongs to the Topic Molecular and Cellular Mechanisms of Diseases: Liver Diseases)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The liver is a multi-potent organ with important metabolic, immunological and endocrine functions. Hepatic physiology is maintained at a balanced state via the delicate actions of different liver-resident cells. Among several factors that modulate hepatic physiology, the harmony between the activity of pro- and anti-inflammatory cytokines is a crucial determinant. However, initiation of inflammatory activity can be detrimental if it goes unresolved, leading to severe consequences such as hepatitis, hepatic fibrosis, cirrhosis or even hepatocellular carcinoma (HCC). Different physiological processes can modulate the hepatic microenvironment; one such factor is a cytosolic protein complex called the inflammasome. Inflammasome activation is a consequence of the cellular encounter with pathogens or products of cellular damage. Once activated, inflammasomes promote the maturation of interleukin-1 family cytokines such as IL-1β and IL-18 via activation of caspase-1. These cytokines have a very potent role in modulating hepatic physiology. Various lines of reports suggest that inflammasome activation and IL-1 cytokines play critical roles in liver diseases, including hepatitis, hepatic fibrosis and HCC. Conversely, inhibition of inflammasome activation and/or IL-1 signaling prevents such effects. This review summarizes the mechanisms leading to inflammasome activation and the role it plays in hepatic physiology.
1. Introduction
Liver physiology is intricately balanced by the coordinated action of various liver-resident cells like hepatocytes, Kuppfer cells, hepatic stellate cells and liver sinusoidal endothelial cells. In addition to these cell types, the liver also harbors several subsets of infiltrating T and B lymphocytes, and hepatic stem cells called oval cells. Under conditions of health, there is an equilibrium between the functioning of these cells to sustain hepatic function. A conflict in hepatic physiology arises when there is the presence of inflammation, toxins, viral and bacterial products, etc. In many cases, inflammation is a starting point in a chain of events leading to the dysfunction of hepatic physiology, because it can modulate diverse signaling mechanisms involved in hepatic damage in a myriad of liver-resident and infiltrating cells. In a majority of cases, the ultimate effect peaks with the loss of hepatocyte function. Hepatocytes account for the largest cell type in terms of sheer cell number and volume. They play a critical role in maintaining liver function via a complex set of mechanisms, including detoxification and metabolic regulation, thereby maintaining a balance in the supply and storage of carbohydrates, lipids and proteins. They also help in maintaining immunological harmony by regulating the activation of autoreactive CD8+ T cells [1]. Hence, any abnormality in the cellular physiology of hepatocytes adversely influences the overall balance and functioning of the liver, with implications for metabolic as well as inflammatory responses.
Inflammation starts a chain of events leading to austere consequences. More importantly, a small degree of inflammation can amplify into a pronounced and severe event at later stages. Amplification of inflammatory signals is often transduced via the cooperative action of a diverse group of proinflammatory cytokines [2]. Additionally, the source of inflammation and the generation of inflammatory cytokines can be any of the liver-resident or infiltrating cells. These cytokines can affect hepatic physiology adversely in an autocrine or paracrine manner [3]. Hence, it can be of paramount importance to limit it in the initial stages. Both the adaptive and innate immune systems can contribute to liver inflammation. Liver-infiltrating T lymphocytes are one of the major players of adaptive immunity in the liver, while innate immune activity can be observed in all liver-resident cells, including hepatocytes [4]. It has been observed that during hepatic inflammation multiple subsets of T and B lymphocytes accumulate in hepatic milieu [5]. Additionally, these cells secrete an assorted group of cytokines, often with a predominance towards a specific cytokine, which provides nomenclature to the cell of its origin. For instance, interleukin-17 secreting CD4+ cells are often termed Th17 cells, although they can secrete other cytokines, including IL-21 and IL-22. Nonetheless, these cells augment and accumulate during hepatic inflammation [6], and their cytokines, particularly IL-17, is reported to contribute significantly in hepatic inflammation and subsequent damage [7]. Interestingly, a critical contributor required for the differentiation of Th17 is IL-1β [8], which is a product of inflammasome activation. IL-1β can act in synergy with other cytokines such as IL-6 and IL-23 to differentiate and stabilize Th17, which can further intensify the damage. Furthermore, IL-18 also has been reported to enhance the cytotoxic activity of liver-resident natural killer T cells (NK-T cells) [9]. These reports clearly indicate an important crossover of diverse immune cells and inflammasome activation in modulating hepatic physiology.
In recent years, a cytosolic multi-protein complex known as the inflammasome has emerged to be an influencer of innate immune responses. Particularly, in regard to the modulation of hepatic physiology, it has received due attention as a signal transducer after it encounters one of several pathogen-associated and damage-associated molecular patterns known, respectively, as PAMPs and DAMPs [10]. This encounter promotes the oligomerization of the inflammasome complex, leading to the recruitment and activation of caspase-1, usually via an adaptor molecule called apoptosis-associated speck like protein (ASC) containing a caspase activation and recruitment domain. Overall, NLRP3 inflammasome is the most studied inflammasome because of its ability to be activated by a wide-ranging set of stimuli. NLRP3 consists of several domains with specific functions. Oligomerization of NLRP3 occurs via interaction with its NACHT domains, while an interaction between the PYD domains of NLRP3 and ASC promotes association between NLRP3 and ASC [11]. Furthermore, association of ASC by its CARD domain to caspase-1 leads to the formation of the inflammasome complex. This assembly of the inflammasome complex plays a critical role in the proximity-dependent autolytic cleavage of caspase-1. Notably, ASC-independent activation of inflammasomes has also been reported for other inflammasomes such as NLRP1, suggesting a redundant role of ASC in some conditions [12]. Activation of caspase-1 is characterized by its cleavage into 20 kDa and 10 kDa cleaved products. Cleaved caspase-1 further promotes the maturation of IL-1β and IL-18 as illustrated in Figure 1. Active caspase-1 acts on different sites of IL-1β and IL-18 to enhance their biological activity. Specifically, it acts on two distinct sites of pro-IL-1β (D26 and D116) to generate 26 kDa and 17 kDa fragments [13]. The biological implications of the 26 kDa fragment are relatively unknown, while the 17 kDa fragment is the one involved in cellular physiological processes such as host defense and cell death pathways, including apoptosis. Likewise, cleavage at D36 of pro-IL-18 generates an 18 kDa mature protein from its 23 kDa pro-form, which possesses enhanced inflammatory property. The aftermath of caspase-1 cleavage is not just the maturation of IL-1 cytokines but also cleavage of gasdermin D [14]. Cleavage of gasdermin D results in translocation of its N-terminal fraction into the plasma membrane to cause pore formation. These pores contribute to the influx of extracellular H2O, as a result of which osmotic balance is disrupted, ultimately leading to cell lysis. Pyroptosis also promotes the release of several cytosolic contents, many of which can act as DAMPS, thus further amplifying the inflammatory response. For instance, engulfment of inflammasome components released due to hepatocyte pyroptosis leads to the activation of hepatic stellate cells [15]. Although the maturation of IL-1β and IL-18 enhances their proinflammatory properties, the biological consequences of IL-33 maturation are controversial. As in an experimental setting, maturation of IL-33 has been observed to result in its reduced activity. Although a wide variety of stimuli trigger NLRP3 activation, an interesting feature commonly observed in regard to the physiological response of these agents is their ability to induce cellular stress [16]. This suggests a potential link between cellular stress and inflammasome activation in cellular fate.
Hepatic physiology can be altered in several pathological conditions like viral hepatitis, alcoholic and non-alcoholic liver disease, and autoimmune liver conditions encompassing autoimmune hepatitis (AIH) and primary biliary cholangitis (PBC), etc as depicted in Figure 2. Incidentally, inflammasome activation has been reported to play a crucial role in these hepatic disorders [17]. Inflammasome activation and its related physiological responses get even more attention because of the liver’s proximity to the digestive system, from where it is subjected to several toxicological insults, including bacterial product lipopolysaccharide (LPS). Although it is well known for its effect as an inducer of the priming step of inflammasome activation, some experimental models suggest it can stimulate an activation step as well. Nonetheless, even if only priming is generated, it still can create a favorable environment for inflammasome-dependent toxicity, especially when there is the presence of another agent likely to have the ability to induce inflammasome activation. Considering that the liver encounters several internal and external factors, including fatty acids, hormones, food and drug borne metabolites, etc., and on a routine basis, this combination is very likely to be noxious to the liver. Such types of effects can be spotted in alcoholic injury and endotoxemia [18].
Several lines of reports suggest that inflammasome-dependent physiological responses are often conveyed by the maturation of two major cytokines: IL-1β and IL-18. Although they bind to their specific receptors, IL-1R1 for IL-1β and IL-1R5 for IL-18, both of them are members of the IL-1 superfamily and are reported to have similar structural confirmations [19]. Both of these cytokines have potent proinflammatory properties. Under ordinary conditions, a balanced production of IL-1Ra and IL-18BP, which are natural inhibitors of IL-1 and IL-18 signaling, respectively, prevent aberrant physiological responses due to these cytokines. However, during inflammatory and pathological conditions the activities of IL-1β and IL-18 supersede the inhibitory capacities of their inhibitors. These cytokines show a pleotropic effect in different liver cells. They can induce hepatocyte death while promoting the activation and proliferation of hepatic stellate cells [20]. Activation of hepatic stellate cells leads to hepatic fibrosis, whereas a loss of the hepatic parenchymal population significantly reduces the functional ability of the liver, thus facilitating liver failure in future. Furthermore, they also enhance the inflammatory activity of liver-resident macrophages called Kupffer cells, consequently further amplifying the inflammatory cascade. Hence, it is of paramount importance to limit the activation of these cytokines for the maintenance of hepatic homeostasis.
The role of inflammasomes in modulating cellular physiology via IL-33-dependent mechanisms is complex. While IL-33 is known to contribute to hepatic fibrosis, especially after secretion from stressed hepatocytes [21], in some experimental conditions such as ischemia-reperfusion induced hepatic injury, IL-33 has provided direct hepatoprotection [22]. In another study, on concanavalin A-mediated hepatic damage, IL-33-knockout mice had increased liver damage compared to their wild-type counterparts [23], suggesting a context-dependent role of IL-33 in modulating hepatic physiology. This is attributed to enhanced generation of TNFα and IL-1β in IL-33 knockout mice. IL-33-mediated hepatoprotective effects were conveyed directly as well as via modulation of the adaptive immune system. In one report, cleavage of IL-33 has been reported to render a reduction in activity compared to its uncleaved form, suggesting that inflammasome activation can also limit the activity of IL-33 in certain situations [24]. Additionally, it can drive T cell polarization towards a Th2 phenotype, thus promoting the upregulation of Th2 cytokines like IL-4 and IL-13 [25]. Since, these Th2 cytokines can behave as anti-inflammatory agents; it is also possible that IL-33 activation via an inflammasome-dependent manner can limit inflammation.
2. Mechanisms Leading to Inflammasome Activation
2.1. Endoplasmic Reticulum Stress (ER Stress)
The endoplasmic reticulum is an important organelle involved in protein folding. In the course of protein folding, mismatches may occur that result in the accumulation of misfolded proteins. The accumulation of misfolded proteins leads to a response termed the unfolded protein response (UPR). Usually, this can be resolved by a temporary halt in protein synthesis. Phosphorylation of eif2α plays a major role in the shutdown of translation; however, some proteins like ATF4 undergo a preferential translation [26], leading to upregulation of CHOP and its downstream proteins such as ERO1, Bax, etc., that play vital roles in apoptosis. ER stress is a response to a disturbance in protein folding that is initiated to reduce potential cellular damage. However, unresolved UPR can lead to ER stress. Three major sensors of ER- PERK, IRE1 and ATF6 relay ER-stress signals. At normal levels, activation of these signal transducers is regulated by chaperone proteins, including Binding Immunoglobulin protein (BiP). Accumulation of misfolded proteins facilitates the release of these chaperone proteins, thus leading to signal transduction via these sensors. ER stress can lead to transcriptional and translational upregulation of inflammasome components, namely NLRP3, ASC, pro-IL-1β and pro-IL-18 [27]. In addition, it can also induce inflammasome activation via the upregulation of reactive oxygen species, enhancement of cytosolic calcium via store-operated entry, and the upregulation and interaction of thioredoxin-interacting protein (TXNIP) with NLRP3 [28].
2.2. Reactive Oxygen Species (ROS)
Reactive oxygen species can be a byproduct of different metabolic processes. Upregulation in ROS can be sensed by the inflammasome sensor proteins as a danger signal. In addition, because of their highly reactive potency, they can induce damage in membrane components akin to lipids in mitochondrial membranes, as a result causing mitochondrial damage [29]. Mitochondrial damage fosters the release of mitochondrial DNA, and is recognized by DNA-sensing inflammasomes, particularly AIM-2 and NLRP3 [30]. This recognition promotes the activation of inflammasomes. Beside this, they can also promote efflux of potassium ions, thereby inducing activation of inflammasomes [31].
It has also been reported that oxidative stress contributes to the phosphorylation of NFκB [32], which leads to its enhanced activity, thus potentially contributing to upregulation of the priming and subsequent activation of inflammasomes. Accumulating evidence suggests that ROS can suppress sirtuin1 (SIRT1) activity by a diverse set of mechanisms [33]. Notably, SIRT1 contributes to a reduction in the transcriptional activity of NFκB via deacetylation of a lysine residue in the NFκB subunit RelA [34]. This indicates an enhanced activation of inflammasomes via NFκB-mediated transcriptional upregulation of inflammasome components by a SIRT1 inhibition pathway. Furthermore, during conditions of inflammation, NFκB can also induce different NADPH oxidases, which are well-known contributors to ROS generation [35]. Additionally, inhibition of NFκB has been demonstrated to suppress ROS production [36]. Cumulatively, these studies hint at a self-perpetuating loop between ROS and NFκB under certain experimental conditions to modulate cell physiology.
2.3. Ions and Inflammasomes
The inflammasome’s assembly and ultimate activation is influenced by the presence or absence of certain ions. The most commonly studied ions in terms of inflammasome activation are K+ and Ca2+, although other ions like Cl−, Zn2+ Mg2+ etc. can also be implicated. Inflammasome activation is promoted by the efflux of K+ and influx of Ca2+ [37]. Several types of inflammasome-activating stimuli induce inflammasome activation with a concomitant reduction in intracellular K+ concentration, suggesting K+ efflux to be a common pathway employed by a variety of inflammasome inducers, including nigericin and gramicidin.
Calcium mobilization by an ER-stress-dependent mechanism has been reported to influence the assembly of the inflammasome complex leading to its activation. Interestingly, it has been witnessed that a reduction in intracellular K+ leads to an increase in Ca2+ [37]. Although the specific mechanisms by which a rise in intracellular calcium promotes inflammasome activation have not been well understood, few potential mechanisms have been suggested. The likely mechanisms include the damaging of mitochondria due to calcium binding, thus promoting the release of mitochondrial DNA, which can be sensed by AIM2 or NLRP3 inflammasomes [38]. Another mechanism involves the downregulation of cAMP by calcium [39]. As cAMP inhibits NLRP3 activation via binding with it, a reduction in cAMP levels is likely to boost inflammasome activation.
2.4. Proteases
Calcium-activated proteases, mainly calpains and cathepsins, crucially contribute to the activation of inflammasomes. Calpains are calcium-dependent proteases, which can lead to the destabilization of lysosomal membranes, thereby increasing the secretion of cathepsin B from the lysosome [40]. Calpains can promote upregulation in mitochondrial ROS and enhance mitochondrial damage to stimulate inflammasome activation [41]. Maturation of cathepsin B leading to its enhanced secretion from lysosomes encourages direct interaction with NLRP3 inflammasomes and ultimately promotes its activation [42]. In recent years, the axis of autophagy and cathepsin B maturation has emerged as a crucial player in the modulation of inflammasome activation in different conditions [43,44].
2.5. Complement System
The complement system is another important part of the innate immune system. Activation of the complement system occurs in a series of steps. The ultimate result due to complement activation is the formation of membrane attack complex (MAC). MAC formation to a high extent is known to induce cell lysis, while sub-lytic MAC formation can lead to the activation of inflammasomes via enhanced cytosolic calcium due to its release from the ER’s IP3R and RyR calcium channels [45]. Interestingly, complement-dependent upregulation of ROS production by cholesterol crystals also has been reported to cause direct activation of inflammasomes, climaxing in the secretion of IL-1β and other cytokines [46]. Additionally, the complement system has also been found to be involved in activation of inflammasome during MERS-CoV infection [47].
3. Involvement of Inflammasome Activation in Liver Diseases
3.1. Acetaminophen (APAP)-Induced Hepatotoxicity
APAP, also called paracetamol, is a widely used non-steroidal anti-inflammatory (NSAID) drug for the management of fever and pain. Although relatively safe in moderate doses, it can have hepatotoxic effects too, especially at high doses, and it is one of the leading causes of drug-induced acute liver failure. It is converted into a toxic intermediate, NAPQI [48], by the cytochrome P450 system under conditions of saturated glucuronidation and sulfation. This ultimately leads to enhanced ROS production. Excess APAP is well documented to be involved in the destruction of liver architecture. There is some controversial information about the type of cell death involved in APAP toxicity, i.e., apoptotic or necrotic. Some evidence suggests significant hepatocyte apoptosis by APAP, while other reports suggest the type of cell death to be predominantly necrosis. Furthermore, a study reports the improper execution of apoptosis being switched to necrotic death by APAP [49]. Nonetheless, its involvement in hepatocyte death [50] and activation of inflammasomes is supported by various studies [51]. Diverse reports suggest that IL-1β is implicated in mediating the toxic effects of APAP. Inhibition of IL-1 signaling by pharmacological means, using IL-1Ra as well as neutralizing antibodies against IL-1β, provided significant hepatoprotection against APAP toxicity [52]. In addition to this, employment of genetic means such as a knockout of the IL-1R1 gene also prevented the toxicity of APAP. Contrary to this, reports where the inhibition of inflammasome activation did not show any hepatoprotection against APAP toxicity have also been reported [53]. These controversial findings warrant further investigation to explore such opposite findings in different studies.
3.2. High-Fat Diet-Induced Hepatotoxicity
Generally, short-term high-fat feeding leads to the development of steatosis but not steatohepatitis, especially in the absence of inflammatory signals, whereas long-term high-fat feeding can lead to NASH [54]. Multiple lines of study highlight the role of IL-1 signaling in the progression of hepatic steatosis, suggesting a crucial role for inflammasome activation in the subsequent process [55,56]. IL-1β can promote insulin resistance, leading to a disruption in the insulin-mediated regulation of lipogenesis, which can result in the accumulation of fat in the liver [57]. Additionally, inflammasome activation can amplify the inflammatory signals in the liver to enhance hepatic damage on administration of high-fat diet [58].
3.3. Hormone-Induced Hepatotoxicity
The liver, being an important metabolic as well as endocrine organ, experiences potent hormonal action. Several hormones account for enhanced inflammatory conditions in the liver. The most well-known among them are the adipokines, namely leptin and visfatin [59,60]. Leptin induces inflammasome activation in diverse cell types, including hepatocytes [43]. Androgens have been reported to stimulate inflammasome activation leading to hepatic fibrosis [61]. Although low testosterone has negative physiological consequences, the administration of exogenous testosterone has been shown to enhance liver damage characterized by hepatocyte ballooning, upregulation of collagen-1 and TGFβ, HSC activation and fibrosis. In a study by Zhang et al., angiotensin II induced EMT of human hepatocytes in an inflammasome-dependent manner characterized by the upregulation of vimentin but downregulation of cadherin, which could promote hepatic fibrosis in the long run [62].
3.4. Alcohol-Induced Hepatotoxicity
In experimental as well as clinical scenarios in humans, alcoholic liver injury is closely associated with inflammasome activation [63]. Alcohol is metabolized primarily in the liver, and this metabolism generates acetaldehyde and ROS, both of which are highly toxic compounds. Aldehyde is also reported to enhance gut permeability, thus increasing the possibility of endotoxin leakage from the intestine [64]. These agents lead to the damage of hepatic membranes, enhancement of inflammation and promotion of hepatocyte death. Hepatic steatosis is also a frequently detected effect of alcohol [65]. Inflammasome activation plays a critical role in the processes of both steatosis and steatohepatitis. Inflammasome-activation-induced IL-1β mediates hepatic injury in both acute and chronic cases of alcohol intake, and inhibition of IL-1β-signaling protected mice from alcohol-induced liver injury [66].
3.5. Endotoxin-Induced Hepatotoxicity
The proximity of the liver to the digestive system and the first-pass effect exposes the liver to both beneficial and harmful products. Notably, one such noxious agent likely to enter liver via digestive tract is the endotoxin LPS. It is a component of gram-negative bacteria. Specifically, in regard to its association with inflammasomes, it is a well-known inducer of the priming step, promoting the upregulation of inflammasome components, including NLRP3, ASC, caspase-1, pro-IL-1β and pro-IL-18 [67]. LPS has a cell-specific role in relation to the activation of inflammasomes, i.e., priming only or also activation. LPS alone at moderate levels may not enhance the direct cytotoxicity of hepatocytes, but through other liver-resident cells it can promote toxic effects in the liver. Interestingly, it has been reported that Kupffer cells mediate the toxic effects of LPS via the upregulation of IL-1β and IL-18 [68]. In addition to this mechanism, despite the lack of direct toxicity to hepatocytes, it can sensitize hepatocytes towards the adverse effect of other harmful agents such as D-galactosamine and promote inflammasome-dependent hepatotoxicity. Interestingly, deletion of hepatocyte-specific IL-1R1 significantly protected murine livers from hepatic damage due to LPS/D-gal, implying a critical role of hepatocyte-specific IL-1R1 in modulating cell death and survival [69].
3.6. Viral Hepatitis
Viral hepatitis is mainly caused by five different strains of hepatitis virus, namely hepatitis A, B, C, D and E. Hepatitis A, along with hepatitis E, accounts for the majority of acute liver failure cases. Reports regarding the effect of hepatitis A virus in inflammasome activation and its implication in hepatic dysfunction is lacking. Nonetheless, a study found an elevation in IL-1β among other cytokines in children infected with hepatitis A [70]. However, it is not well understood whether the enhancement of IL-1β alone or in combination with other cytokines induced any adverse effects. Hepatitis B virus can lie dormant for a period and resurface. Upregulation of AIM2 inflammasomes and IL-18 have been reported in cases of hepatitis B virus (HBV) [71]. Once it has entered into the body, hepatitis C virus (HCV) replicates in hepatocytes after internalization facilitated by endocytosis. HCV induces activation of inflammasomes and the subsequent generation of IL-1β with the involvement hepatic macrophages [72]. While it is believed that hepatitis D virus (HDV) associates with HBV to exert its pathogenic effects, information about HDV in regard to inflammasome activation is lacking. A recent report has highlighted the fact that hepatitis E virus (HEV) induces inflammasome activation via NFκB-dependent mechanisms, leading to enhanced replication in macrophages [73]. Furthermore, inhibition of inflammasomes promoted decreased viral replication. Additionally, in mice infected with murine coronavirus, IL-1 deficiency showed a protective role with survival of affected mice shown to be similar to that of wild-type mice, but IL-18 deficient mice showed reduced survival compared to their wild-type counterparts, suggesting that a protective effect during hepatitis virus infection is likely derived from IL-18-dependent mechanisms rather than IL-1β-dependent pathways [74].
3.7. Autoimmune Liver Disease
Autoimmune liver disease is an umbrella term for various liver diseases that arise due to aberrant activation of the immune system, leading to attacks on hepatocytes and bile ducts. CD4+ T cells, mainly of Th1 and Th17 lineages, are the pivotal contributors to autoimmune liver disease [75]. It often presents with interface hepatitis, portal inflammation and the upregulation of IgG, ANA, LKM1 and ASGPR autoantibodies [76]. If unresolved, it can ultimately cause liver failure. The most common autoimmune liver diseases are autoimmune hepatitis (AIH), primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC). In a model of concanavalin A-induced AIH, administration of IL-1Ra provided significant protection against AIH, which was characterized by a reduction in liver injury, lower serum enzymes, decreased hepatic pyroptosis and a reduction in serum as well as liver IL-17, which can act as a major player in AIH [77]. Additionally, in another model of TCE-induced liver injury, inhibition of oxidative stress suppressed inflammasome activation and the upregulation of antibody producing B cells as well as hepatic damage [78]. This insinuates the possibility of a correlation between inflammasome activation and autoimmune hepatitis. Additionally, in a model of PBC, NLRP3 inflammasome activation and IL-1β signaling were found to be related to the upregulation of IL-17, which is a critical modulator of AIH [79].
4. Inflammasome-Dependent Therapies in Liver Disease
4.1. Anakinra (IL-1Ra)
IL-1 receptor antagonist is an endogenous inhibitor of IL-1. It is also known as anakinra by its trade name and acts via binding to the IL-1 receptor for the prevention in IL-1 cytokine-dependent signaling, thus reducing the signal transduction via IL-1 receptor. While it has been reported that the dosage of IL-1Ra required to inhibit IL-1β-dependent effects is over one hundred fold than that of IL-1β [80], nonetheless IL-1Ra has shown promising effects in inhibiting several models of experimental liver disease, including NASH, fibrosis and hepatitis [81,82,83]. It has been approved by the FDA for the treatment of rheumatoid arthritis and cryopyrin-associated periodic syndromes. However, administration of IL-1Ra in combination with pentoxifylline and zinc showed no significant increase in protection compared to methylprednisolone in a randomized clinical trial in a model of severe alcoholic hepatitis, although endotoxin levels were reduced by the combined treatment of IL-1Ra [84]. In the study, IL-1Ra did not show any adverse effects; hence, this strategy could still be a viable alternative in cases of allergy to corticosteroids, although further research is required for confirmation.
4.2. Canakinumab
Canakinumab is a monoclonal antibody developed to specifically target IL-1β. The FDA has approved canakinumab for the treatment of Still’s disease and it is sold under the brand name Ilaris. A study reported that treatment with canakinumab in combination with colchicine showed improvement in hepatic dysfunction due to familial Mediterranean fever [85]. Although it has shown promising effects in the management of cardiovascular disorders, its significant efficacy in the specific management of hepatic disorders has not been observed yet. In addition, a multicenter randomized clinical trial in a model of AH reported slight improvement in hepatic histology but not to significant levels as deduced by regression models between canakinumab-treated patients and placebo-treated patients [86].
4.3. MCC950
MCC950 is a selective NLRP3 inhibitor that binds to the central crater of the NACHT domain to prevent its oligomerization [87]. During the activation of inflammasomes, oligomerization of NLRP3 is required for the formation of the assembly complex with ASC that recruits caspase-1, thus promoting its cleavage. This complex of ASC fibrils and NLRP3 is also known as ASC specks. MCC950, via its selective action on NLRP3, disrupts this speck formation [88]. MCC950 has been reported to prevent several hepatic diseases in experimental settings, including NASH-associated fibrosis [89].
4.4. Ac-YVAD
Ac-YVAD is a tetrapeptide with sequence homology to the IL-1β cleavage site of caspase-1. It has a very high specificity towards caspase-1 and, in a competitive manner, it inhibits the activity of caspase-1 to prevent the maturation of IL-1β. It has a long history of protective effects against liver toxicity in a variety of experimental conditions, including hepatic apoptosis [90]. Additionally, it has shown promising effects in the management of insulin resistance and associated NASH [91,92].
4.5. Gasdermin D Inhibitors
Additionally, inhibitors of gasdermin D like disulfiram, which is also known to inhibit aldehyde dehydrogenase, have been observed to play a protective role in liver disease [93]. Dimethyl fumarate, which has been observed to inhibit pyroptosis in some studies, has shown protective effects against ethanol-induced steatosis by preventing liver inflammation and the impairment of gut-barrier integrity [94]. Additionally, in a genetic model of mice deficient with gasdermin D due to gene knockout, less steatosis was observed that in their wild-type counterparts [95], which can be attributed to its role as a pyroptosis executioner leading to enhanced cytokine secretion.
4.6. IL-18 Inhibitors
A natural inhibitor of IL-18 (IL-18BP) is documented to prevent liver disease in a model of LPS-induced lethality [96]. Some other reports suggest a protective role of IL-18BP in preventing hepatic fibrosis [97]. Similar effects were also observed in IL-18 knockout mice, further strengthening the protective role of IL-18 inhibition in hepatic fibrosis. Although IL-18 upregulation is reported in human subjects with hepatic fibrosis, no study has emerged suggesting a protective effect of IL-18 inhibitors in human.
4.7. Melatonin
Melatonin is a hormone secreted by the pineal gland that helps in maintaining a circadian rhythm in mammals. In addition to its beneficial role in sleep regulation, several lines of evidence suggest that it acts as an anti-inflammatory agent as well. Particularly, in the liver, it has been observed to inhibit inflammasome activation and limit hepatic damage due to cadmium. It acts on hepatocytes to limit cadmium-induced TXNIP upregulation, leading to reduced interaction with NLRP3 and preventing inflammasome activation and subsequent hepatic damage [98]. Additionally, a meta-analysis has reported that melatonin could improve features of hepatic damage, including steatosis, steatohepatitis and elevation of liver enzymes [99]. Interestingly, melatonin has also been reported in different studies to regulate NLRP3 inflammasomes and exert a protective effect in the heart that helps to maintain its structural and functional integrity [100,101,102].
5. Inflammasome Activation Is Not Always Detrimental
The beneficial roles that inflammasome activation plays in certain scenarios has suggested it to have an ambivalent role in the modulation of liver physiology. For instance, the activation of inflammasomes have been shown to act as a defensive mechanism in the cellular fight against certain bacterial pathogens. NLRP3 activation induced by inhibition of mammalian target of rapamycin (mTOR) has polarized macrophages towards an M1-phenotype, thereby limiting parasite load [103]. Furthermore, it is required for better clearance of pathogens on infection with Trypanosoma cruzi. Not only this, during infection with murine coronavirus, mice with defective inflammasome signaling because of the genetic knockout of inflammasome components had a lower chance of survival than their wild-type counterparts. In this study, the lack of IL-18 showed more detrimental effects than the lack of IL-1β, suggesting that IL-18 plays a more cytoprotective role than IL-1β following inflammasome activation [74]. Furthermore, under conditions of hypoxia/reoxygenation, caspase-1 induces a protective effect in hepatocytes, as caspase-1 knockout hepatocytes had higher rate of cell death than wild-type hepatocytes in [104]. Additionally, the downregulation in inflammasome components had an inverse relationship with the stages of hepatocellular carcinoma [105], suggesting a possibility for inflammasome activation to limit the development and growth of HCC. Furthermore, in recent years, the role of the NLRP6 inflammasome, which was relatively unknown, has emerged as protective in some studies. The overexpression of NLRP6 using adeno-virus protected liver from hepatic steatosis and inflammation in a model of alcoholic hepatitis [106]. In yet another study, genetic removal of NLRP6 using LoxP/Cre technology potentiated hepatic steatosis and inflammation in NLRP6 knockout mice more than was seen in than their wild-type littermates fed with a high-fat diet [107].
6. Conclusions
Inflammasome activation seems to modulate hepatic physiology in a diverse way with complex cross talk between innate and adaptive immune responses. Although, a lot of information has been obtained about the regulation of extensively studied inflammasomes comprising NLRP3 and AIM2, the existence of relatively unexplored ones like NLRP6 that have a significant role in modulating hepatic physiology has emerged. This demands more fundamental research in the area of inflammasome study. In addition, while some encouraging results have been observed under experimental conditions as summarized in Figure 3 by targeting inflammasome-dependent mechanisms in the treatment of various liver disorders, the best approach for the transformation of laboratory findings into clinical settings is still an enigma, which can only be clarified by future research into the topic.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
| AIM2 | Absent in melanoma 2 |
| ANA | Antinuclear antibodies |
| ASGPR | Asialogylcoprotein receptor |
| ASC | Apoptosis-associated speck like protein containing a CARD |
| cAMP | Cyclic adenosine monophosphate |
| CARD | Caspase activation and recruitment domain |
| DAMPs | Damage-Associated Molecular Patterns |
| FDA | Food and Drug Administration |
| IP3R | Inositol triphosphate receptor |
| LKM1 | Liver kidney microsomal antibody type 1 |
| LPS | Lipopolysaccharide |
| MAC | Membrane attack complex |
| NASH | Non-alcoholic steatohepatitis |
| NLRP3 | Nucleotide-binding domain, leucine-rich-containing family, pyrin domain-3 |
| PAMPs | Pathogen-Associated Molecular Patterns |
| PYD | Pyrin domain |
| ROS | Reactive oxygen species |
| RyR | Ryanodine receptor |
| TCE | Trichloroethane |
References
- Benseler, V.; Warren, A.; Vo, M.; Holz, L.E.; Tay, S.S.; Le Couteur, D.G.; Breen, E.; Allison, A.C.; van Rooijen, N.; McGuffog, C.; et al. Hepatocyte entry leads to degradation of autoreactive CD8 T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16735–16740. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cytokine amplification and inhibition of immune and inflammatory responses. J. Viral Hepat. 1997, 4 (Suppl. S2), 6–15. [Google Scholar] [CrossRef] [PubMed]
- Andus, T.; Bauer, J.; Gerok, W. Effects of cytokines on the liver. Hepatology 1991, 13, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Xu, M.J.; Gao, B. Hepatocytes: A key cell type for innate immunity. Cell. Mol. Immunol. 2016, 13, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Zhang, Z.; Lin, F.; Zou, Z.-S.; Xu, R.-N.; Jin, L.; Fu, J.-L.; Shi, F.; Shi, M.; Wang, H.-F.; et al. Interleukin-17–producing CD4+ T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology 2010, 51, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 2011, 166, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.; Chang, S.H.; Martinez, G.J.; Yang, X.O.; Nurieva, R.; Kang, H.S.; Ma, L.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 2009, 30, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.; Mehal, W.Z.; Crispe, I.N. IL-18 augments perforin-dependent cytotoxicity of liver NK-T cells. J. Immunol. 1998, 161, 2217–2222. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.D.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Van Opdenbosch, N.; Gurung, P.; Vande Walle, L.; Fossoul, A.; Kanneganti, T.D.; Lamkanfi, M. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat. Commun 2014, 5, 3209. [Google Scholar] [CrossRef] [PubMed]
- Afonina, I.S.; Müller, C.; Martin, S.J.; Beyaert, R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity 2015, 42, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Xue, W.; Wei, H.; Fan, Q.; Li, X.; Qiu, Y.; Cui, D. Research Progress of Pyroptosis in Fatty Liver Disease. Int. J. Mol. Sci. 2023, 24, 13065. [Google Scholar] [CrossRef] [PubMed]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Ju, D. Inflammasome: A Double-Edged Sword in Liver Diseases. Front. Immunol. 2018, 9, 2201. [Google Scholar] [CrossRef]
- Schaffert, C.S.; Duryee, M.J.; Hunter, C.D.; Hamilton, B.C., 3rd; DeVeney, A.L.; Huerter, M.M.; Klassen, L.W.; Thiele, G.M. Alcohol metabolites and lipopolysaccharide: Roles in the development and/or progression of alcoholic liver disease. World J. Gastroenterol. 2009, 15, 1209–1218. [Google Scholar] [CrossRef]
- Tapia, V.S.; Daniels, M.J.D.; Palazón-Riquelme, P.; Dewhurst, M.; Luheshi, N.M.; Rivers-Auty, J.; Green, J.; Redondo-Castro, E.; Kaldis, P.; Lopez-Castejon, G.; et al. The three cytokines IL-1β, IL-18, and IL-1α share related but distinct secretory routes. J. Biol. Chem. 2019, 294, 8325–8335. [Google Scholar] [CrossRef]
- Charan, H.V.; Dwivedi, D.K.; Khan, S.; Jena, G. Mechanisms of NLRP3 inflammasome-mediated hepatic stellate cell activation: Therapeutic potential for liver fibrosis. Genes Dis. 2023, 10, 480–494. [Google Scholar] [CrossRef]
- Tan, Z.; Liu, Q.; Jiang, R.; Lv, L.; Shoto, S.S.; Maillet, I.; Quesniaux, V.; Tang, J.; Zhang, W.; Sun, B.; et al. Interleukin-33 drives hepatic fibrosis through activation of hepatic stellate cells. Cell. Mol. Immunol. 2018, 15, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Van Sweringen, H.L.; Quillin, R.C.; Schuster, R.; Blanchard, J.; Burns, J.M.; Tevar, A.D.; Edwards, M.J.; Lentsch, A.B. Interleukin-33 is hepatoprotective during liver ischemia/reperfusion in mice. Hepatology 2012, 56, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Volarevic, V.; Mitrovic, M.; Milovanovic, M.; Zelen, I.; Nikolic, I.; Mitrovic, S.; Pejnovic, N.; Arsenijevic, N.; Lukic, M.L. Protective role of IL-33/ST2 axis in Con A-induced hepatitis. J. Hepatol. 2012, 56, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. USA 2009, 106, 9021–9026. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Baird, T.D.; Zhou, D.; Palam, L.R.; Spandau, D.F.; Wek, R.C. Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J. Biol. Chem. 2010, 285, 33165–33174. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wen, Y.; Lv, L.L.; Liu, H.; Tang, R.N.; Ma, K.L.; Liu, B.C. Involvement of endoplasmic reticulum stress in angiotensin II-induced NLRP3 inflammasome activation in human renal proximal tubular cells in vitro. Acta Pharmacol. Sin. 2015, 36, 821–830. [Google Scholar] [CrossRef]
- Li, W.; Cao, T.; Luo, C.; Cai, J.; Zhou, X.; Xiao, X.; Liu, S. Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. Appl. Microbiol. Biotechnol. 2020, 104, 6129–6140. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Kolliputi, N.; Shaik, R.S.; Waxman, A.B. The inflammasome mediates hyperoxia-induced alveolar cell permeability. J. Immunol. 2010, 184, 5819–5826. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Yi, T.; Chen, C. NF-κB-Gasdermin D (GSDMD) Axis Couples Oxidative Stress and NACHT, LRR and PYD Domains-Containing Protein 3 (NLRP3) Inflammasome-Mediated Cardiomyocyte Pyroptosis Following Myocardial Infarction. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 6044–6052. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.-g. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, K.; Zhang, K.; Zhou, F.; Zhu, L. Induction of oxidative and nitrosative stresses in human retinal pigment epithelial cells by all-trans-retinal. Exp. Cell Res. 2016, 348, 87–94. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Negash, A.A.; Olson, R.M.; Griffin, S.; Gale, M., Jr. Modulation of calcium signaling pathway by hepatitis C virus core protein stimulates NLRP3 inflammasome activation. PLOS Pathog. 2019, 15, e1007593. [Google Scholar] [CrossRef]
- Lee, G.-S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef]
- Yamashima, T.; Kohda, Y.; Tsuchiya, K.; Ueno, T.; Yamashita, J.; Yoshioka, T.; Kominami, E. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074: A novel strategy for neuroprotection based on ‘calpain-cathepsin hypothesis’. Eur. J. Neurosci. 1998, 10, 1723–1733. [Google Scholar] [CrossRef]
- Liu, X.; Li, M.; Chen, Z.; Yu, Y.; Shi, H.; Yu, Y.; Wang, Y.; Chen, R. Mitochondrial calpain-1 activates NLRP3 inflammasome by cleaving ATP5A1 and inducing mitochondrial ROS in CVB3-induced myocarditis. 2022, 117, 40. [CrossRef]
- Chevriaux, A.; Pilot, T.; Derangère, V.; Simonin, H.; Martine, P.; Chalmin, F.; Ghiringhelli, F.; Rébé, C. Cathepsin B Is Required for NLRP3 Inflammasome Activation in Macrophages, Through NLRP3 Interaction. Front. Cell Dev. Biol. 2020, 8, 167. [Google Scholar] [CrossRef] [PubMed]
- Baral, A.; Park, P.-H. Leptin Induces Apoptotic and Pyroptotic Cell Death via NLRP3 Inflammasome Activation in Rat Hepatocytes. Int. J. Mol. Sci. 2021, 22, 12589. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Mo, S.T. Caspase-8 inactivation drives autophagy-dependent inflammasome activation in myeloid cells. Sci. Adv. 2022, 8, eabn9912. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Hughes, T.R.; Triantafilou, M.; Morgan, B.P. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 2013, 126, 2903–2913. [Google Scholar] [CrossRef] [PubMed]
- Samstad, E.O.; Niyonzima, N.; Nymo, S.; Aune, M.H.; Ryan, L.; Bakke, S.S.; Lappegård, K.T.; Brekke, O.L.; Lambris, J.D.; Damås, J.K.; et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J. Immunol. 2014, 192, 2837–2845. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, J.; Teng, Y.; Sun, H.; Tian, G.; He, L.; Li, P.; Chen, Y.; Guo, Y.; Li, J.; et al. Complement Receptor C5aR1 Inhibition Reduces Pyroptosis in hDPP4-Transgenic Mice Infected with MERS-CoV. Viruses 2019, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenetics Genom. 2015, 25, 416–426. [Google Scholar] [CrossRef] [PubMed]
- El-Hassan, H.; Anwar, K.; Macanas-Pirard, P.; Crabtree, M.; Chow, S.C.; Johnson, V.L.; Lee, P.C.; Hinton, R.H.; Price, S.C.; Kass, G.E. Involvement of mitochondria in acetaminophen-induced apoptosis and hepatic injury: Roles of cytochrome c, Bax, Bid, and caspases. Toxicol. Appl. Pharmacol. 2003, 191, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Kil, Y.-S.; Baral, A.; Jeong, B.-S.; Laatikainen, P.; Liu, Y.; Han, A.-R.; Hong, M.-J.; Kim, J.-B.; Choi, H.; Park, P.-H.; et al. Combining NMR and MS to Describe Pyrrole-2-Carbaldehydes in Wheat Bran of Radiation. J. Agric. Food Chem. 2022, 70, 13002–13014. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Jaeschke, H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J. Hepatol. 2017, 66, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Farhood, A.; Jaeschke, H. Role of caspase-1 and interleukin-1beta in acetaminophen-induced hepatic inflammation and liver injury. Toxicol. Appl. Pharmacol. 2010, 247, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Velázquez, K.T.; Enos, R.T.; Bader, J.E.; Sougiannis, A.T.; Carson, M.S.; Chatzistamou, I.; Carson, J.A.; Nagarkatti, P.S.; Nagarkatti, M.; Murphy, E.A. Prolonged high-fat-diet feeding promotes non-alcoholic fatty liver disease and alters gut microbiota in mice. World J. Hepatol. 2019, 11, 619–637. [Google Scholar] [CrossRef] [PubMed]
- Kamari, Y.; Shaish, A.; Vax, E.; Shemesh, S.; Kandel-Kfir, M.; Arbel, Y.; Olteanu, S.; Barshack, I.; Dotan, S.; Voronov, E.; et al. Lack of interleukin-1α or interleukin-1β inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J. Hepatol. 2011, 55, 1086–1094. [Google Scholar] [CrossRef]
- Gehrke, N.; Hofmann, L.J.; Straub, B.K.; Rühle, F. Hepatic interleukin-1 receptor type 1 signalling regulates insulin sensitivity in the early phases of nonalcoholic fatty liver disease. Clin. Transl. Med. 2022, 12, e1048. [Google Scholar] [CrossRef]
- Mirea, A.M.; Tack, C.J.; Chavakis, T.; Joosten, L.A.B.; Toonen, E.J.M. IL-1 Family Cytokine Pathways Underlying NAFLD: Towards New Treatment Strategies. Trends Mol. Med. 2018, 24, 458–471. [Google Scholar] [CrossRef]
- Vargas-Pozada, E.E.; Ramos-Tovar, E.; Rodriguez-Callejas, J.D.; Cardoso-Lezama, I.; Galindo-Gómez, S.; Gil-Becerril, K.; Vásquez-Garzón, V.R.; Arellanes-Robledo, J.; Tsutsumi, V.; Villa-Treviño, S.; et al. Activation of the NLRP3 inflammasome by CCl(4) exacerbates hepatopathogenic diet-induced experimental NASH. Ann. Hepatol. 2023, 28, 100780. [Google Scholar] [CrossRef]
- Xia, M.; Boini, K.M.; Abais, J.M.; Xu, M.; Zhang, Y.; Li, P.L. Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am. J. Pathol. 2014, 184, 1617–1628. [Google Scholar] [CrossRef]
- Koka, S.; Xia, M.; Zhang, C.; Zhang, Y.; Li, P.L.; Boini, K.M. Podocyte NLRP3 Inflammasome Activation and Formation by Adipokine Visfatin. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2019, 53, 355–365. [Google Scholar] [CrossRef]
- Ma, X.; Zhou, Y.; Qiao, B.; Jiang, S.; Shen, Q.; Han, Y.; Liu, A.; Chen, X.; Wei, L.; Zhou, L.; et al. Androgen aggravates liver fibrosis by activation of NLRP3 inflammasome in CCl(4)-induced liver injury mouse model. 2020, 318, E817–E829. [CrossRef]
- Zhang, L.L.; Huang, S.; Ma, X.X.; Zhang, W.Y.; Wang, D.; Jin, S.Y.; Zhang, Y.P.; Li, Y.; Li, X. Angiotensin(1-7) attenuated Angiotensin II-induced hepatocyte EMT by inhibiting NOX-derived H2O2-activated NLRP3 inflammasome/IL-1β/Smad circuit. Free Radic. Biol. Med. 2016, 97, 531–543. [Google Scholar] [CrossRef]
- Sheriff, L.; Lalor, P.F. The Impact of the NLRP3 Pathway in the Pathogenesis of Non-Alcoholic Fatty Liver Disease and Alcohol-Related Liver Disease. Livers 2021, 1, 68–81. [Google Scholar] [CrossRef]
- Purohit, V.; Bode, J.C.; Bode, C.; Brenner, D.A.; Choudhry, M.A.; Hamilton, F.; Kang, Y.J.; Keshavarzian, A.; Rao, R.; Sartor, R.B.; et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: Summary of a symposium. Alcohol 2008, 42, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Donohue, T.M., Jr. Alcohol-induced steatosis in liver cells. World J. Gastroenterol. 2007, 13, 4974–4978. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef]
- Ganz, M.; Csak, T.; Nath, B.; Szabo, G. Lipopolysaccharide induces and activates the Nalp3 inflammasome in the liver. World J. Gastroenterol. 2011, 17, 4772–4778. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Nishiguchi, S. Importance of Kupffer Cells in the Development of Acute Liver Injuries in Mice. Int. J. Mol. Sci. 2014, 15, 7711–7730. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, N.; Hövelmeyer, N.; Waisman, A.; Straub, B.K.; Weinmann-Menke, J.; Wörns, M.A.; Galle, P.R.; Schattenberg, J.M. Hepatocyte-specific deletion of IL1-RI attenuates liver injury by blocking IL-1 driven autoinflammation. J. Hepatol. 2018, 68, 986–995. [Google Scholar] [CrossRef]
- Budarina, N.A.; Belaia, O.F.; Chulanov, V.P.; Paĭmanov, N.V.; Pak, S.G. [Characteristics of cellular immunity in children with acute viral hepatitis A]. Ter. Arkhiv 2003, 75, 31–35. [Google Scholar]
- Pan, X.; Xu, H.; Zheng, C.; Li, M.; Zou, X.; Cao, H.; Xu, Q. Human hepatocytes express absent in melanoma 2 and respond to hepatitis B virus with interleukin-18 expression. Virus Genes 2016, 52, 445–452. [Google Scholar] [CrossRef]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.Y.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1β Production through the NLRP3 Inflammasome by Hepatic Macrophages Links Hepatitis C Virus Infection with Liver Inflammation and Disease. PLOS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef]
- Li, Y.; Yu, P.; Kessler, A.L.; Shu, J.; Liu, X.; Liang, Z.; Liu, J.; Li, Y.; Li, P.; Wang, L.; et al. Hepatitis E virus infection activates NOD-like receptor family pyrin domain-containing 3 inflammasome antagonizing interferon response but therapeutically targetable. Hepatology 2022, 75, 196–212. [Google Scholar] [CrossRef]
- Zalinger, Z.B.; Elliott, R.; Weiss, S.R. Role of the inflammasome-related cytokines Il-1 and Il-18 during infection with murine coronavirus. J. Neurovirology 2017, 23, 845–854. [Google Scholar] [CrossRef]
- Chen, H.; Han, Z.; Fan, Y.; Chen, L.; Peng, F.; Cheng, X.; Wang, Y.; Su, J.; Li, D. CD4+ T-cell subsets in autoimmune hepatitis: A review. Hepatol. Commun. 2023, 7, e0269. [Google Scholar] [CrossRef] [PubMed]
- Floreani, A.; Restrepo-Jiménez, P.; Secchi, M.F.; De Martin, S.; Leung, P.S.C.; Krawitt, E.; Bowlus, C.L.; Gershwin, M.E.; Anaya, J.M. Etiopathogenesis of autoimmune hepatitis. J. Autoimmun. 2018, 95, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Zhang, X.; Wang, S.; Li, Y.; Fan, J.; Chen, W.; Zai, W.; Wang, S.; Wang, Y.; Chen, M.; et al. NOD-Like Receptor Protein 3 Inflammasome-Dependent IL-1β Accelerated ConA-Induced Hepatitis. Front. Immunol. 2018, 9, 758. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, G.; Liang, Y.; Du, X.; Boor, P.J.; Sun, J.; Khan, M.F. Redox regulation of hepatic NLRP3 inflammasome activation and immune dysregulation in trichloroethene-mediated autoimmunity. Free Radic. Biol. Med. 2019, 143, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Yang, G.; Chen, H.Y.; Hsu, D.K.; Tomilov, A.; Olson, K.A.; Dehnad, A.; Fish, S.R.; Cortopassi, G.; Zhao, B.; et al. Galectin-3 regulates inflammasome activation in cholestatic liver injury. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 4202–4213. [Google Scholar] [CrossRef] [PubMed]
- Arend, W.P. Interleukin-1 receptor antagonist. Adv. Immunol. 1993, 54, 167–227. [Google Scholar] [CrossRef]
- Mancini, R.; Benedetti, A.; Jezequel, A.M. An interleukin-1 receptor antagonist decreases fibrosis induced by dimethylnitrosamine in rat liver. Virchows Arch. 1994, 424, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.Z.; Xiang, D.; Xie, C.; Li, J.J.; Hu, J.J.; He, H.L.; Yuan, Y.S.; Gao, J.; Han, W.; Yu, Y. Protective effect of recombinant human IL-1Ra on CCl4-induced acute liver injury in mice. World J. Gastroenterol. 2010, 16, 2771–2779. [Google Scholar] [CrossRef]
- Hu, J.; Yan, D.; Gao, J.; Xu, C.; Yuan, Y.; Zhu, R.; Xiang, D.; Weng, S.; Han, W.; Zang, G.; et al. rhIL-1Ra reduces hepatocellular apoptosis in mice with acetaminophen-induced acute liver failure. Lab. Investig. 2010, 90, 1737–1746. [Google Scholar] [CrossRef]
- Szabo, G.; Mitchell, M. IL-1 receptor antagonist plus pentoxifylline and zinc for severe alcohol-associated hepatitis. Hepatology 2022, 76, 1058–1068. [Google Scholar] [CrossRef]
- Massaro, M.G.; Pompili, M.; Sicignano, L.L.; Pizzolante, F.; Verrecchia, E.; Vecchio, F.M.; Rigante, D.; Manna, R. Improvement of Liver Involvement in Familial Mediterranean Fever After the Introduction of Canakinumab: A Case Report. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020059. [Google Scholar] [CrossRef] [PubMed]
- Vergis, N.; Patel, V.C.; Bogdanowicz, K.; Czyzewska-Khan, J.; Keshinro, R.; Fiorentino, F.; Day, E.; Middleton, P.; Atkinson, S.; Cross, M.; et al. OS034—Il-1beta Signal Inhibition in acute alcoholic hepatitis: A multicentre, randomised, double-blind, placebo-controlled phase 2 trial of canakinumab therapy (ISAIAH). J. Hepatol. 2022, 77, S34–S35. [Google Scholar] [CrossRef]
- Vande Walle, L.; Stowe, I.B.; Šácha, P. MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PLOS Biol. 2019, 17, e3000354. [Google Scholar] [CrossRef] [PubMed]
- Nizami, S.; Millar, V.; Arunasalam, K.; Zarganes-Tzitzikas, T.; Brough, D.; Tresadern, G.; Brennan, P.E.; Davis, J.B.; Ebner, D.; Di Daniel, E. A phenotypic high-content, high-throughput screen identifies inhibitors of NLRP3 inflammasome activation. Sci. Rep. 2021, 11, 15319. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Rouquet, N.; Pagès, J.C.; Molina, T.; Briand, P.; Joulin, V. ICE inhibitor YVADcmk is a potent therapeutic agent against in vivo liver apoptosis. Curr. Biol. 1996, 6, 1192–1195. [Google Scholar] [CrossRef]
- Morrison, M.C.; Mulder, P.; Salic, K.; Verheij, J.; Liang, W.; van Duyvenvoorde, W.; Menke, A.; Kooistra, T.; Kleemann, R.; Wielinga, P.Y. Intervention with a caspase-1 inhibitor reduces obesity-associated hyperinsulinemia, non-alcoholic steatohepatitis and hepatic fibrosis in LDLR−/−.Leiden mice. Int. J. Obes. 2016, 40, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Wang, Q.; Dong, Y.; Ma, W.; Zhang, Y.; Zhao, Y.; Bian, F.; Chen, L. High Glucose-Aggravated Hepatic Insulin Resistance: Role of the NLRP3 Inflammasome in Kupffer Cells. Obesity 2020, 28, 1270–1282. [Google Scholar] [CrossRef]
- Lei, Y.; Tang, L.; Chen, Q.; Wu, L.; He, W.; Tu, D.; Wang, S.; Chen, Y.; Liu, S.; Xie, Z.; et al. Disulfiram ameliorates nonalcoholic steatohepatitis by modulating the gut microbiota and bile acid metabolism. Nat. Commun. 2022, 13, 6862. [Google Scholar] [CrossRef]
- Sangineto, M.; Grabherr, F.; Adolph, T.E. Dimethyl fumarate ameliorates hepatic inflammation in alcohol related liver disease. Liver Int. 2020, 40, 1610–1619. [Google Scholar] [CrossRef]
- Xu, B.; Jiang, M.; Chu, Y.; Wang, W.; Chen, D.; Li, X.; Zhang, Z.; Zhang, D.; Fan, D.; Nie, Y.; et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2018, 68, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Faggioni, R.; Cattley, R.C.; Guo, J.; Flores, S.; Brown, H.; Qi, M.; Yin, S.; Hill, D.; Scully, S.; Chen, C.; et al. IL-18-binding protein protects against lipopolysaccharide-induced lethality and prevents the development of Fas/Fas ligand-mediated models of liver disease in mice. J. Immunol. 2001, 167, 5913–5920. [Google Scholar] [CrossRef] [PubMed]
- Knorr, J.; Kaufmann, B.; Inzaugarat, M.E.; Holtmann, T.M.; Geisler, L.; Hundertmark, J.; Kohlhepp, M.S.; Boosheri, L.M.; Chilin-Fuentes, D.R.; Birmingham, A. Interleukin-18 signaling promotes activation of hepatic stellate cells in mouse liver fibrosis. Hepatology 2023, 77, 1968–1982. [Google Scholar] [CrossRef]
- Cao, Z.; Fang, Y.; Lu, Y.; Tan, D.; Du, C.; Li, Y.; Ma, Q.; Yu, J.; Chen, M.; Zhou, C.; et al. Melatonin alleviates cadmium-induced liver injury by inhibiting the TXNIP-NLRP3 inflammasome. J. Pineal Res. 2017, 62, e12389. [Google Scholar] [CrossRef]
- Mansoori, A.; Salimi, Z.; Hosseini, S.A.; Hormoznejad, R.; Jafarirad, S.; Bahrami, M.; Asadi, M. The effect of melatonin supplementation on liver indices in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis of randomized clinical trials. Complement. Ther. Med. 2020, 52, 102398. [Google Scholar] [CrossRef]
- Fernández-Ortiz, M.; Sayed, R.K.A. Age and Chronodisruption in Mouse Heart: Effect of the NLRP3 Inflammasome and Melatonin Therapy. Int. J. Mol. Sci. 2022, 23, 6846. [Google Scholar] [CrossRef] [PubMed]
- Volt, H.; García, J.A.; Doerrier, C.; Díaz-Casado, M.E.; Guerra-Librero, A.; López, L.C.; Escames, G.; Tresguerres, J.A.; Acuña-Castroviejo, D. Same molecule but different expression: Aging and sepsis trigger NLRP3 inflammasome activation, a target of melatonin. J. Pineal Res. 2016, 60, 193–205. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Najafi, M. Anti-Inflammatory Activity of Melatonin: A Focus on the Role of NLRP3 Inflammasome. Inflammation 2021, 44, 1207–1222. [Google Scholar] [CrossRef] [PubMed]
- Rojas Márquez, J.D.; Ana, Y.; Baigorrí, R.E.; Stempin, C.C.; Cerban, F.M. Mammalian Target of Rapamycin Inhibition in Trypanosoma cruzi-Infected Macrophages Leads to an Intracellular Profile That Is Detrimental for Infection. Front. Immunol. 2018, 9, 313. [Google Scholar] [CrossRef]
- Sun, Q.; Gao, W.; Loughran, P.; Shapiro, R.; Fan, J.; Billiar, T.R.; Scott, M.J. Caspase 1 activation is protective against hepatocyte cell death by up-regulating beclin 1 protein and mitochondrial autophagy in the setting of redox stress. J. Biol. Chem. 2013, 288, 15947–15958. [Google Scholar] [CrossRef]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Li, L.; Lu, P.; Li, X.; Tian, D.; Liu, M. NLRP6 exerts a protective role via NF-kB with involvement of CCL20 in a mouse model of alcoholic hepatitis. Biochem. Biophys. Res. Commun. 2020, 528, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liu, Q.; Tang, Q.; Jing, X.; Wu, T.; Zhang, J.; Zhang, G.; Zhou, J.; Zhang, Z.; Zhao, Y.; et al. Hepatocyte-specific deletion of Nlrp6 in mice exacerbates the development of non-alcoholic steatohepatitis. Free Radic. Biol. Med. 2021, 169, 110–121. [Google Scholar] [CrossRef] [PubMed]
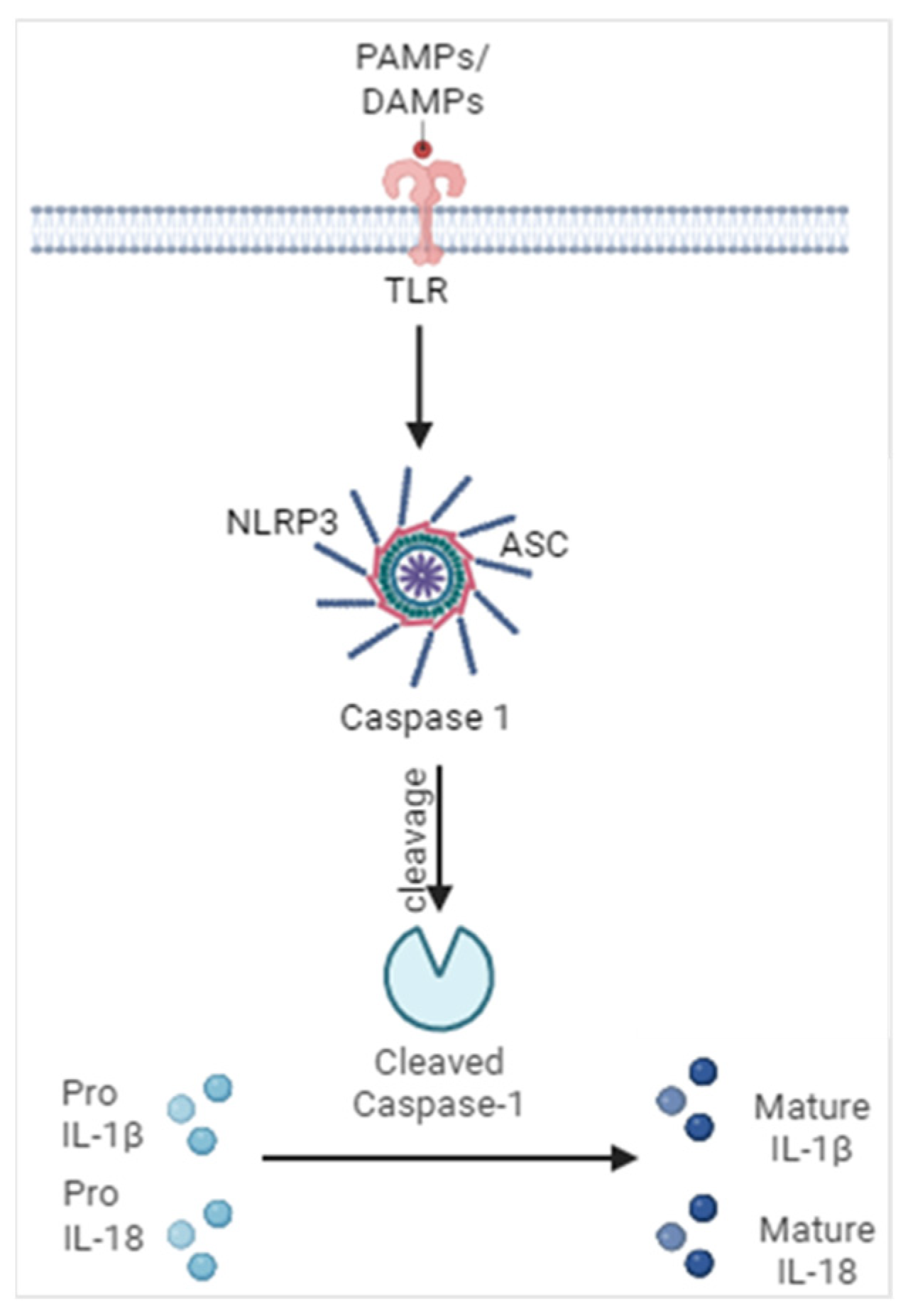
Figure 1.
Activation of inflammasomes. Encountering of pathogens and their products as well as products of cellular damage generates a signal leading to the oligomerization of inflammasome components such as a sensor protein (NLRP3), adaptor protein (ASC) and caspase-1. This leads to the cleavage of caspase-1 into its active form, ultimately leading to the maturation and activation of IL-1 family cytokines IL-1β and IL-18. Illustration created with BioRender.com.
Figure 1.
Activation of inflammasomes. Encountering of pathogens and their products as well as products of cellular damage generates a signal leading to the oligomerization of inflammasome components such as a sensor protein (NLRP3), adaptor protein (ASC) and caspase-1. This leads to the cleavage of caspase-1 into its active form, ultimately leading to the maturation and activation of IL-1 family cytokines IL-1β and IL-18. Illustration created with BioRender.com.
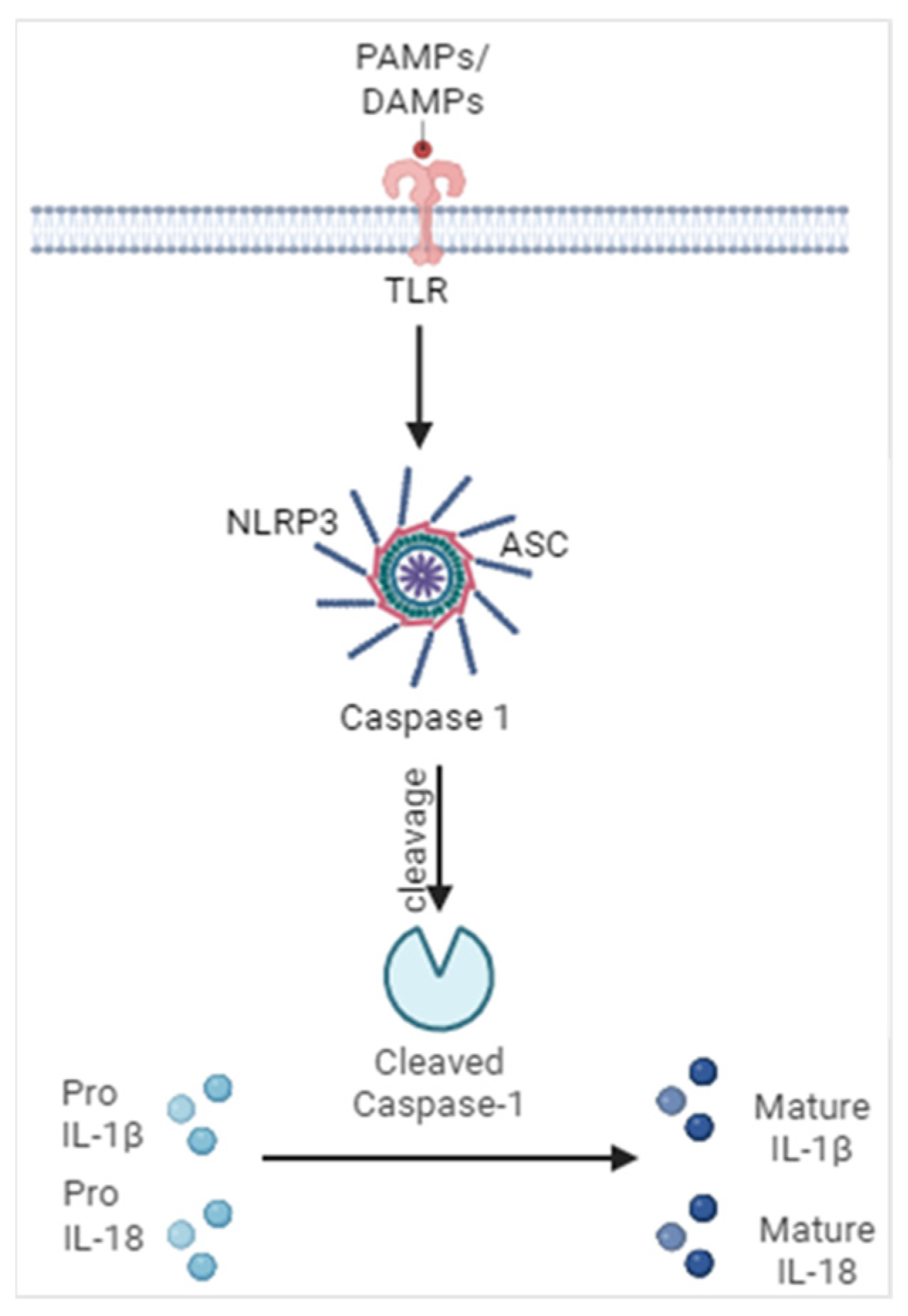
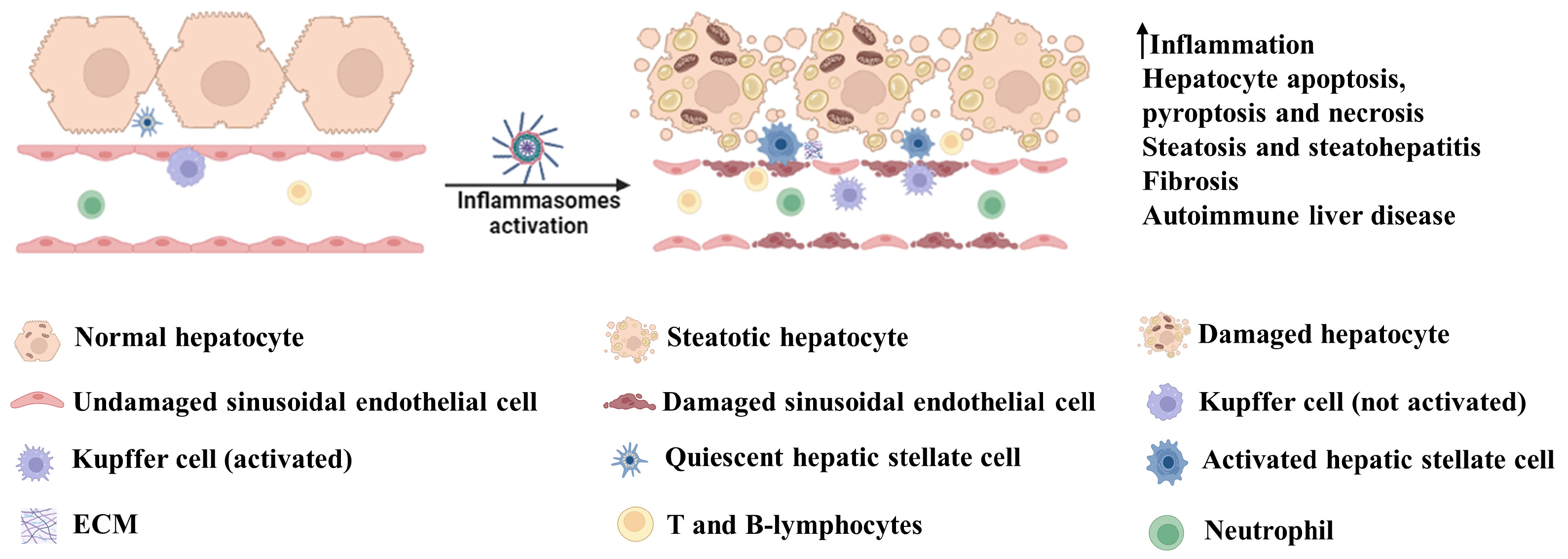
Figure 2.
Involvement of inflammasomes in liver disease. Activation of inflammasomes leads to an increase in inflammation via enhanced secretion of IL-1β and IL-18. These cytokines play critical roles in the induction of hepatocyte death by apoptotic, pyroptotic and necrotic pathways. They also promote hepatic steatosis and steatohepatitis and, in the long run, engulfment of inflammasome products or cytokine-dependent signaling leads to hepatic fibrosis. IL-1β-dependent activation of CD4+ T cells and B cells can lead to autoimmune hepatic disease. Illustration created with BioRender.com.
Figure 2.
Involvement of inflammasomes in liver disease. Activation of inflammasomes leads to an increase in inflammation via enhanced secretion of IL-1β and IL-18. These cytokines play critical roles in the induction of hepatocyte death by apoptotic, pyroptotic and necrotic pathways. They also promote hepatic steatosis and steatohepatitis and, in the long run, engulfment of inflammasome products or cytokine-dependent signaling leads to hepatic fibrosis. IL-1β-dependent activation of CD4+ T cells and B cells can lead to autoimmune hepatic disease. Illustration created with BioRender.com.
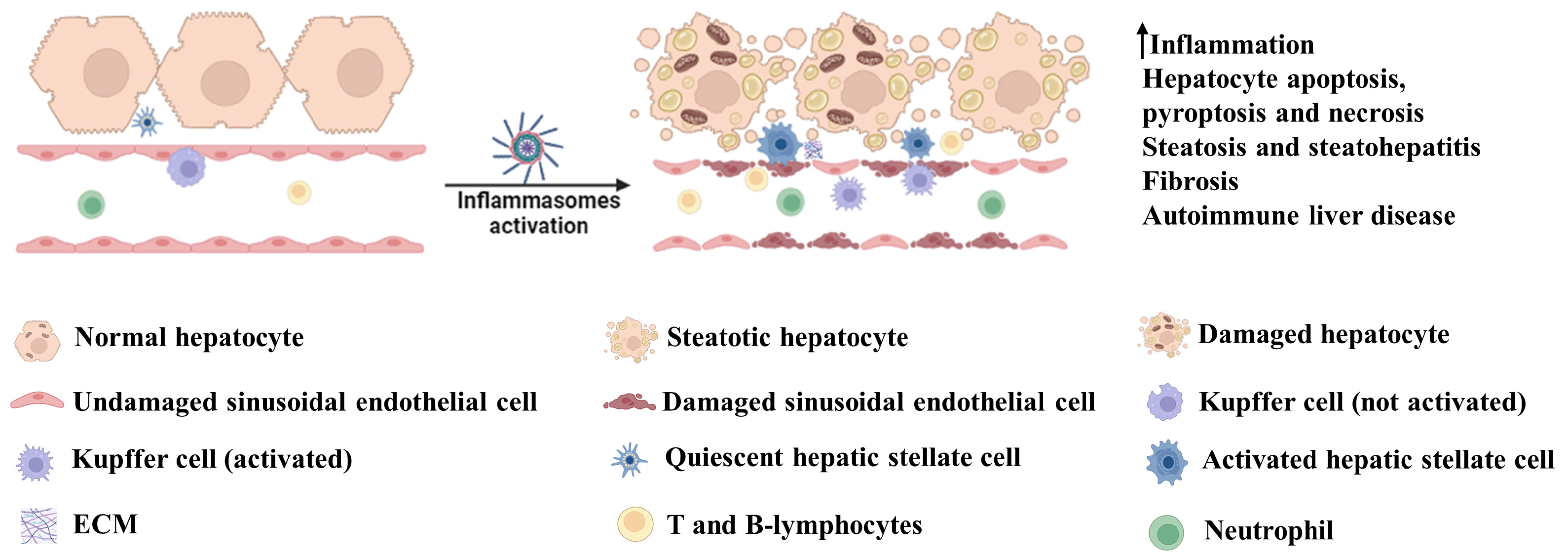
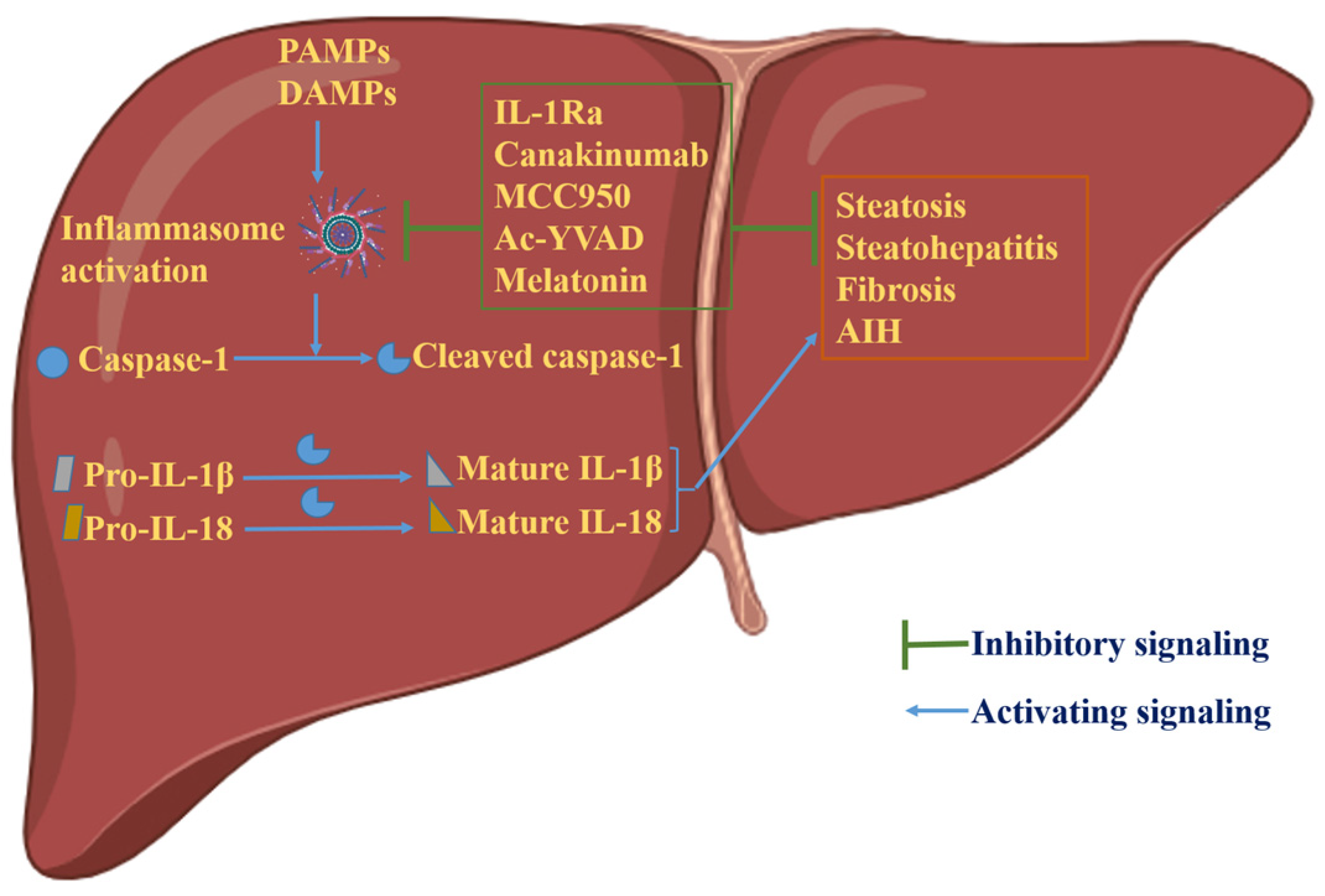
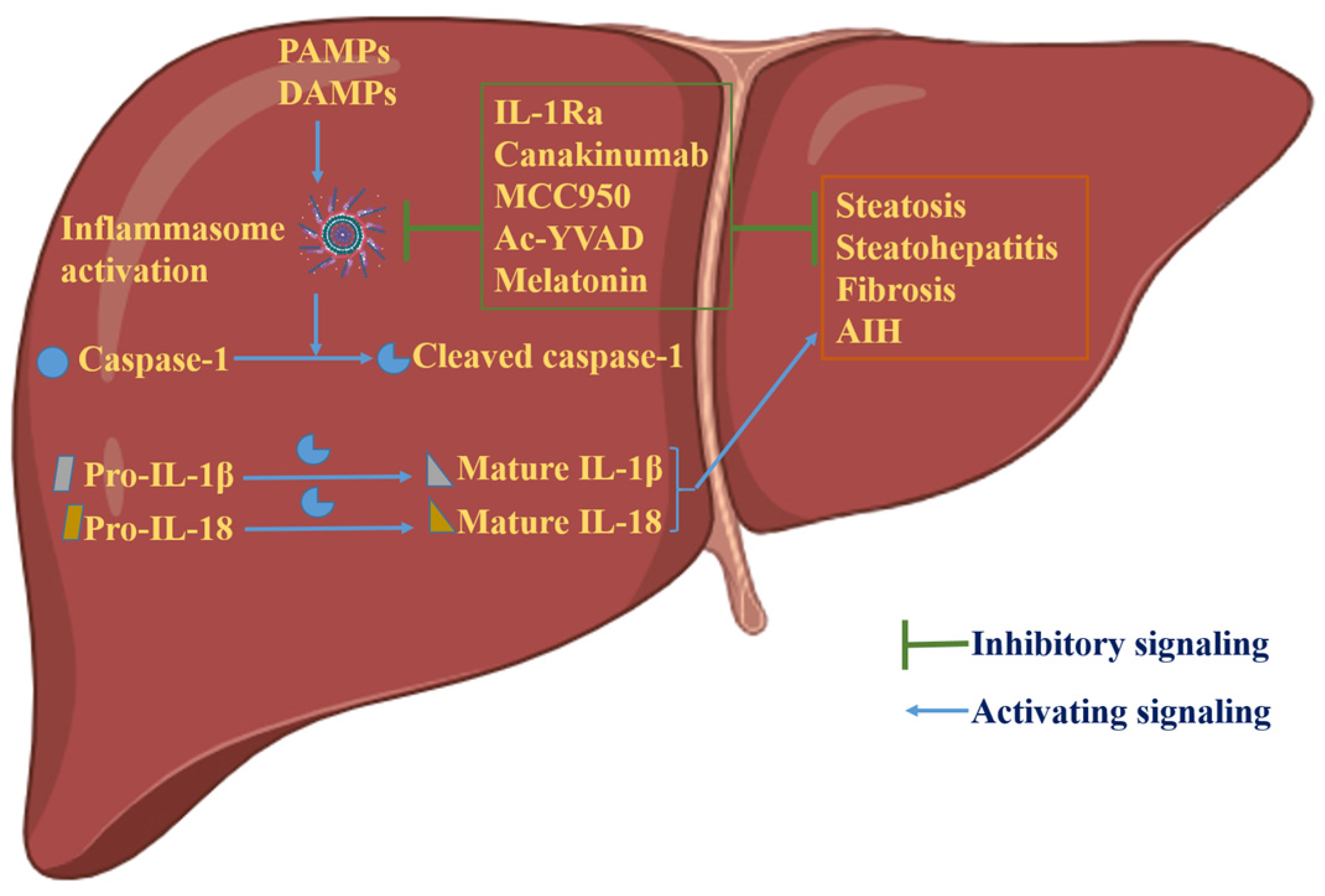
Figure 3.
Graphical representation of the role of inflammasome activation in hepatic physiology. Activation of inflammasomes generates mature and biologically active cytokines, IL-1β and IL-18, via caspase-1 cleavage. These cytokines lead to hepatic disorders, including steatosis, steatohepatitis, fibrosis and AIH. In experimental settings, inhibition of inflammasome activation has shown encouraging results in the treatment of hepatic disorders due to aberrant inflammasome activation. Illustration created with BioRender.com.
Figure 3.
Graphical representation of the role of inflammasome activation in hepatic physiology. Activation of inflammasomes generates mature and biologically active cytokines, IL-1β and IL-18, via caspase-1 cleavage. These cytokines lead to hepatic disorders, including steatosis, steatohepatitis, fibrosis and AIH. In experimental settings, inhibition of inflammasome activation has shown encouraging results in the treatment of hepatic disorders due to aberrant inflammasome activation. Illustration created with BioRender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Baral, A. Mechanisms of Inflammasome Activation and Involvement in Liver Disease. J. Mol. Pathol. 2024, 5, 171-186. https://doi.org/10.3390/jmp5020011
AMA Style
Baral A. Mechanisms of Inflammasome Activation and Involvement in Liver Disease. Journal of Molecular Pathology. 2024; 5(2):171-186. https://doi.org/10.3390/jmp5020011
Chicago/Turabian StyleBaral, Ananda. 2024. "Mechanisms of Inflammasome Activation and Involvement in Liver Disease" Journal of Molecular Pathology 5, no. 2: 171-186. https://doi.org/10.3390/jmp5020011