Abstract
Iatrogenic amyloidosis results from medical therapeutic interventions, leading to the misfolding and aggregation of proteins into amyloid fibrils or to their direct deposition in different tissues. This review aims to provide a comprehensive overview of the iatrogenic amyloidosis pathology, underlying the possible molecular mechanisms, associated pathological manifestations, and clinical implications within modern medicine. By conducting a systematic analysis of the current literature, this paper highlights the diverse instances of iatrogenic amyloidosis triggered by medical procedures such as dialysis, organ and tissue transplantation, and therapeutic drugs. Exploring the intricate molecular pathways and contributing factors involved in protein misfolding and amyloidogenesis, and uncovering the pathological consequences observed in various tissues and organs, allows us to establish appropriate nomenclature and to gain a more profound understanding of the condition, working towards improved medical interventions and treatments.
1. Introduction
Proteins demonstrate a remarkable ability for self-assembly, orchestrating the formation of intricate structures vital to biological systems. This intrinsic process initiates with the folding of individual protein molecules and progresses to the assembly of more complex architectures, spanning from multi-subunit globular proteins to filaments and virus capsids [1,2,3,4]. Among the diverse outcomes of protein self-assembly, the emergence of amyloid fibrils is notable as an important and intriguing phenomenon, often associated with pathological deposits observed in various species [5].
Amyloidosis constitutes a group of complex diseases characterized by pathological misfolding and aggregation of certain proteins, resulting in the accumulation of amyloid fibrils in various tissues and organs [6]. These amyloid fibrils can disrupt normal tissue architecture and functionality, leading to a wide range of clinical manifestations and incurable and fatal diseases [7,8,9]. To date, several proteins with the propensity to form amyloids, both in vivo and in vitro, have been identified. Examples include amyloid-β, transthyretin, α-synuclein, and prion proteins. These proteins are linked to distinct pathologies such as Alzheimer’s disease, Parkinson’s disease, Transthyretin amyloidosis, and Creutzfeldt–Jakob disease (CJD) [2,7,10].
While primary amyloidosis, associated with the generation or overproduction of a specific amyloidogenic protein in a specific disease, has been extensively studied, the subcategory known as iatrogenic amyloidosis remains relatively enigmatic. Iatrogenic amyloidosis is a rare and distinct form of amyloidosis that emerges as an unintended consequence of medical interventions, therapeutic procedures, or drug treatments, often unrelated to underlying amyloidogenic processes (Figure 1). It has been observed primarily in patients exposed to infectious prion proteins (iatrogenic Creutzfeldt–Jakob disease), long-term dialysis patients, and recipients of “domino” transplants [11,12,13,14,15]. This condition has remained relatively obscure due to its infrequency and intricate nature; however, as new and unique cases come to light [7], there is a growing need for a deeper comprehension of its pathogenic mechanisms, alongside more precise diagnostic techniques and therapeutic interventions in its management.
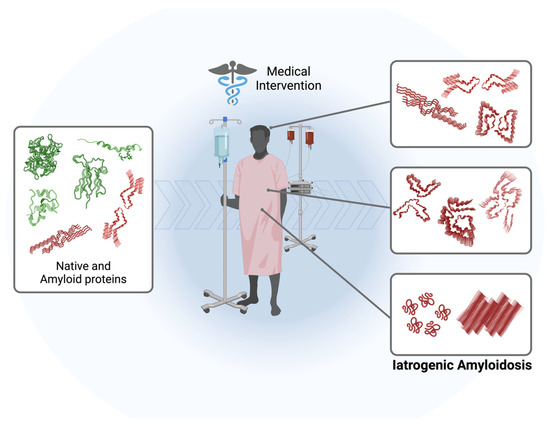
Figure 1.
Schematic representation of the pathway leading to the development of iatrogenic amyloidosis in patients subjected to various medical interventions such as dialysis, organ transplantation, and therapeutic drugs. The amyloidogenic cascade can be initiated by the administration of exogenous proteins/peptides, activation of endogenous native proteins, or the transplantation/transmission of amyloid oligomers and fibrils. The molecular structures used for illustration purposes can be checked using PDB: 5ENA, 1LDS, 8ADE, 7ZKY, 5APD, 6GK3, 6W0O, 4TLT, 6LNI, and 5OQV.
This comprehensive review aims to provide an in-depth analysis of the molecular pathology of iatrogenic amyloidosis, covering its to date known causal factors, underlying pathogenic mechanisms, predilection for specific organs and tissues, some diagnostic challenges, and existing therapeutic approaches. Furthermore, this review serves to inform clinicians, scientists, and healthcare professionals, promoting early diagnosis and effective management of the disease and stimulating better therapeutic strategies and innovative research.
2. Molecular Mechanism of the Amyloid Formation Process
The process of amyloid fibril formation is characterized by its distinctive β-cross structure. It typically follows a nucleation-dependent polymerization mechanism consisting of two pivotal stages: nucleation and elongation [16]. Recent research endeavors have focused on elucidating the nucleation phase, seeking to unravel the intricate process by which individual protein molecules converge to form nuclei, analogous to a crystallization process. In this manner, misfolded soluble monomers give rise to oligomeric species that subsequently expand through additional monomer incorporation, ultimately forming insoluble mature fibrils. The growth pattern of this process is typically sigmoidal, indicating the greater facility of adding monomers to existing aggregates in contrast to the de novo formation of new oligomers directly from monomers. Consequently, the overall reaction rate accelerates when a substantial number of aggregates are present in solution, leading to an initial lag phase (i.e., nucleation) followed by a rapid growth phase (i.e., elongation), culminating in a plateau region (i.e., mature amyloid fibril) [6,17,18]. One of the crucial points in some cases of iatrogenic amyloidosis may be the possibility of accelerating and triggering this amyloidogenic cascade, where a phenomenon known as seeding has a direct effect. Amyloid seeds (preformed fibrils) catalyze the formation of mature amyloid fibrils by shortening or overlapping the lag phase [19,20]. This process can occur through the amyloid seeds of the same protein (homologous seeding) or of other proteins (heterologous seeding or cross-seeding) [19].
Amyloidogenesis comprises a multitude of properties and factors, encompassing protein structural and sequence characteristics, mutations, and the involvement of other molecules such as ions, proteins, and lipids, as well as several biophysical and biochemical forces [21,22,23,24]. The primary sequence of the protein plays a fundamental role in amyloid formation, with specific amino acid sequences predisposing proteins to amyloidogenic behavior. Additionally, mutations in proteins can enhance or decrease their propensity to aggregate into amyloid fibrils, leading to the onset of amyloid diseases [24,25]. Moreover, the presence of specific cofactors or ions can modulate amyloid formation kinetics and stability; for example, metal ions such as copper and zinc have been demonstrated to influence amyloid formation by promoting protein aggregation or by stabilizing amyloid fibrils [22,23]. Additionally, small molecules or ligands can interact with proteins and either inhibit or enhance amyloid formation, contingent on their binding affinity and mechanism of action [26]. Biophysical forces, including hydrophobic interactions, electrostatic interactions, and hydrogen bonding, also play critical roles in amyloidogenesis. Hydrophobic regions within proteins can interact to initiate fibril formation, while electrostatic interactions and hydrogen bonding contribute to the stabilization of the amyloid structure [18,27]. Furthermore, environmental factors such as pH, temperature, and protein concentration can significantly impact the kinetics and thermodynamics of amyloid formation [28]. This illustrates that the amyloidogenic cascade is multifaceted, heterogeneous, and complex.
3. Iatrogenic Amyloidosis Nomenclature
Given its rarity, there is currently no established nomenclature to accurately describe the different cases, involved proteins, and affected tissues in iatrogenic amyloidosis. As previously mentioned, initial cases of iatrogenic amyloidosis are directly associated with medical interventions such as dialysis, blood transfusions, transplants, and neurosurgeries; however, more recently, instances of amyloidosis triggered by the local (i.e., subcutaneous) administration of peptide- and polypeptide-derived drugs have introduced a new category of interventions that lead to iatrogenic amyloidosis [29].
For a better understanding, a classification subdivided according to the nature of the medical intervention and its subsequent impact on amyloid proteins has been determined here. Essentially, the different cases of iatrogenic amyloidosis have been classified in relation to the medical intervention/treatment and the form in which the protein is found. Accordingly, exogenous proteins administered in their native and soluble forms and subsequently undergoing amyloidogenic processes were classified as “generated-iatrogenic amyloidosis”. This category predominantly encompasses peptide medications and those commonly associated with cutaneous amyloidosis. In turn, the class of endogenous or tissue-specific proteins, which have their amyloidogenic cascade activated by medical interference, were classified as “triggered-iatrogenic amyloidosis”. Finally, the class of proteins that are already in a pre-existing amyloid state (i.e., oligomers and fibrils) and are transmitted or transplanted through different surgical procedures were termed “transmitted/transplanted-iatrogenic amyloidosis” (Figure 2, Table 1). In the following sections, these categories will be analyzed in detail.
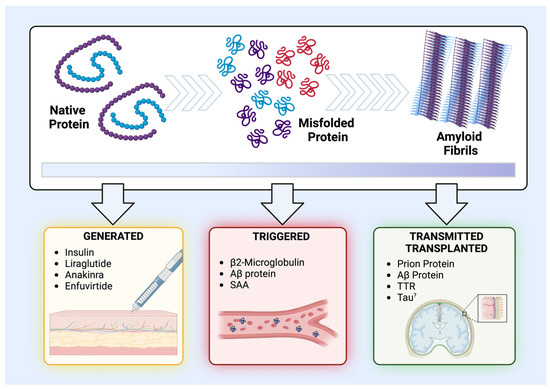
Figure 2.
Schematics and classification of the different types of iatrogenic amyloidosis. The upper panel illustrates the stages of the amyloidogenic process of certain proteins, starting from their normal and functional state, passing through to a stage of misfolding and aggregation that generates oligomers and protein aggregates, and, finally, reaching the state of amyloid fibrils. The lower panel demonstrates the three classes of iatrogenic amyloidosis as well as the structural state of the protein being separated into “generated-iatrogenic amyloidosis” (in yellow), “triggered-iatrogenic amyloidosis” (in red), and “transmitted/transplanted-iatrogenic amyloidosis” (in green).

Table 1.
Proteins and Peptides Involved in Iatrogenic Amyloidosis.
3.1. Generated-Iatrogenic Amyloidosis
As mentioned earlier, the generated-iatrogenic amyloidosis category consists of medical interventions; more specifically, the subcutaneous administration of exogenous peptide-derived drugs at different application sites (Figure 2) and the subsequent generation of the amyloid forms of this exogenous peptide. It is important to underscore that the physical stability and fibrillation of peptides present a common challenge in formulating and developing therapeutic peptides during both manufacturing and storage as well as in maintaining stability post-administration (e.g., in acidic skin pH conditions). Additionally, peptides have found extensive use in medicine, with an estimation of over 150 peptides undergoing clinical trials and potentially around 500 others being evaluated in preclinical studies. Currently, the number of therapeutic peptides approved and utilized in clinical settings may range from 70 to 114 [30,31,32].
Although considered rare, and with no available data related to the prevalence of cases, the first report was made in 1983 by Storkel et al. [33], in which they demonstrated a case of iatrogenic amyloidosis caused by the continuous administration of insulin in a diabetic patient as well as in an animal model. Subsequently, cases were termed as iatrogenic localized insulin-derived amyloidosis (LIDA) or localized insulin amyloid (AIns), characterized by firm, painless subcutaneous nodules (lumps, masses, tumors) in the arms, abdomen, thighs, breasts, and inguinal lymph nodes, with the majority of cases located in the abdomen, which can be resolved with surgical resection [34,35,36]. It is important to note that, upon analyzing these amyloid nodules through tandem mass spectrometry, it was found that there was a varied concentration of the protein in these nodules, and that detection was exclusively of the insulin A and B chains only, without the C-peptide (a component of human pro-insulin), suggesting that this protein is indeed derived from the deposition of recombinant insulin [35]. The scenario of amyloid nodules was also mimicked in mice when Chinisaz et al. generated insulin amyloid fibrils in vitro and injected them into animals for 21 days, resulting in firm lumps positive for Congo red and containing connective and adipose tissue [37]. Although the ability to mimic the pathology is important, the in situ amyloidogenic process was not generated, and there is a lack of further understanding regarding possible molecular and mechanical interactions and potential targets that could prevent, reduce, or induce the amyloidogenic process.
Therefore, the molecular mechanisms of insulin conversion into amyloid fibrils directly at the injection site have not yet been elucidated. In addition, although several studies have analyzed and studied the amyloidogenic process of insulin in vitro, mainly due to its amyloidogenic predisposition under conditions such as high temperatures, agitation, co-solvents, and low pH, little is known about its possible structural polymorphisms in the amyloid form and how this interferes with the pathological condition.
Insulin is a globular protein composed of an A (21 aa) and B (30 aa) chain (Table 1) and it can be found as hexamers, dimers, and monomers, with monomers being the possible form of the initiation of the amyloidogenic cascade (nucleation). Both the A and B chains have amyloidogenic capacity, with the B chain appearing to adopt a β-sheet-like interaction with another B chain from another insulin monomer, leading to nucleation and increased amyloid fibril elongation and formation (i.e., nucleation-driven polymerization) [38,39,40]. However, the exact mechanism of insulin fibrillogenesis still appears to be dependent on many variables, with other mechanisms being hypothesized, such as the formation of a secondary nucleus where nucleation for the formation of a new amyloid fibril is initiated from the surface of another existing fibril [40,41]. Additionally, it is not known to what extent protein oligomers influence this amyloidogenic process and fibril elongation. The structural morphology of insulin amyloid fibrils is presented as a left-handed helix composed of protofilaments rich in β-sheet orientation perpendicular to the fiber, which undergo a series of inter-chain interactions during their assembly process that can be propagated during fibrillar growth [42,43]. Despite this, the morphology of insulin amyloid fibrils appears to be polymorphic and heterogeneous, depending on their formulation and sequence [44].
A better understanding of this insulin-induced amyloidosis is of great importance due to the increasing number of cases reported in the literature [34,45,46,47,48,49,50,51,52,53,54]. A better understanding of the in vivo amyloidogenic process is necessary mainly because these amyloid nodules can lead to the impairment of insulin absorption at the injection site, and due to the unpredictable release of accumulated insulin in these nodules, which results in detrimental effects on glycemic control [55]. Nakamura et al. generated in vitro insulin amyloid fibrils using low pH, temperature, and shaking conditions. After this step, they subcutaneously injected high concentrations of these fibrils daily for 7 days into mice, and observed massive nodules. These nodules reduced the absorption of insulin in its globular form after application, demonstrating a high adherence between amyloid and native insulin forms. Additionally, the authors demonstrated in vitro that insulin amyloid has seeding capacity at physiological pH, which activates and increases the production of amyloid proteins from the natural form of insulin [56]. In turn, Yuzu et al., using specific fluorophores, demonstrated a wide variety of polymorphic amyloid aggregates in samples of iatrogenic insulin nodules. Furthermore, these authors analyzed nine insulin preparations in vitro and showed that organized fibrils (of a needle-like shape), such as those generated by insulin glargine, are more cytotoxic than less organized filaments [57].
It is important, however, to highlight that not all cases of iatrogenic insulin amyloidosis present with a subcutaneous nodule or lump. In such cases, amyloid plaques are detected after an abdominal fat biopsy and tested for suspicious systemic amyloidosis [48,58]. Thus, these findings may indicate that a considerable number of other patients suffer from the same condition and are not properly diagnosed, which can result in poor glycemic control and disease management.
Another important case that has been raising curiosity is iatrogenic amyloidosis caused by the administration of Liraglutide (Table 1). Liraglutide is an analog of human glucagon-like peptide-1 (GLP-1) used for the management of type 2 diabetes mellitus and obesity and for the prevention of cardiovascular diseases. It is administered through daily subcutaneous injections [59]. So far, only three cases have been documented in the literature [60,61,62]. Similar to amyloidosis caused by insulin, the iatrogenic Liraglutide amyloidosis is also focal, subcutaneous, localized directly at the injection site, and is positive for Congo red staining, in addition to the protein being detected by mass spectrometry, confirming the sequence as exogenous and not derived from the endogenous protein, and immunohistochemistry [61,62,63].
Further, in common with insulin, mature GLP-1 protein is generated from the post-translational processing of proglucagon by the enzyme prohormone convertase subtilisin/kexin type 1 or 3 (PCSK1 or PCSK3, respectively). Additionally, both proteins have multiple bioactive forms, with insulin having the A and B forms (previously mentioned), while GLP-1 has the glycine-extended (GLP1 7–37) and amidated (GLP1 7–36) forms [64,65]. GLP-1 physiological function is closely linked to glucose regulation and metabolism, exerting potent metabolic and endocrine effects. It is secreted by the enteroendocrine cells of the intestinal mucosa in response to nutrient ingestion [64,65]. One of the key features of GLP-1 is its short plasma half-life and rapid degradation; therefore, to become an effective drug, modifications need to be made to its structure, such as attaching a fatty acid chain that enables reversible binding to albumin [64], as seen with Liraglutide (Table 1).
In in vitro studies, the ability of the GLP-1 peptide (31 aa) and of a GLP-1-like peptide (29 aa) to form amyloid fibrils was observed. This dynamic process of protein polymerization resulted in an increase in β-sheet structures and fibrillation, exhibiting a distinct polymerization lag time, elongation, and stationary phases at various pH levels (i.e., 5.8, 7.4, 7.5, 8.2, and 8.5), buffering, incubation time, temperature and protein concentration [66,67,68,69]. Despite these studies providing significant insights into the possible molecular mechanisms of GLP-1 amyloid fibril generation, there is still a lack of effective data regarding the drugs themselves, as well as in vivo data and information about potential molecules that could effectively activate or inhibit this cascade (e.g., dipeptidyl peptidase IV, heparin, or surfactants). Therefore, with the significant increase in the use of GLP-1-receptor agonist drugs (GLP-1Ra), these cases should be closely studied, although more advanced drugs that only require weekly applications and/or are administered orally are entering the market [70,71,72].
Another curious case is that of the drug Anakinra (Table 1), which is a recombinant protein of the human interleukin-1 (IL-1) receptor antagonist (IL-1Ra). This drug binds to the IL-1 receptor, competing with and inhibiting the activity of IL-1 α and β. It is used for the treatment of neonatal onset multisystem inflammatory disease (NOMID), which is caused by autosomal dominant gain-of-function mutations in the NLRP3 gene [73].
In two documented cases, male patients developed firm nodules at the site of the Anakinra injection (daily treatment duration of 18 and 13 years), which tested positive for Congo red, with the protein positive identified (IL1RA) by tandem mass spectrometry [74]. The same group also documented the case of a 41-year-old woman undergoing daily treatment with Anakinra since the age of 28 (daily treatment duration of 13 years), with firm nodules at the injection sites testing positive for Congo red. However, more importantly, after stomach and kidney biopsies, researchers detected the presence of amyloid deposits in these organs confirmed by Congo red staining, mass spectrometry, and transmission electron microscopy [75], thus demonstrating, for the first time, the possibility of iatrogenic systemic amyloidosis, in this case, due to Anakinra. Similarly, they presented another case of a 28-year-old woman undergoing daily treatment with Anakinra since the age of 9 (a treatment duration of 19 years), with subcutaneous amyloid deposits at the injection site and in the kidneys [76]. So far, the molecular mechanism involved in the amyloidogenic process of this recombinant protein, and the possible molecules that may be involved in triggering the amyloid cascade, are not known; however, these case reports shed light on the importance of understanding the basic biology involved in this amyloid protein scenario, as well as accurate diagnostic and detection methods, given the significant health risks for patients and their quality of life.
Other cases involving another peptide-derived drug have also been reported: Enfuvirtide (Table 1), which is used as an HIV-1 gp41 fusion inhibitor in CD4 cells for patients experiencing HIV-1 replication who are already undergoing treatment with other antiretrovirals [77]. Hemorrhagic nodules were found on the patients’ arms and abdomen, positively stained with Congo red and confirmed by tandem mass spectrometry confirmed that the amyloid protein was from the drug itself (100% coverage) and not from the endogenous protein [35,78,79,80]. The reconstitution of lyophilized Enfuvirtide is performed with Sterile Water and excipients such as mannitol and sodium carbonate buffer (pH around 9) due to the drug’s poor solubility under acidic conditions [81]. Morales et al. monitored the in vitro amyloidogenic process of different concentrations of Enfuvirtide for 5 days at 37 °C and pH 7, 6.5, and 6. The results obtained demonstrated a clear pH dependency in the generation process of the amyloid protein, as well as a dose-dependent manner [82]. Additionally, a significant increase in β-sheet structures after 45 h of incubation and visualization by transmission electron microscopy demonstrated the generation of mature amyloid fibrils.
Nevertheless, the precise molecular mechanism underlying subcutaneous amyloid generation (i.e., generated-iatrogenic amyloidosis) in all these patients is still unknown. Several potential causes can be hypothesized, ranging from a potential decrease in the pH of the skin to the absence or reduction of the function of specific cleavage enzymes. Interaction with extracellular matrix components or even an increase in the fibrotic process (scarring) at the injection site could entrap and increase the concentration of the peptide-drug, initiating the amyloidogenic process. Additionally, the seeding hypothesis may also play an important role. Accordingly, further in vitro and in vivo studies are needed to comprehensively elucidate these mechanisms.
3.2. Triggered-Iatrogenic Amyloidosis
In certain specific instances, a medical procedure or intervention can directly trigger the amyloidogenic cascade (misfolding followed by aggregation, nucleation, and fibrillation) of endogenous proteins (Figure 2). This is exemplified by the better-documented dialysis-related amyloidosis (DRA) or hemodialysis-associated amyloidosis (HAA). This type of “triggered-iatrogenic amyloidosis” affects chronic kidney disease patients undergoing long-term hemodialysis (>10 years) due to the increased concentration of native protein and the deposition of the amyloid form of the β2-microglobulin protein in osteoarticular, musculoskeletal, and gastrointestinal tissues [83]. Therefore, this amyloidosis is a serious complication of medical intervention (long-term dialysis), and is considered a real threat to the quality of life and survival of these patients who are already in a critical state [83].
β2-microglobulin (β2m) is a protein component of the major histocompatibility complex (MHC) class I expressed on the surface of all nucleated cells (Table 1). This protein is found in its monomeric form when MHCs dissociate, releasing into the bloodstream. It undergoes glomerular filtration, followed by reabsorption via the proximal tubule receptor megalin, and subsequent catabolism. In a scenario of chronic renal failure treated with hemodialysis, β2m concentrations increase significantly (up to ~50–60-fold) due to the lack of normal protein filtration/degradation. Consequently, the amyloidogenic cascade is triggered, leading to the amyloid form of β2m and its deposition in target tissues. It is worth noting that the exact molecular mechanism of amyloid protein generation in these cases is still unknown, but the accumulation of large protein concentrations, stability, and a lack of catabolism are key factors [21,84,85,86,87,88]. In addition, other elements may be involved, such as the participation of other chaperones and/or proteins and a systemic inflammatory process, which need to be explored further. Nevertheless, it is crucial to underscore that the incidence of β2m-associated amyloidosis has seen a significant decline in recent decades. This decline can likely be attributed to the adoption of novel protocols and hemodialysis methods (e.g., high- and low-flux membranes) [88].
Wild-type β2m comprises a β-sandwich fold with a seven antiparallel β-strand arrangement (A–G, with the E-strand being a region of high amyloidogenicity), connected by a single disulfide bond (Cys25-Cys80) [89,90]. Like other proteins with amyloidogenic properties, β2m has the ability to form amyloid fibrils in vitro under various conditions such as low pH, temperature, agitation, truncation, incubation with Cu(II), and the addition of trifluoroethanol [91,92,93]. In previous studies, it was hypothesized that the formation of β2m amyloid fibrils is a phenomenon involving the aggregation of the denatured protein into crystal-like structures, occurring above the solubility limit upon breaking supersaturation [94]. Furthermore, these amyloidogenic processes take place in a complex free-energy situation, and the mechanisms of amyloid fibril generation also depend on the conditions and factors involved. These alone, and other situations (e.g., mutations and posttranslational modifications), can cause significant heterogeneity and polymorphism in the architecture of the amyloid fibril [95].
At a structural level, Iadanza et al. generated β2m amyloid fibrils in vitro and characterized them as having two identical protofilaments, subunit stabilization by hydrophobic packing, and an essential disulfide bond. Additionally, Iadanza et al. demonstrated that different fibril polymorphs can be formed from a common subunit structure [96]. In addition, Wilkinson et al. demonstrated the polymorphism of β2m fibrils derived from variants (β2m-ΔN6, β2m-D76N, and β2m-V27M) that also afflict patients with DRA. Through cryo-EM, they determined that the fibrils of these variants contain differing numbers of protofilaments and β2m subunits per layer of the amyloid fibril structure. They also concluded that the polymorphism of these fibrils arises from the assembly of a common building block that is remarkably homogeneous and monomorphic [97].
Another intriguing case involves long-term immunosuppressive therapy (e.g., corticosteroids), which has been linked to a higher incidence of systemic amyloidosis caused by the serum amyloid A (SAA) protein (Table 1) [98,99,100]. Although there are not many studies investigating how certain medical therapies can trigger the amyloid formation of SAA, its systemic form is caused typically by chronic inflammatory conditions such as tuberculosis, leprosy, and rheumatoid arthritis. This amyloidosis poses risks both locally and systemically, with the spleen and liver and, primarily, kidneys and vasculature being the most affected organs/tissues [101,102]. In addition, its regional and temporal context has been explored (i.e., it was common in Western countries until the mid-20th century) [101,103,104]. This amyloidosis is a common systemic amyloidosis among humans, as well as rats, birds, and other animals [105]. It is important to emphasize that, despite being incurable and fatal, novel treatment approaches aimed at suppressing the underlying inflammatory disease have proven effective in managing the condition; moreover, there has been a global decline in cases of AA amyloidosis, although the reasons behind this trend remain unknown [106].
The SAA1 gene encodes a member of the serum amyloid A family of apolipoproteins (SAA family composed of SAA1, SAA2, and SAA3). This SAA1 protein (Table 1) is highly expressed in response to inflammatory processes and tissue injury [107]. It is proposed that the generation of the SAA1 amyloidogenic process occurs intracellularly in lysosomes (i.e., low pH) [108], with its mature fibrils being twisted left- or right-handed, and containing two unequal or equal stacks of fibrils depending on the affected tissue (i.e., vascular and glomerular, respectively) [109]. This polymorphism increases the complexity of the disease itself and complicates the search for new treatments.
3.3. Transmitted/Transplanted-Iatrogenic Amyloidosis
This third category of iatrogenic amyloidosis has been studied and documented for decades. It is observed in specific cases of organ/tissue transplantation or contaminated instruments containing amyloid forms of proteins (i.e., “transplanted-iatrogenic amyloidosis”), as well as in the administration of drugs containing amyloid fibrils (i.e., “transmitted-iatrogenic amyloidosis”) (Figure 2).
A notable case is iatrogenic Creutzfeldt–Jakob disease (iCJD), also known as transmissible spongiform encephalopathy (TSE), an incurable and fatal illness. Over 200 cases have been documented involving the administration of human growth hormone (hGH) or human gonadotropin (hGN) derived from human cadaveric pituitaries contaminated with transmissible prion proteins (PrP) (Table 1) for the treatment of growth deficiency in children [110,111]. These proteins form amyloid fibrils, leading to brain damage and progressive neurodegenerative disorders. However, other iatrogenic forms of PrP transmission have been documented, such as corneal transplantation, contaminated neurosurgical instruments and electrodes, blood and blood products, and dura mater graft transplantation (in this case, over 200 cases) [110,112,113].
Understanding the amyloidogenic process of PrP, its propagation, and transmissibility has proven to be challenging and requires the employment of various biochemical and biophysical techniques [114]. From a structural perspective, Glynn et al. utilized cryo-EM to elucidate the architecture of in vitro-generated human PrP fibrils from an 85-residue recombinant form. These fibrils feature two intertwined and symmetrical protofilaments. Each protofilament is comprised by a hydrophobic and markedly fibrillogenic disease-associated segment (residues 106 to 145) of the prion protein [115]. In turn, also employing cryo-EM, Wang et al. generated in vitro human PrP fibrils from full-length human PrP (residues 23–231). They determined that the amyloid fibril core was composed of residues 170–229, where the fibril consisted of two intertwined protofilaments (left-hand twist) [116]. Additionally, murine PrP amyloid fibrils exhibit significant polymorphism [117,118], and further efforts are needed to understand this multifaceted disease.
Certainly, the cases that raise the most discussion in the field of iatrogenic amyloidosis are those involving the transmission of the β-amyloid (Aβ) proteins (Table 1) involved in the pathogenesis of Alzheimer’s disease (AD). Recently, Ritchie et al., analyzing samples from cases of iCJD caused by the use of contaminated hGH in the UK, identified that approximately 50% of recipient patients presented Aβ deposits resulting in an AD-like phenotype and cerebral amyloid angiopathy (CAA) regardless of the occurrence of CJD. The authors concluded that Aβ derived from hGH acted as a seeding amyloid, causing a “transmitted-iatrogenic amyloidosis” of AD-like disease [119]. Similarly, Duyckaerts et al., studying cases of contaminated hGH in France, detected the concentration of Aβ40 or Aβ42, and tau protein, above the detection limit in 17 of 26 vials (i.e., 65%) produced from 1974 to 1988. However, of the 24 deceased patients with iCJD, only one patient showed Aβ deposits [120]. Several other studies have demonstrated similar results [121,122,123,124,125]. Moreover, various studies have tested this hypothesis of Aβ transmission/transplantation in animal models [123,126,127,128,129,130,131]. It is worth noting that the use of hGH extracted from human cadavers is prohibited in many countries.
In addition to potential transmission, Aβ fibrils can be transplanted to the recipient, thus defining the occurrence as “transplanted-iatrogenic amyloidosis”. In this way, Aβ fibrils can be transplanted in surgeries involving cadaveric dura mater grafts or contaminated instruments, resulting in iatrogenic cerebral amyloid angiopathy (iCAA) disease, also known as “acquired CAA”, which is Aβ deposition in the cerebral vessels causing hemorrhages and stroke [14]. Several cases have been described in adults (<55 years old) with a history of neurosurgeries with or without dura mater grafts in early childhood or teenage years [14,132,133,134,135,136,137,138]. These cases highlight the need to establish methods to prevent the transplantation of cerebral β-amyloidosis among individuals.
The Aβ protein originates from the cleavage of the amyloid precursor protein (APP gene) by β- and γ-secretases, while α-secretase is responsible for the cleavage of the non-amyloidogenic form [139]. The most abundant forms of Aβ are 40 and 42 amino acids in length (Aβ40 and Aβ42) (Table 1). Like other amyloid fibrils, Aβ also exhibits polymorphic structures, both in the case of fibrils extracted from the brains of AD patients (i.e., Aβ40) [140] and those generated in vitro (i.e., Aβ42) [141], which feature two twisted protofilaments composed of molecules stacked in a parallel, cross-β structure. Understanding these structures is necessary to better comprehend the pathophysiology of the disease and to develop new treatments for these currently incurable conditions.
Another case that continues to spark significant debate and needs further investigations is about the tau protein. As previously mentioned, this protein was detected at levels exceeding the detection threshold in batches of hGH [120]. While transmissibility has been demonstrated in animal models [142,143] and has also been detected in patients who received injections of contaminated hGH, the definitive confirmation of iatrogenic transmission in humans remains elusive due to the complexity of cases (e.g., induction by previous radiotherapy treatment or PrP inducing tau amyloidogenesis) [119,120].
Cases of iatrogenic amyloidosis have also been documented following domino liver transplantation (DLT), also known as sequential liver transplantation (SLT). In these cases, livers excised from patients with mutations in the TTR gene, which encodes the transthyretin protein, are transplanted into patients with severe hepatic pathologies or liver failure [144]. DLT was created because the first therapeutic strategy used to address transthyretin amyloidosis (ATTR) was liver transplantation. Thus, the procedure entails the transplantation of the liver removed from a patient with hereditary ATTR amyloidosis into a third person suffering from end-stage liver disease [145].
Transthyretin (TTR) is primarily produced in the liver, being released into the bloodstream where it performs its function of transporting retinol and thyroxine. Thus far, over 130 mutations in the TTR gene have been identified, with the majority being pathogenic and capable of causing ATTR. Over the years, these mutations have undergone extensive study due to their distinct characteristics, where certain mutations have a propensity for causing more neuropathies (e.g., Val30Met), while others may induce more cardiomyopathies (e.g., Val122Ile) and/or mixed phenotypes [146,147,148,149].
Patients receiving liver grafts from individuals with TTR mutations usually begin to exhibit symptoms of amyloidosis within 10 years of transplantation, presenting symptoms such as sensory disturbance, autonomic dysfunction, neuropathic pain, skin denervation, intestinal abnormalities, and cardiac involvement. Additionally, amyloid deposits are found in different tissues such as the heart, kidneys, duodenum, tongue, sciatic nerve, and peripheral nerve [150,151,152,153,154,155,156,157,158,159,160,161]. In an attempt to understand the onset of the amyloidogenic process in patients who underwent transplantation, Yoshinaga et al. detected initial lesions and small amyloid deposits in the gastroduodenal mucosa in two asymptomatic patients who had received DLT (from Val30Met and Val30Leu donors) after 14 months and 12 years. The authors demonstrated by tandem mass spectrometry that the amyloid deposit was predominantly composed of mutant protein derived from the transplanted tissue and only a small amount of wild-type TTR (i.e., ratio ~95:5). Both patients were placed on specific medications to stabilize the TTR tetramer (i.e., Tafamidis and Diflunisal). The authors concluded that blocking the amyloid formation cascade in the early postoperative stage may possibly prevent the development and progression of this iatrogenic amyloidosis [162].
The amyloidogenic process of the TTR protein has been extensively studied for decades [163,164,165,166,167,168,169]; however, with the advancement of microscopy techniques, a true assessment of the amyloid fibril structure has become possible in recent years. The wild-type TTR protein, which in certain cases (mechanisms yet to be determined) also becomes amyloid, is responsible for ATTR with a higher predominance of cardiac involvement. Since DLT cases arise from liver grafts from individuals with different mutations in the TTR gene, their amyloid fibrils possibly exhibit a higher degree of polymorphism, which can directly influence tissue tropism and disease phenotype. TTR amyloid fibrils have a spearhead-like shape and can be classified into Type A (cleaved TTR) and Type B (full-length TTR). Type A fibers are more abundant and common, where the fibril is formed by the N- and C-terminal segments encompassing residues from Pro11 to Lys35 and Gly57 to Thr123/Asn124, respectively (Table 1) [170,171,172,173,174]. However, their structural conformation may vary due to a plethora of factors.
It is worth noting that, while there is one FDA-approved drug (i.e., Tafamidis) [175] and off-label drugs (i.e., Diflunisal) for stabilizing the protein in its tetrameric form, this treatment alone does not result in the disruption and clearance of amyloid fibrils. Nonetheless, despite the complexity of the disease and its being incurable and fatal, there are ongoing efforts involving testing new drugs (i.e., antibodies anti-TTR fibrils) aimed at facilitating tissue fibril clearance [176]. Another therapeutic possibility post-DLT would be the use of TTR-gene-expression-silencing drugs (i.e., RNA interference agents), commonly employed in patients with ATTR polyneuropathy [177,178].
4. Conclusions
In conclusion, amyloidosis is a complex and multifaceted phenomenon, with various underlying mechanisms involved, and iatrogenic amyloidosis further complicates the picture. The lack of standardized nomenclature to classify different cases, the involvement of different proteins and tissues, and the pathological complexity involved in amyloid diseases, all represent a significant challenge for further understanding and managing iatrogenic amyloidosis. Therefore, through this comprehensive review, iatrogenic amyloidosis has been categorized into three main classes: generated-iatrogenic, triggered-iatrogenic, and transmitted/transplanted-iatrogenic, based on the nature of the medical intervention and its impact on amyloid proteins. Each class presents unique challenges and considerations, demanding further research to elucidate the underlying molecular mechanisms and to develop effective therapeutic strategies or improved medical methods and interventions.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Dobson, C.M. Protein Folding and Misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Ezzat, K.; Pernemalm, M.; Pålsson, S.; Roberts, T.C.; Järver, P.; Dondalska, A.; Bestas, B.; Sobkowiak, M.J.; Levänen, B.; Sköld, M.; et al. The Viral Protein Corona Directs Viral Pathogenesis and Amyloid Aggregation. Nat. Commun. 2019, 10, 2331. [Google Scholar] [CrossRef]
- Hammarström, P.; Nyström, S. Viruses and Amyloids—A Vicious Liaison. Prion 2023, 17, 82–104. [Google Scholar] [CrossRef]
- Woldemeskel, M. A Concise Review of Amyloidosis in Animals. Vet. Med. Int. 2012, 2012, 427296. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The Amyloid State and Its Association with Protein Misfolding Diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Buxbaum, J.N.; Dispenzieri, A.; Eisenberg, D.S.; Fändrich, M.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Westermark, P. Amyloid Nomenclature 2022: Update, Novel Proteins, and Recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid 2022, 29, 213–219. [Google Scholar] [CrossRef]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid Nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef]
- Picken, M.M. The Pathology of Amyloidosis in Classification: A Review. Acta Haematol. 2020, 143, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Biza, K.V.; Nastou, K.C.; Tsiolaki, P.L.; Mastrokalou, C.V.; Hamodrakas, S.J.; Iconomidou, V.A. The Amyloid Interactome: Exploring Protein Aggregation. PLoS ONE 2017, 12, e0173163. [Google Scholar] [CrossRef] [PubMed]
- Billette de Villemeur, T.; Gelot, A.; Deslys, J.P.; Dormont, D.; Duyckaerts, C.; Jardin, L.; Denni, J.; Robain, O. Iatrogenic Creutzfeldt-Jakob Disease in Three Growth Hormone Recipients: A Neuropathological Study. Neuropathol. Appl. Neurobiol. 1994, 20, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Feeney, C.; Scott, G.P.; Cole, J.H.; Sastre, M.; Goldstone, A.P.; Leech, R. Seeds of Neuroendocrine Doubt. Nature 2016, 535, E1–E2. [Google Scholar] [CrossRef]
- Lladó, L.; Baliellas, C.; Casasnovas, C.; Ferrer, I.; Fabregat, J.; Ramos, E.; Castellote, J.; Torras, J.; Xiol, X.; Rafecas, A. Risk of Transmission of Systemic Transthyretin Amyloidosis after Domino Liver Transplantation. Liver Transpl. 2010, 16, 1386–1392. [Google Scholar] [CrossRef]
- Kaushik, K.; van Etten, E.S.; Siegerink, B.; Kappelle, L.J.; Lemstra, A.W.; Schreuder, F.H.B.M.; Klijn, C.J.M.; Peul, W.C.; Terwindt, G.M.; van Walderveen, M.A.A.; et al. Iatrogenic Cerebral Amyloid Angiopathy Post Neurosurgery: Frequency, Clinical Profile, Radiological Features, and Outcome. Stroke 2023, 54, 1214–1223. [Google Scholar] [CrossRef]
- Scarpioni, R.; Ricardi, M.; Albertazzi, V.; De Amicis, S.; Rastelli, F.; Zerbini, L. Dialysis-Related Amyloidosis: Challenges and Solutions. Int. J. Nephrol. Renov. Dis. 2016, 9, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Lansbury, P.T. Amyloid Fibril Formation Requires a Chemically Discriminating Nucleation Event: Studies of an Amyloidogenic Sequence from the Bacterial Protein OsmB. Biochemistry 1992, 31, 12345–12352. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Nucleated Polymerization with Secondary Pathways. II. Determination of Self-Consistent Solutions to Growth Processes Described by Non-Linear Master Equations. J. Chem. Phys. 2011, 135, 065106. [Google Scholar] [CrossRef]
- Cremades, N.; Dobson, C.M. The Contribution of Biophysical and Structural Studies of Protein Self-Assembly to the Design of Therapeutic Strategies for Amyloid Diseases. Neurobiol. Dis. 2018, 109, 178–190. [Google Scholar] [CrossRef]
- Morales, R.; Green, K.M.; Soto, C. Cross Currents in Protein Misfolding Disorders: Interactions and Therapy. CNS Neurol. Disord. Drug Targets 2009, 8, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Subedi, S.; Sasidharan, S.; Nag, N.; Saudagar, P.; Tripathi, T. Amyloid Cross-Seeding: Mechanism, Implication, and Inhibition. Molecules 2022, 27, 1776. [Google Scholar] [CrossRef]
- Hoop, C.L.; Zhu, J.; Bhattacharya, S.; Tobita, C.A.; Radford, S.E.; Baum, J. Collagen I Weakly Interacts with the β-Sheets of β2-Microglobulin and Enhances Conformational Exchange To Induce Amyloid Formation. J. Am. Chem. Soc. 2020, 142, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, L.D.C.; Lima, L.M.T.R.; Freire, J.B.B.; Bleicher, L.; Polikarpov, I.; Almeida, F.C.L.; Foguel, D. Novel Zn2+-Binding Sites in Human Transthyretin: Implications for Amyloidogenesis and Retinol-Binding Protein Recognition. J. Biol. Chem. 2010, 285, 31731–31741. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, H.; Wu, J.; Romankov, V.; Daffé, N.; Dreiser, J. Amyloid-Beta-Copper Interaction Studied by Simultaneous Nitrogen K and Copper L2,3 -Edge Soft X-Ray Absorption Spectroscopy. iScience 2021, 24, 103465. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Brayne, C. Understanding the Roles of Mutations in the Amyloid Precursor Protein in Alzheimer Disease. Mol. Psychiatry 2018, 23, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Hammarström, P.; Schneider, F.; Kelly, J.W. Trans-Suppression of Misfolding in an Amyloid Disease. Science 2001, 293, 2459–2462. [Google Scholar] [CrossRef] [PubMed]
- Velander, P.; Wu, L.; Henderson, F.; Zhang, S.; Bevan, D.R.; Xu, B. Natural Product-Based Amyloid Inhibitors. Biochem. Pharmacol. 2017, 139, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.I.; Lin, Y.; Lee, Y.-H.; Zheng, J.; Ramamoorthy, A. Biophysical Processes Underlying Cross-Seeding in Amyloid Aggregation and Implications in Amyloid Pathology. Biophys. Chem. 2021, 269, 106507. [Google Scholar] [CrossRef] [PubMed]
- Morel, B.; Varela, L.; Azuaga, A.I.; Conejero-Lara, F. Environmental Conditions Affect the Kinetics of Nucleation of Amyloid Fibrils and Determine Their Morphology. Biophys. J. 2010, 99, 3801–3810. [Google Scholar] [CrossRef] [PubMed]
- Dogan, A.; Mereuta, O.M. Iatrogenic Pharmaceutical Amyloidosis Associated with Insulin and Enfuvirtide Administration. In Amyloid and Related Disorders; Picken, M.M., Herrera, G.A., Dogan, A., Eds.; Current Clinical Pathology; Springer International Publishing: Cham, Switzerland, 2015; pp. 481–485. ISBN 978-3-319-19293-2. [Google Scholar]
- Barman, P.; Joshi, S.; Sharma, S.; Preet, S.; Sharma, S.; Saini, A. Strategic Approaches to Improvise Peptide Drugs as Next Generation Therapeutics. Int. J. Pept. Res. Ther. 2023, 29, 61. [Google Scholar] [CrossRef]
- D’Aloisio, V.; Dognini, P.; Hutcheon, G.A.; Coxon, C.R. PepTherDia: Database and Structural Composition Analysis of Approved Peptide Therapeutics and Diagnostics. Drug Discov. Today 2021, 26, 1409–1419. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorganic. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Störkel, S.; Schneider, H.M.; Müntefering, H.; Kashiwagi, S. Iatrogenic, Insulin-Dependent, Local Amyloidosis. Lab. Investig. 1983, 48, 108–111. [Google Scholar] [PubMed]
- Ansari, A.M.; Osmani, L.; Matsangos, A.E.; Li, Q.K. Current Insight in the Localized Insulin-Derived Amyloidosis (LIDA): Clinico-Pathological Characteristics and Differential Diagnosis. Pathol. Res. Pract. 2017, 213, 1237–1241. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.; Theis, J.D.; Vrana, J.A.; Dogan, A. Pharmaceutical Amyloidosis Associated with Subcutaneous Insulin and Enfuvirtide Administration. Amyloid 2014, 21, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Lytle, A.; Darvishian, F.; Ozerdem, U. Localized Amyloidosis: A Diagnostic Pitfall in Breast Pathology. Pathol. Res. Pract. 2019, 215, 152699. [Google Scholar] [CrossRef] [PubMed]
- Chinisaz, M.; Ebrahim-Habibi, A.; Yaghmaei, P.; Parivar, K.; Dehpour, A.-R. Generating Local Amyloidosis in Mice by the Subcutaneous Injection of Human Insulin Amyloid Fibrils. Exp. Ther. Med. 2014, 8, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.C. Understanding Insulin and Its Receptor from Their Three-Dimensional Structures. Mol. Metab. 2021, 52, 101255. [Google Scholar] [CrossRef] [PubMed]
- Van Lierop, B.; Ong, S.C.; Belgi, A.; Delaine, C.; Andrikopoulos, S.; Haworth, N.L.; Menting, J.G.; Lawrence, M.C.; Robinson, A.J.; Forbes, B.E. Insulin in Motion: The A6-A11 Disulfide Bond Allosterically Modulates Structural Transitions Required for Insulin Activity. Sci. Rep. 2017, 7, 17239. [Google Scholar] [CrossRef] [PubMed]
- Fagihi, M.H.A.; Bhattacharjee, S. Amyloid Fibrillation of Insulin: Amelioration Strategies and Implications for Translation. ACS Pharmacol. Transl. Sci. 2022, 5, 1050–1061. [Google Scholar] [CrossRef]
- Foderà, V.; Librizzi, F.; Groenning, M.; Van De Weert, M.; Leone, M. Secondary Nucleation and Accessible Surface in Insulin Amyloid Fibril Formation. J. Phys. Chem. B 2008, 112, 3853–3858. [Google Scholar] [CrossRef]
- Jiménez, J.L.; Nettleton, E.J.; Bouchard, M.; Robinson, C.V.; Dobson, C.M.; Saibil, H.R. The Protofilament Structure of Insulin Amyloid Fibrils. Proc. Natl. Acad. Sci. USA 2002, 99, 9196–9201. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, B.; Groenning, M.; Roessle, M.; Kastrup, J.S.; van de Weert, M.; Flink, J.M.; Frokjaer, S.; Gajhede, M.; Svergun, D.I. A Helical Structural Nucleus Is the Primary Elongating Unit of Insulin Amyloid Fibrils. PLoS Biol. 2007, 5, e134. [Google Scholar] [CrossRef] [PubMed]
- Selivanova, O.M.; Galzitskaya, O.V. Structural Polymorphism and Possible Pathways of Amyloid Fibril Formation on the Example of Insulin Protein. Biochemistry 2012, 77, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Kranc, C.; Wagner, R.; Joy, N.M.; Feldman, J.; Reid, D.C. Cutaneous Insulin-Derived Amyloidosis Presenting as Hyperkeratotic Nodules. Cutis 2021, 107, E6–E9. [Google Scholar] [CrossRef] [PubMed]
- Shikama, Y.; Kitazawa, J.-I.; Yagihashi, N.; Uehara, O.; Murata, Y.; Yajima, N.; Wada, R.; Yagihashi, S. Localized Amyloidosis at the Site of Repeated Insulin Injection in a Diabetic Patient. Intern. Med. 2010, 49, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.A.; Gedik, R.; Haddady, S. An Atypical Presentation of Insulin Amyloidosis: An Uncommon but Important Complication of Insulin Therapy. AACE Clin. Case Rep. 2018, 4, 80–83. [Google Scholar] [CrossRef]
- Nilsson, M.R. Insulin Amyloid at Injection Sites of Patients with Diabetes. Amyloid 2016, 23, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Samlaska, C.; Reber, S.; Murry, T. Insulin-Derived Amyloidosis: The Insulin Ball, Amyloidoma. JAAD Case Rep. 2020, 6, 351–353. [Google Scholar] [CrossRef]
- Hrudka, J.; Sticová, E.; Krbcová, M.; Schwarzmannová, K. Localized Insulin-Derived Amyloidosis in Diabetes Mellitus Type 1 Patient: A Case Report. Diagnostics 2023, 13, 2415. [Google Scholar] [CrossRef]
- Godse, R.; Rauck, C.; Woods, R.; Steele, K.T.; Elenitsas, R. Two Cases of Insulin-Derived Amyloidosis with Acanthosis Nigricans-Like Changes. Am. J. Dermatopathol. 2022, 44, 979–980. [Google Scholar] [CrossRef]
- Dubernet, A.; Toulmonde, M.; Colombat, M.; Hartog, C.; Riviere, E. Insulin Amyloidosis: A Case Report. Front. Med. 2023, 10, 1064832. [Google Scholar] [CrossRef] [PubMed]
- Iwaya, K.; Zako, T.; Fukunaga, J.; Sörgjerd, K.M.; Ogata, K.; Kogure, K.; Kosano, H.; Noritake, M.; Maeda, M.; Ando, Y.; et al. Toxicity of Insulin-Derived Amyloidosis: A Case Report. BMC Endocr. Disord. 2019, 19, 61. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-C.; Lee, D.-D. Atypical Presentation of Localized Insulin-Derived Amyloidosis as Protruding Brownish Skin Tumours. Clin. Exp. Dermatol. 2020, 45, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Nagase, T.; Iwaya, K.; Iwaki, Y.; Kotake, F.; Uchida, R.; Oh-i, T.; Sekine, H.; Miwa, K.; Murakami, S.; Odaka, T.; et al. Insulin-Derived Amyloidosis and Poor Glycemic Control: A Case Series. Am. J. Med. 2014, 127, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Estep, A.; Uhrig, J.; Bower, C.; Jarrett, R. Impact of Insulin-Derived Amyloidosis on Glycemic Control and Insulin Dosing. Clin. Diabetes 2022, 40, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Misumi, Y.; Nomura, T.; Oka, W.; Isoguchi, A.; Kanenawa, K.; Masuda, T.; Yamashita, T.; Inoue, Y.; Ando, Y.; et al. Extreme Adhesion Activity of Amyloid Fibrils Induces Subcutaneous Insulin Resistance. Diabetes 2019, 68, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Yuzu, K.; Lindgren, M.; Nyström, S.; Zhang, J.; Mori, W.; Kunitomi, R.; Nagase, T.; Iwaya, K.; Hammarström, P.; Zako, T. Insulin Amyloid Polymorphs: Implications for Iatrogenic Cytotoxicity. RSC Adv. 2020, 10, 37721–37727. [Google Scholar] [CrossRef] [PubMed]
- D’souza, A.; Theis, J.D.; Vrana, J.A.; Buadi, F.; Dispenzieri, A.; Dogan, A. Localized Insulin-derived Amyloidosis: A Potential Pitfall in the Diagnosis of Systemic Amyloidosis by Fat Aspirate. Am. J. Hematol. 2012, 87, E131–E132. [Google Scholar] [CrossRef] [PubMed]
- Pi-Sunyer, X.; Astrup, A.; Fujioka, K.; Greenway, F.; Halpern, A.; Krempf, M.; Lau, D.C.W.; Le Roux, C.W.; Violante Ortiz, R.; Jensen, C.B.; et al. A Randomized, Controlled Trial of 3.0 Mg of Liraglutide in Weight Management. N. Engl. J. Med. 2015, 373, 11–22. [Google Scholar] [CrossRef]
- Martins, C.O.; Lezcano, C.; Yi, S.S.; Landau, H.J.; Chapman, J.R.; Dogan, A. Novel Iatrogenic Amyloidosis Caused by Peptide Drug Liraglutide: A Clinical Mimic of AL Amyloidosis. Haematologica 2018, 103, e610–e612. [Google Scholar] [CrossRef]
- Muhammad, S.; McPhail, E.D.; Tobin, W.O.; Dasari, S.; Theis, J.; Vrana, J.A.; Naddaf, E. A Second Case of Liraglutide-Type Localised Amyloidosis. Amyloid 2023, 30, 244–245. [Google Scholar] [CrossRef] [PubMed]
- Aujla, S.K.; Jasti, P. A Rare Case of Liraglutide-Induced Iatrogenic Amyloidosis. J. Immunol. 2022, 208, 48.13. [Google Scholar] [CrossRef]
- Andersen, A.; Lund, A.; Knop, F.K.; Vilsbøll, T. Glucagon-like Peptide 1 in Health and Disease. Nat. Rev. Endocrinol. 2018, 14, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, D.A.; D’Alessio, D.A. Physiology of Proglucagon Peptides: Role of Glucagon and GLP-1 in Health and Disease. Physiol. Rev. 2015, 95, 513–548. [Google Scholar] [CrossRef] [PubMed]
- Poon, S.; Birkett, N.; Fowler, S.; Luisi, B.; Dobson, C.; Zurdo, J. Amyloidogenicity and Aggregate Cytotoxicity of Human Glucagon-Like Peptide-1 (hGLP-1). Protein Pept. Lett. 2009, 16, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Zapadka, K.L.; Becher, F.J.; Uddin, S.; Varley, P.G.; Bishop, S.; Gomes Dos Santos, A.L.; Jackson, S.E. A pH-Induced Switch in Human Glucagon-like Peptide-1 Aggregation Kinetics. J. Am. Chem. Soc. 2016, 138, 16259–16265. [Google Scholar] [CrossRef] [PubMed]
- Egbu, R.; Van Der Walle, C.F.; Brocchini, S.; Williams, G.R. Inhibiting the Fibrillation of a GLP-1-like Peptide. Int. J. Pharm. 2020, 574, 118923. [Google Scholar] [CrossRef] [PubMed]
- Jha, N.N.; Anoop, A.; Ranganathan, S.; Mohite, G.M.; Padinhateeri, R.; Maji, S.K. Characterization of Amyloid Formation by Glucagon-like Peptides: Role of Basic Residues in Heparin-Mediated Aggregation. Biochemistry 2013, 52, 8800–8810. [Google Scholar] [CrossRef]
- Wharton, S.; Blevins, T.; Connery, L.; Rosenstock, J.; Raha, S.; Liu, R.; Ma, X.; Mather, K.J.; Haupt, A.; Robins, D.; et al. Daily Oral GLP-1 Receptor Agonist Orforglipron for Adults with Obesity. N. Engl. J. Med. 2023, 389, 877–888. [Google Scholar] [CrossRef]
- Lincoff, A.M.; Brown-Frandsen, K.; Colhoun, H.M.; Deanfield, J.; Emerson, S.S.; Esbjerg, S.; Hardt-Lindberg, S.; Hovingh, G.K.; Kahn, S.E.; Kushner, R.F.; et al. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N. Engl. J. Med. 2023, 389, 2221–2232. [Google Scholar] [CrossRef]
- Jastreboff, A.M.; Kaplan, L.M.; Frías, J.P.; Wu, Q.; Du, Y.; Gurbuz, S.; Coskun, T.; Haupt, A.; Milicevic, Z.; Hartman, M.L. Triple–Hormone-Receptor Agonist Retatrutide for Obesity—A Phase 2 Trial. N. Engl. J. Med. 2023, 389, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Goldbach-Mansky, R.; Dailey, N.J.; Canna, S.W.; Gelabert, A.; Jones, J.; Rubin, B.I.; Kim, H.J.; Brewer, C.; Zalewski, C.; Wiggs, E.; et al. Neonatal-Onset Multisystem Inflammatory Disease Responsive to Interleukin-1β Inhibition. N. Engl. J. Med. 2006, 355, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Alehashemi, S.; Dasari, S.; De Jesus, A.A.; Cowen, E.W.; Lee, C.-C.R.; Goldbach-Mansky, R.; McPhail, E.D. Anakinra-Associated Amyloidosis. JAMA Dermatol. 2022, 158, 1454. [Google Scholar] [CrossRef] [PubMed]
- Alehashemi, S.; Dasari, S.; Metpally, A.; Uss, K.; Castelo-Soccio, L.A.; Heller, T.; Kellman, P.; Chen, M.Y.; Ahlman, M.; Kim, J.; et al. Anakinra-Associated Systemic Amyloidosis. Arthritis Rheumatol. 2024, 76, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Nasr, S.H.; Alehashemi, S.; Dasari, S.; Waldman, M.; Afzali, B.; Chiu, A.; Bolanos, J.; Goldbach-Mansky, R.; McPhail, E.D. Anakinra-Associated Renal Amyloidosis. Kidney Int. 2024, 105, 395–396. [Google Scholar] [CrossRef] [PubMed]
- Lalezari, J.P.; Henry, K.; O’Hearn, M.; Montaner, J.S.G.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron, J.J.; et al. Enfuvirtide, an HIV-1 Fusion Inhibitor, for Drug-Resistant HIV Infection in North and South America. N. Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Naujokas, A.; Vidal, C.I.; Mercer, S.E.; Harp, J.; Kurtin, P.J.; Fox, L.P.; Thompson, M.M. A Novel Form of Amyloid Deposited at the Site of Enfuvirtide Injection. J. Cutan. Pathol. 2012, 39, 220–221. [Google Scholar] [CrossRef] [PubMed]
- Morilla, M.E.; Kocher, J.; Harmaty, M. Localized Amyloidosis at the Site of Enfuvirtide Injection. Ann. Intern. Med. 2009, 151, 515. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.; Theis, J.D.; Vrana, J.A.; Dogan, A. Drug-Induced Amyloidosis: A Proteomic Insight Into 52 Cases. Blood 2013, 122, 1871. [Google Scholar] [CrossRef]
- Fuzeon, Drugs at FDA. In FDA Fact Sheet; Roche Laboratories Inc.: Indianapolis, IN, USA, 2003; pp. 1–34.
- Martinez Morales, M.; van der Walle, C.F.; Derrick, J.P. Modulation of the Fibrillation Kinetics and Morphology of a Therapeutic Peptide by Cucurbit[7]Uril. Mol. Pharm. 2023, 20, 3559–3569. [Google Scholar] [CrossRef]
- Tsuruya, K.; Arima, H.; Iseki, K.; Hirakata, H.; Kyushu Dialysis-Related Amyloidosis Study Group. Association of Dialysis-Related Amyloidosis with Lower Quality of Life in Patients Undergoing Hemodialysis for More than 10 Years: The Kyushu Dialysis-Related Amyloidosis Study. PLoS ONE 2021, 16, e0256421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Fu, X.; Sawashita, J.; Yao, J.; Zhang, B.; Qian, J.; Tomozawa, H.; Mori, M.; Ando, Y.; Naiki, H.; et al. Mouse Model to Study Human A beta2M Amyloidosis: Generation of a Transgenic Mouse with Excessive Expression of Human Beta2-Microglobulin. Amyloid 2010, 17, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Yamaguchi, I.; Hasegawa, K.; Tsutsumi, S.; Goto, Y.; Gejyo, F.; Naiki, H. Glycosaminoglycans Enhance the Trifluoroethanol-Induced Extension of Beta 2-Microglobulin-Related Amyloid Fibrils at a Neutral pH. J. Am. Soc. Nephrol. 2004, 15, 126–133. [Google Scholar] [CrossRef]
- Niwa, T.; Katsuzaki, T.; Momoi, T.; Miyazaki, T.; Ogawa, H.; Saito, A.; Miyazaki, S.; Maeda, K.; Tatemichi, N.; Takei, Y. Modification of Beta 2m with Advanced Glycation End Products as Observed in Dialysis-Related Amyloidosis by 3-DG Accumulating in Uremic Serum. Kidney Int. 1996, 49, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Ikizler, T.A.; Hoover, R.L.; Nakamoto, M.; Yasunaga, C.; Pupim, L.B.; Hakim, R.M. Transforming Growth Factor-Beta Is Involved in the Pathogenesis of Dialysis-Related Amyloidosis. Kidney Int. 2000, 57, 697–708. [Google Scholar] [CrossRef]
- Portales-Castillo, I.; Yee, J.; Tanaka, H.; Fenves, A.Z. Beta-2 Microglobulin Amyloidosis: Past, Present, and Future. Kidney360 2020, 1, 1447–1455. [Google Scholar] [CrossRef]
- Becker, J.W.; Reeke, G.N. Three-Dimensional Structure of Beta 2-Microglobulin. Proc. Natl. Acad. Sci. USA 1985, 82, 4225–4229. [Google Scholar] [CrossRef]
- Eichner, T.; Radford, S.E. A Diversity of Assembly Mechanisms of a Generic Amyloid Fold. Mol. Cell 2011, 43, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Naiki, H.; Hashimoto, N.; Suzuki, S.; Kimura, H.; Nakakuki, K.; Gejyo, F. Establishment of a Kinetic Model of Dialysis-Related Amyloid Fibril Extension in Vitro. Amyloid 1997, 4, 223–232. [Google Scholar] [CrossRef]
- Connors, L.H.; Shirahama, T.; Skinner, M.; Fenves, A.; Cohen, A.S. In Vitro Formation of Amyloid Fibrils from Intact Beta 2-Microglobulin. Biochem. Biophys. Res. Commun. 1985, 131, 1063–1068. [Google Scholar] [CrossRef]
- Marcinko, T.M.; Liang, C.; Savinov, S.; Chen, J.; Vachet, R.W. Structural Heterogeneity in the Preamyloid Oligomers of β-2-Microglobulin. J. Mol. Biol. 2020, 432, 396–409. [Google Scholar] [CrossRef]
- Nakajima, K.; Yamaguchi, K.; Noji, M.; Aguirre, C.; Ikenaka, K.; Mochizuki, H.; Zhou, L.; Ogi, H.; Ito, T.; Narita, I.; et al. Macromolecular Crowding and Supersaturation Protect Hemodialysis Patients from the Onset of Dialysis-Related Amyloidosis. Nat. Commun. 2022, 13, 5689. [Google Scholar] [CrossRef]
- Loureiro, R.J.S.; Faísca, P.F.N. The Early Phase of Β2-Microglobulin Aggregation: Perspectives from Molecular Simulations. Front. Mol. Biosci. 2020, 7, 578433. [Google Scholar] [CrossRef]
- Iadanza, M.G.; Silvers, R.; Boardman, J.; Smith, H.I.; Karamanos, T.K.; Debelouchina, G.T.; Su, Y.; Griffin, R.G.; Ranson, N.A.; Radford, S.E. The Structure of a Β2-Microglobulin Fibril Suggests a Molecular Basis for Its Amyloid Polymorphism. Nat. Commun. 2018, 9, 4517. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.; Gallardo, R.U.; Martinez, R.M.; Guthertz, N.; So, M.; Aubrey, L.D.; Radford, S.E.; Ranson, N.A. Disease-Relevant Β2-Microglobulin Variants Share a Common Amyloid Fold. Nat. Commun. 2023, 14, 1190. [Google Scholar] [CrossRef] [PubMed]
- HogenEsch, H.; Niewold, T.A.; Higuchi, K.; Tooten, P.C.; Gruys, E.; Radl, J. Gastrointestinal AAPOAII and Systemic AA-Amyloidosis in Aged C57BL/Ka Mice. Amyloid-Type Dependent Effect of Long-Term Immunosuppressive Treatment. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1993, 64, 37–43. [Google Scholar] [CrossRef]
- Kanoh, T.; Yogo, K.; Ohnaka, T. Rapid progression of systemic amyloidosis after high-dose corticosteroid therapy in multiple myeloma. Rinsho Ketsueki 1990, 31, 1736–1739. [Google Scholar] [PubMed]
- Zilko, P.J.; Dawkins, R.L. Amyloidosis Associated with Dermatomyositis and Features of Multiple Myeloma. The Progression of Amyloidosis Associated with Corticosteroid and Cytotoxic Drug Therapy. Am. J. Med. 1975, 59, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, H.J.; Goodman, H.J.B.; Gilbertson, J.A.; Gallimore, J.R.; Sabin, C.A.; Gillmore, J.D.; Hawkins, P.N. Natural History and Outcome in Systemic AA Amyloidosis. N. Engl. J. Med. 2007, 356, 2361–2371. [Google Scholar] [CrossRef]
- Westermark, G.T.; Fändrich, M.; Westermark, P. AA Amyloidosis: Pathogenesis and Targeted Therapy. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 321–344. [Google Scholar] [CrossRef]
- Sack, G.H. Serum Amyloid A—A Review. Mol. Med. 2018, 24, 46. [Google Scholar] [CrossRef]
- Westermark, P.; Nilsson, G.T. Demonstration of Amyloid Protein AA in Old Museum Specimens. Arch. Pathol. Lab. Med. 1984, 108, 217–219. [Google Scholar]
- Murakami, T.; Ishiguro, N.; Higuchi, K. Transmission of Systemic AA Amyloidosis in Animals. Vet. Pathol. 2014, 51, 363–371. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Hawkins, P.N. Pathophysiology and Treatment of Systemic Amyloidosis. Nat. Rev. Nephrol. 2013, 9, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Sack, G.H. Serum Amyloid A (SAA) Proteins. In Vertebrate and Invertebrate Respiratory Proteins, Lipoproteins and Other Body Fluid Proteins; Hoeger, U., Harris, J.R., Eds.; Subcellular Biochemistry; Springer International Publishing: Cham, Switzerland, 2020; Volume 94, pp. 421–436. ISBN 978-3-030-41768-0. [Google Scholar]
- Claus, S.; Meinhardt, K.; Aumüller, T.; Puscalau-Girtu, I.; Linder, J.; Haupt, C.; Walther, P.; Syrovets, T.; Simmet, T.; Fändrich, M. Cellular Mechanism of Fibril Formation from Serum Amyloid A1 Protein. EMBO Rep. 2017, 18, 1352–1366. [Google Scholar] [CrossRef]
- Banerjee, S.; Baur, J.; Daniel, C.; Pfeiffer, P.B.; Hitzenberger, M.; Kuhn, L.; Wiese, S.; Bijzet, J.; Haupt, C.; Amann, K.U.; et al. Amyloid Fibril Structure from the Vascular Variant of Systemic AA Amyloidosis. Nat. Commun. 2022, 13, 7261. [Google Scholar] [CrossRef] [PubMed]
- Knight, R. Infectious and Sporadic Prion Diseases. Prog. Mol. Biol. Transl. Sci. 2017; 150, 293–318. [Google Scholar] [CrossRef]
- Appleby, B.S.; Lu, M.; Bizzi, A.; Phillips, M.D.; Berri, S.M.; Harbison, M.D.; Schonberger, L.B. Iatrogenic Creutzfeldt-Jakob Disease from Commercial Cadaveric Human Growth Hormone. Emerg. Infect. Dis. 2013, 19, 682–684. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, J.D.F.; Collinge, J. Molecular Basis of Prion Diseases. Basic Neurochem. 2012; 872–885. [Google Scholar] [CrossRef]
- Douet, J.-Y.; Huor, A.; Cassard, H.; Lugan, S.; Aron, N.; Mesic, C.; Vilette, D.; Barrio, T.; Streichenberger, N.; Perret-Liaudet, A.; et al. Prion Strains Associated with Iatrogenic CJD in French and UK Human Growth Hormone Recipients. Acta Neuropathol. Commun. 2021, 9, 145. [Google Scholar] [CrossRef]
- Yamaguchi, K.-I.; Kuwata, K. Formation and Properties of Amyloid Fibrils of Prion Protein. Biophys. Rev. 2018, 10, 517–525. [Google Scholar] [CrossRef]
- Glynn, C.; Sawaya, M.R.; Ge, P.; Gallagher-Jones, M.; Short, C.W.; Bowman, R.; Apostol, M.; Zhou, Z.H.; Eisenberg, D.S.; Rodriguez, J.A. Cryo-EM Structure of a Human Prion Fibril with a Hydrophobic, Protease-Resistant Core. Nat. Struct. Mol. Biol. 2020, 27, 417–423. [Google Scholar] [CrossRef]
- Wang, L.-Q.; Zhao, K.; Yuan, H.-Y.; Wang, Q.; Guan, Z.; Tao, J.; Li, X.-N.; Sun, Y.; Yi, C.-W.; Chen, J.; et al. Cryo-EM Structure of an Amyloid Fibril Formed by Full-Length Human Prion Protein. Nat. Struct. Mol. Biol. 2020, 27, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Jaroniec, C.P.; Surewicz, W.K. Cryo-EM Structure of Disease-Related Prion Fibrils Provides Insights into Seeding Barriers. Nat. Struct. Mol. Biol. 2022, 29, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.H.-L.; Kao, H.-W.; Lee, C.-H.; Huang, J.Y.C.; Wu, K.-P.; Chen, R.P.-Y. 2.2 Å Cryo-EM Tetra-Protofilament Structure of the Hamster Prion 108–144 Fibril Reveals an Ordered Water Channel in the Center. J. Am. Chem. Soc. 2022, 144, 13888–13894. [Google Scholar] [CrossRef]
- Ritchie, D.L.; Adlard, P.; Peden, A.H.; Lowrie, S.; Le Grice, M.; Burns, K.; Jackson, R.J.; Yull, H.; Keogh, M.J.; Wei, W.; et al. Amyloid-β Accumulation in the CNS in Human Growth Hormone Recipients in the UK. Acta Neuropathol. 2017, 134, 221–240. [Google Scholar] [CrossRef] [PubMed]
- Duyckaerts, C.; Sazdovitch, V.; Ando, K.; Seilhean, D.; Privat, N.; Yilmaz, Z.; Peckeu, L.; Amar, E.; Comoy, E.; Maceski, A.; et al. Neuropathology of Iatrogenic Creutzfeldt–Jakob Disease and Immunoassay of French Cadaver-Sourced Growth Hormone Batches Suggest Possible Transmission of Tauopathy and Long Incubation Periods for the Transmission of Abeta Pathology. Acta Neuropathol. 2018, 135, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Boone, C.; Goodwin, C.R.; Anderson, W.S. Iatrogenic Alzheimer Disease? Amyloid-β Protein Transmission Between Humans. Neurosurgery 2016, 78, N17–N18. [Google Scholar] [CrossRef] [PubMed]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.F.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for Human Transmission of Amyloid-β Pathology and Cerebral Amyloid Angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Purro, S.A.; Farrow, M.A.; Linehan, J.; Nazari, T.; Thomas, D.X.; Chen, Z.; Mengel, D.; Saito, T.; Saido, T.; Rudge, P.; et al. Transmission of Amyloid-β Protein Pathology from Cadaveric Pituitary Growth Hormone. Nature 2018, 564, 415–419. [Google Scholar] [CrossRef]
- Cali, I.; Cohen, M.L.; Haik, S.; Parchi, P.; Giaccone, G.; Collins, S.J.; Kofskey, D.; Wang, H.; McLean, C.A.; Brandel, J.-P.; et al. Iatrogenic Creutzfeldt-Jakob Disease with Amyloid-β Pathology: An International Study. Acta Neuropathol. Commun. 2018, 6, 5. [Google Scholar] [CrossRef]
- Banerjee, G.; Farmer, S.F.; Hyare, H.; Jaunmuktane, Z.; Mead, S.; Ryan, N.S.; Schott, J.M.; Werring, D.J.; Rudge, P.; Collinge, J. Iatrogenic Alzheimer’s Disease in Recipients of Cadaveric Pituitary-Derived Growth Hormone. Nat. Med. 2024, 30, 394–402. [Google Scholar] [CrossRef]
- Singh, C.S.B.; Johns, K.M.; Kari, S.; Munro, L.; Mathews, A.; Fenninger, F.; Pfeifer, C.G.; Jefferies, W.A. Conclusive Demonstration of Iatrogenic Alzheimer’s Disease Transmission in a Model of Stem Cell Transplantation. Stem Cell Rep. 2024, 19, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Duran-Aniotz, C.; Bravo-Alegria, J.; Estrada, L.D.; Shahnawaz, M.; Hu, P.-P.; Kramm, C.; Morales-Scheihing, D.; Urayama, A.; Soto, C. Infusion of Blood from Mice Displaying Cerebral Amyloidosis Accelerates Amyloid Pathology in Animal Models of Alzheimer’s Disease. Acta Neuropathol. Commun. 2020, 8, 213. [Google Scholar] [CrossRef] [PubMed]
- Gary, C.; Lam, S.; Hérard, A.-S.; Koch, J.E.; Petit, F.; Gipchtein, P.; Sawiak, S.J.; Caillierez, R.; Eddarkaoui, S.; Colin, M.; et al. Encephalopathy Induced by Alzheimer Brain Inoculation in a Non-Human Primate. Acta Neuropathol. Commun. 2019, 7, 126. [Google Scholar] [CrossRef] [PubMed]
- Hérard, A.-S.; Petit, F.; Gary, C.; Guillermier, M.; Boluda, S.; Garin, C.M.; Lam, S.; Dhenain, M.; Brainbank Neuro-CEB Neuropathology Network. Induction of Amyloid-β Deposits from Serially Transmitted, Histologically Silent, Aβ Seeds Issued from Human Brains. Acta Neuropathol. Commun. 2020, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Kim, J.H.; Hasegawa, A.; Goto, R.; Sakai, K.; Ono, K.; Itoh, Y.; Yamada, M. Exogenous Aβ Seeds Induce Aβ Depositions in the Blood Vessels Rather than the Brain Parenchyma, Independently of Aβ Strain-Specific Information. Acta Neuropathol. Commun. 2021, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.; Petit, F.; Hérard, A.-S.; Boluda, S.; Eddarkaoui, S.; Guillermier, M.; Buée, L.; Duyckaerts, C.; Haïk, S.; Brain Bank Neuro-C. E. B. Neuropathology Network; et al. Transmission of Amyloid-Beta and Tau Pathologies Is Associated with Cognitive Impairments in a Primate. Acta Neuropathol. Commun. 2021, 9, 165. [Google Scholar] [CrossRef]
- Fandler-Höfler, S.; Kneihsl, M.; Beitzke, M.; Enzinger, C.; Gattringer, T. Intracerebral Haemorrhage Caused by Iatrogenic Cerebral Amyloid Angiopathy in a Patient with a History of Neurosurgery 35 Years Earlier. Lancet 2023, 402, 411. [Google Scholar] [CrossRef]
- Storti, B.; Canavero, I.; Gabriel, M.M.; Capozza, A.; Rifino, N.; Stanziano, M.; Tagliabue, L.; Bersano, A. Iatrogenic Cerebral Amyloid Angiopathy: An Illustrative Case of a Newly Introduced Disease. Eur. J. Neurol. 2023, 30, 3397–3399. [Google Scholar] [CrossRef]
- Banerjee, G.; Adams, M.E.; Jaunmuktane, Z.; Alistair Lammie, G.; Turner, B.; Wani, M.; Sawhney, I.M.S.; Houlden, H.; Mead, S.; Brandner, S.; et al. Early Onset Cerebral Amyloid Angiopathy Following Childhood Exposure to Cadaveric Dura: Banerjee: Early Onset CAA. Ann. Neurol. 2019, 85, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Caroppo, P.; Marucci, G.; Maccagnano, E.; Gobbo, C.L.; Bizzozero, I.; Tiraboschi, P.; Redaelli, V.; Catania, M.; Di Fede, G.; Caputi, L.; et al. Cerebral Amyloid Angiopathy in a 51-Year-Old Patient with Embolization by Dura Mater Extract and Surgery for Nasopharyngeal Angiofibroma at Age 17. Amyloid 2021, 28, 142–143. [Google Scholar] [CrossRef]
- Giaccone, G.; Maderna, E.; Marucci, G.; Catania, M.; Erbetta, A.; Chiapparini, L.; Indaco, A.; Caroppo, P.; Bersano, A.; Parati, E.; et al. Iatrogenic Early Onset Cerebral Amyloid Angiopathy 30 Years after Cerebral Trauma with Neurosurgery: Vascular Amyloid Deposits Are Made up of Both Aβ40 and Aβ42. Acta Neuropathol. Commun. 2019, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Raposo, N.; Planton, M.; Siegfried, A.; Calviere, L.; Payoux, P.; Albucher, J.-F.; Viguier, A.; Delisle, M.-B.; Uro-Coste, E.; Chollet, F.; et al. Amyloid-β Transmission through Cardiac Surgery Using Cadaveric Dura Mater Patch. J. Neurol. Neurosurg. Psychiatry 2020, 91, 440–441. [Google Scholar] [CrossRef] [PubMed]
- Yoshiki, K.; Hirose, G.; Kumahashi, K.; Kohda, Y.; Ido, K.; Shioya, A.; Misaki, K.; Kasuga, K. Follow-up Study of a Patient with Early Onset Cerebral Amyloid Angiopathy Following Childhood Cadaveric Dural Graft. Acta Neurochir. 2021, 163, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, U.; Thurber, K.R.; Yau, W.-M.; Tycko, R. Molecular Structure of a Prevalent Amyloid-β Fibril Polymorph from Alzheimer’s Disease Brain Tissue. Proc. Natl. Acad. Sci. USA 2021, 118, e2023089118. [Google Scholar] [CrossRef] [PubMed]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril Structure of Amyloid-β(1–42) by Cryo–Electron Microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and Spreading of Tauopathy in Transgenic Mouse Brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Audouard, E.; Houben, S.; Masaracchia, C.; Yilmaz, Z.; Suain, V.; Authelet, M.; De Decker, R.; Buée, L.; Boom, A.; Leroy, K.; et al. High-Molecular-Weight Paired Helical Filaments from Alzheimer Brain Induces Seeding of Wild-Type Mouse Tau into an Argyrophilic 4R Tau Pathology in Vivo. Am. J. Pathol. 2016, 186, 2709–2722. [Google Scholar] [CrossRef] [PubMed]
- Stangou, A.J.; Heaton, N.D.; Hawkins, P.N. Transmission of Systemic Transthyretin Amyloidosis by Means of Domino Liver Transplantation. N. Engl. J. Med. 2005, 352, 2356. [Google Scholar] [CrossRef]
- Grande-Trillo, A.; Baliellas, C.; Lladó, L.; Casasnovas, C.; Franco-Baux, J.V.; Gracia-Sánchez, L.; Gómez-Bravo, M.Á.; González-Vilatarsana, E.; Caballero-Gullón, L.; Echeverri, E.; et al. Transthyretin Amyloidosis with Cardiomyopathy after Domino Liver Transplantation: Results of a Cross-Sectional Study. Am. J. Transplant. 2021, 21, 372–381. [Google Scholar] [CrossRef]
- Adams, D.; Koike, H.; Slama, M.; Coelho, T. Hereditary Transthyretin Amyloidosis: A Model of Medical Progress for a Fatal Disease. Nat. Rev. Neurol. 2019, 15, 387–404. [Google Scholar] [CrossRef]
- Maurer, M.S.; Hanna, M.; Grogan, M.; Dispenzieri, A.; Witteles, R.; Drachman, B.; Judge, D.P.; Lenihan, D.J.; Gottlieb, S.S.; Shah, S.J.; et al. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J. Am. Coll. Cardiol. 2016, 68, 161–172. [Google Scholar] [CrossRef]
- Bonilauri, B.; Shin, H.S.; Htet, M.; Yan, C.D.; Witteles, R.M.; Sallam, K.; Wu, J.C. Generation of Two Induced Pluripotent Stem Cell Lines from Patients with Cardiac Amyloidosis Carrying Heterozygous Transthyretin (TTR) Mutation. Stem Cell Res. 2023, 72, 103215. [Google Scholar] [CrossRef] [PubMed]
- Melesio, J.; Bonilauri, B.; Li, A.; Pang, P.D.; Liao, R.; Witteles, R.M.; Wu, J.C.; Sallam, K. Generation of Two Induced Pluripotent Stem Cell Lines from Hereditary Amyloidosis Patients with Polyneuropathy Carrying Heterozygous Transthyretin (TTR) Mutation. Stem Cell Res. 2024, 74, 103265. [Google Scholar] [CrossRef]
- Guttmann, S.; Röcken, C.; Schmidt, M.; Grünewald, I.; Zibert, A.; Stypmann, J.; Schilling, M.; Schmidt, H. De Novo Hereditary (Familial) Amyloid Polyneuropathy (FAP) in a FAP Liver Recipient. Amyloid 2017, 24, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Misumi, Y.; Oshima, T.; Ueda, M.; Yamashita, T.; Tasaki, M.; Masuda, T.; Obayashi, K.; Ando, Y. Occurrence Factors and Clinical Picture of Iatrogenic Transthyretin Amyloidosis after Domino Liver Transplantation. Amyloid 2017, 24, 123–124. [Google Scholar] [CrossRef]
- Adams, D.; Lacroix, C.; Antonini, T.; Lozeron, P.; Denier, C.; Kreib, A.M.; Epelbaum, S.; Blandin, F.; Karam, V.; Azoulay, D.; et al. Symptomatic and Proven de Novo Amyloid Polyneuropathy in Familial Amyloid Polyneuropathy Domino Liver Recipients. Amyloid 2011, 18 (Suppl. S1), 174–177. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Yamashita, T.; Ueda, M.; Ohshima, S.; Yoneyama, K.; Nakamura, M.; Nanjo, H.; Asonuma, K.; Inomata, Y.; Watanabe, S.; et al. Iatrogenic Amyloid Neuropathy in a Japanese Patient after Sequential Liver Transplantation. Am. J. Transpl. 2006, 6, 2512–2515. [Google Scholar] [CrossRef]
- Masuda, T.; Ueda, M.; Suenaga, G.; Misumi, Y.; Tasaki, M.; Izaki, A.; Yanagisawa, Y.; Inoue, Y.; Motokawa, H.; Matsumoto, S.; et al. Early Skin Denervation in Hereditary and Iatrogenic Transthyretin Amyloid Neuropathy. Neurology 2017, 88, 2192–2197. [Google Scholar] [CrossRef]
- Ohya, Y.; Tasaki, M.; Hayashida, S.; Katayama, N.; Tsuchida, T.; Kuriwaki, K.; Ueda, M.; Inomata, Y. Carpal Tunnel Syndrome Due to Iatrogenic Amyloidosis After Domino Liver Transplantation from Hereditary Transthyretin Amyloidosis: A Case Report. Transpl. Proc. 2021, 53, 1313–1316. [Google Scholar] [CrossRef]
- Ball, H.A.; Stevens, J.; Gillmore, J.D. Peripheral Neuropathy Secondary to a “domino” Liver Transplant: A Case Report. J. Med. Case Rep. 2023, 17, 291. [Google Scholar] [CrossRef]
- Tsuda, Y.; Misumi, Y.; Ueda, M.; Tasaki, M.; Huang, G.; Masuda, T.; Suenaga, G.; Kinoshita, Y.; Obayashi, K.; Yamashita, T.; et al. Iatrogenic Systemic Transthyretin Amyloid Deposits in a Case with Domino Liver Transplantation: An Autopsy Case Study. Amyloid 2017, 24, 125. [Google Scholar] [CrossRef]
- Mnatsakanova, D.; Živković, S.A. Iatrogenic Amyloid Polyneuropathy after Domino Liver Transplantation. World J. Hepatol. 2017, 9, 126–130. [Google Scholar] [CrossRef]
- Costa, R.; Rodrigues, P.; Felix, R.; Oliveira, M.; Frias, A.; Campinas, A.; Santos, M.; Reis, H.; Torres, S. Iatrogenic Transthyretin Cardiac Amyloidosis after Sequential Liver Transplantation. Eur. Heart J. 2020, 41, ehaa946.2123. [Google Scholar] [CrossRef]
- Takei, Y.; Gono, T.; Yazaki, M.; Ikeda, S.; Ikegami, T.; Hashikura, Y.; Miyagawa, S.; Hoshii, Y. Transthyretin-Derived Amyloid Deposition on the Gastric Mucosa in Domino Recipients of Familial Amyloid Polyneuropathy Liver. Liver Transpl. 2007, 13, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.M.; Ferrão, J.; Fernandes, R.; Guimarães, A.; Geraldes, J.B.; Perdigoto, R.; Tomé, L.; Mota, O.; Negrão, L.; Furtado, A.L.; et al. Deposition and Passage of Transthyretin through the Blood-Nerve Barrier in Recipients of Familial Amyloid Polyneuropathy Livers. Lab. Investig. 2004, 84, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, T.; Yazaki, M.; Sekijima, Y.; Kametani, F.; Miyashita, K.; Hachiya, N.; Tanaka, T.; Kokudo, N.; Higuchi, K.; Ikeda, S.-I. The Pathological and Biochemical Identification of Possible Seed-Lesions of Transmitted Transthyretin Amyloidosis after Domino Liver Transplantation. J. Pathol. Clin. Res. 2016, 2, 72–79. [Google Scholar] [CrossRef]
- Morfino, P.; Aimo, A.; Vergaro, G.; Sanguinetti, C.; Castiglione, V.; Franzini, M.; Perrone, M.A.; Emdin, M. Transthyretin Stabilizers and Seeding Inhibitors as Therapies for Amyloid Transthyretin Cardiomyopathy. Pharmaceutics 2023, 15, 1129. [Google Scholar] [CrossRef] [PubMed]
- Saelices, L.; Chung, K.; Lee, J.H.; Cohn, W.; Whitelegge, J.P.; Benson, M.D.; Eisenberg, D.S. Amyloid Seeding of Transthyretin by Ex Vivo Cardiac Fibrils and Its Inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E6741–E6750. [Google Scholar] [CrossRef]
- Raimondi, S.; Mangione, P.P.; Verona, G.; Canetti, D.; Nocerino, P.; Marchese, L.; Piccarducci, R.; Mondani, V.; Faravelli, G.; Taylor, G.W.; et al. Comparative Study of the Stabilities of Synthetic in Vitro and Natural Ex Vivo Transthyretin Amyloid Fibrils. J. Biol. Chem. 2020, 295, 11379–11387. [Google Scholar] [CrossRef]
- Colon, W.; Kelly, J.W. Partial Denaturation of Transthyretin Is Sufficient for Amyloid Fibril Formation in Vitro. Biochemistry 1992, 31, 8654–8660. [Google Scholar] [CrossRef] [PubMed]
- Miroy, G.J.; Lai, Z.; Lashuel, H.A.; Peterson, S.A.; Strang, C.; Kelly, J.W. Inhibiting Transthyretin Amyloid Fibril Formation via Protein Stabilization. Proc. Natl. Acad. Sci. USA 1996, 93, 15051–15056. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Colón, W.; Kelly, J.W. The Acid-Mediated Denaturation Pathway of Transthyretin Yields a Conformational Intermediate That Can Self-Assemble into Amyloid. Biochemistry 1996, 35, 6470–6482. [Google Scholar] [CrossRef]
- Yee, A.W.; Aldeghi, M.; Blakeley, M.P.; Ostermann, A.; Mas, P.J.; Moulin, M.; De Sanctis, D.; Bowler, M.W.; Mueller-Dieckmann, C.; Mitchell, E.P.; et al. A Molecular Mechanism for Transthyretin Amyloidogenesis. Nat. Commun. 2019, 10, 925. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Wiese, S.; Adak, V.; Engler, J.; Agarwal, S.; Fritz, G.; Westermark, P.; Zacharias, M.; Fändrich, M. Cryo-EM Structure of a Transthyretin-Derived Amyloid Fibril from a Patient with Hereditary ATTR Amyloidosis. Nat. Commun. 2019, 10, 5008. [Google Scholar] [CrossRef]
- Steinebrei, M.; Baur, J.; Pradhan, A.; Kupfer, N.; Wiese, S.; Hegenbart, U.; Schönland, S.O.; Schmidt, M.; Fändrich, M. Common Transthyretin-Derived Amyloid Fibril Structures in Patients with Hereditary ATTR Amyloidosis. Nat. Commun. 2023, 14, 7623. [Google Scholar] [CrossRef] [PubMed]
- Steinebrei, M.; Gottwald, J.; Baur, J.; Röcken, C.; Hegenbart, U.; Schönland, S.; Schmidt, M. Cryo-EM Structure of an ATTRwt Amyloid Fibril from Systemic Non-Hereditary Transthyretin Amyloidosis. Nat. Commun. 2022, 13, 6398. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.A.; Singh, V.; Afrin, S.; Yakubovska, A.; Wang, L.; Ahmed, Y.; Pedretti, R.; Fernandez-Ramirez, M.D.C.; Singh, P.; Pękała, M.; et al. Structural Polymorphism of Amyloid Fibrils in ATTR Amyloidosis Revealed by Cryo-Electron Microscopy. Nat. Commun. 2024, 15, 581. [Google Scholar] [CrossRef] [PubMed]
- Shintani-Domoto, Y.; Ode, K.L.; Nomura, S.; Abe, H.; Ueda, H.R.; Sakatani, T.; Ohashi, R. Elucidation of the Mechanism of Amyloid A and Transthyretin Formation Using Mass Spectrometry-Based Absolute Quantification. Virchows Arch. 2023. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Aus Dem Siepen, F.; Donal, E.; Lairez, O.; Van Der Meer, P.; Kristen, A.V.; Mercuri, M.F.; Michalon, A.; Frost, R.J.A.; Grimm, J.; et al. Phase 1 Trial of Antibody NI006 for Depletion of Cardiac Transthyretin Amyloid. N. Engl. J. Med. 2023, 389, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
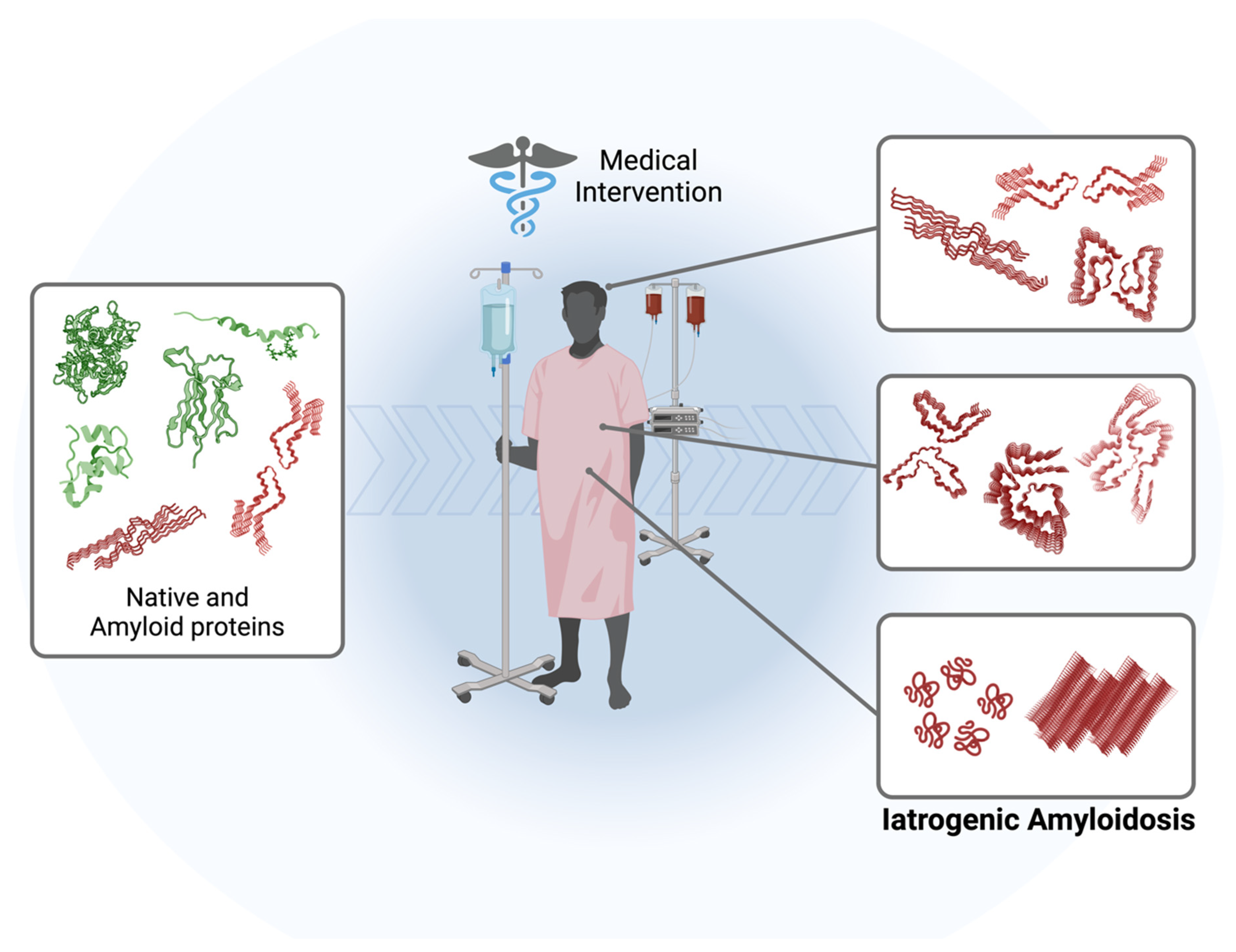
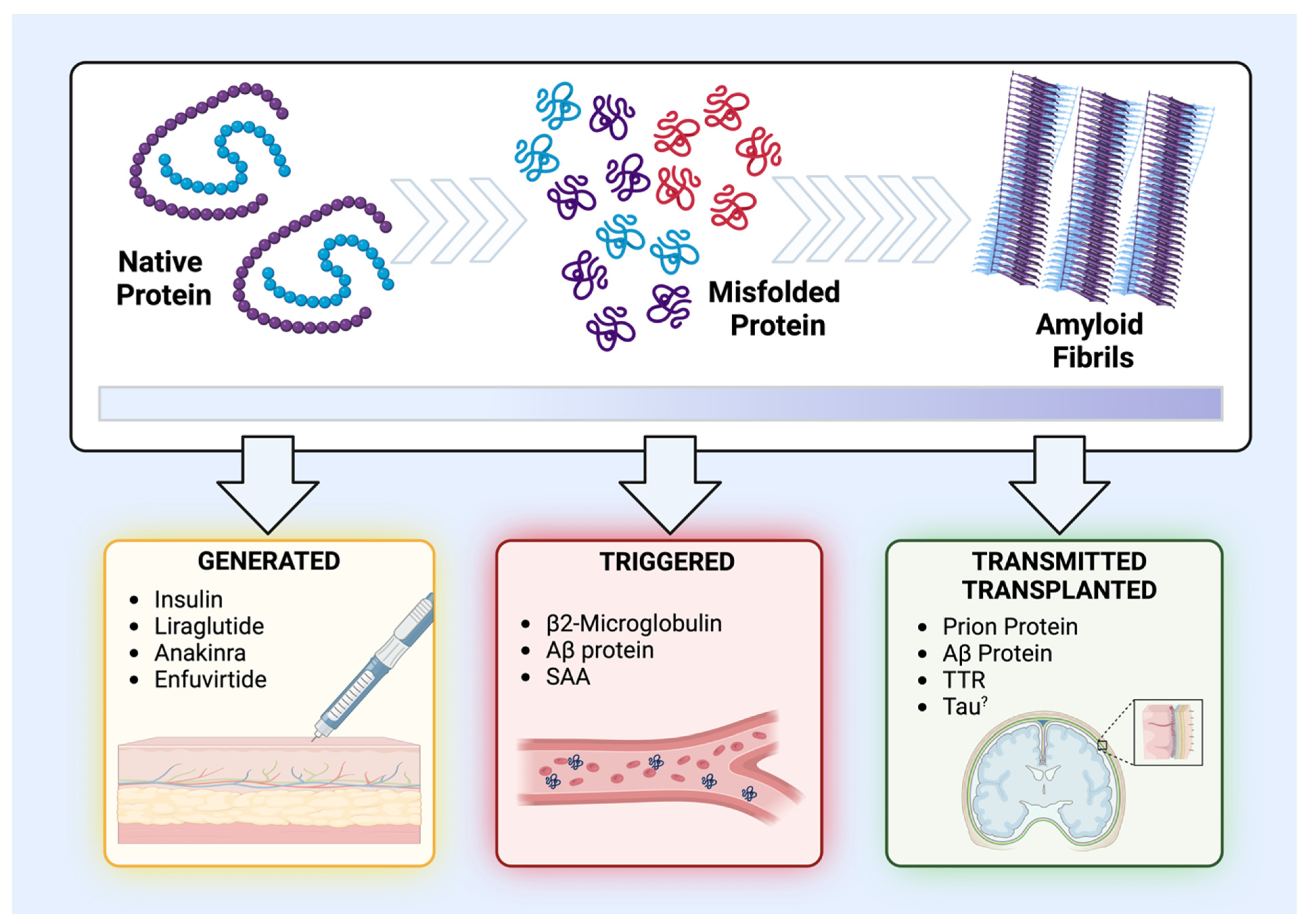








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).