
The Role of Leukemia Inhibitory Factor in Counteracting the Immunopathology of Acute and Chronic Lung Inflammatory Diseases

 , ,
, , 
{kind=link}
Abstract
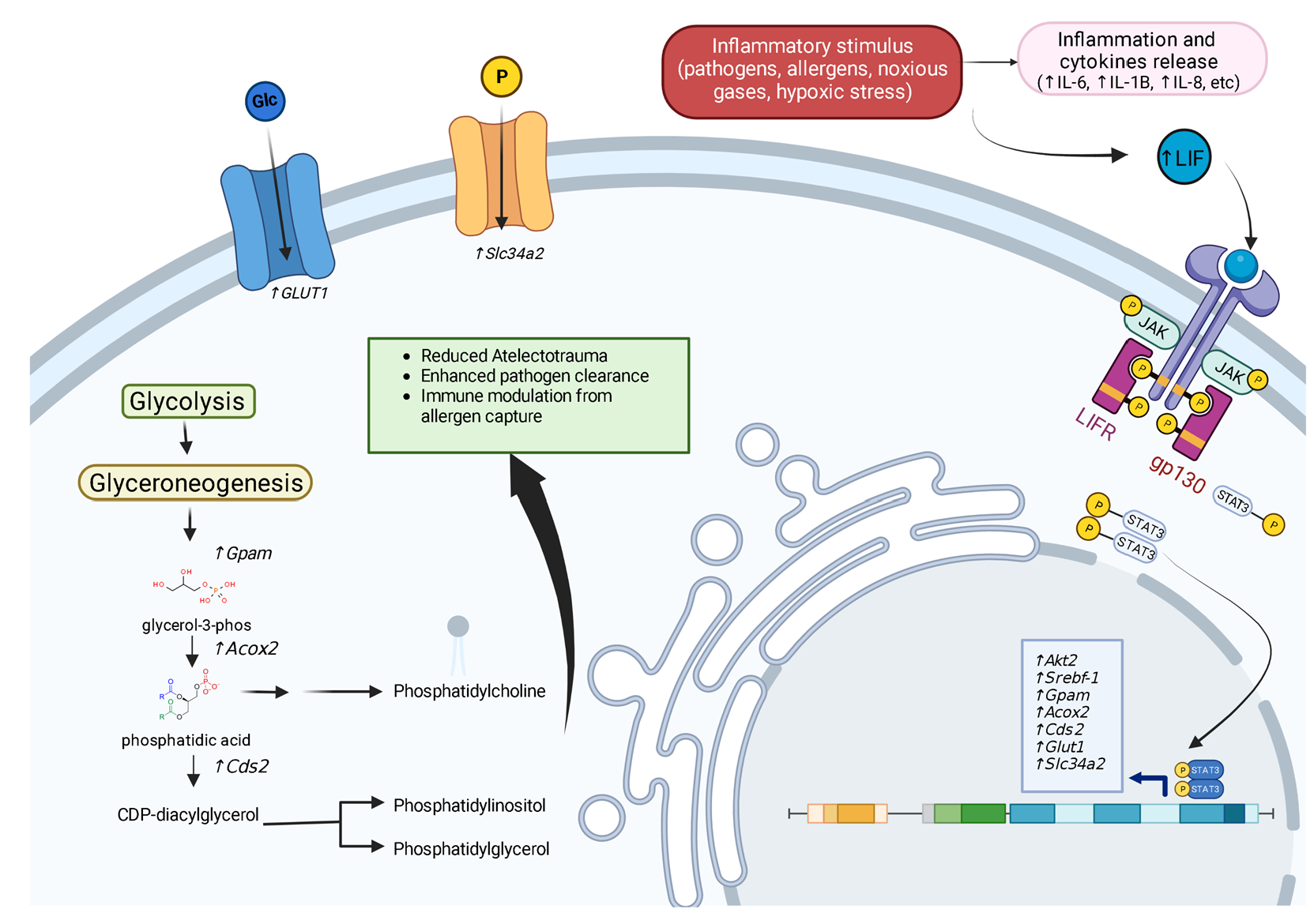
1. Introduction
2. LIF Gene Regulation
3. LIF Protein Structure
Receptor Binding
4. LIF Downstream Pathways
4.1. STAT3
4.2. PI(3)Kinase Signaling
4.3. MAPK Signaling
5. Regulators of LIF Gene Expression
5.1. Hypoxia-Inducible Factors
5.2. IL-1β and IL-6
5.3. TGF-β
5.4. NF-κB RelA
5.5. P53
5.6. Activating Transcription Factor (AT) 3
5.7. TLR5
6. LIF Signaling in Pulmonary Diseases
6.1. Non-Small Cell Lung Cancer (NSCLC)
6.2. Acute Viral Lung Injury
6.3. Acute COPD Exacerbation
6.4. Acute Respiratory Distress Syndrome (ARDS)
6.5. Idiopathic Pulmonary Fibrosis (IPF)
6.6. Asthma
7. Immune System Interaction
7.1. Promoting Surfactant Synthesis
7.2. Acute Inflammatory Response Regulation
7.3. Innate Immunity Regulation
7.4. Adaptive Immunity Regulation
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gearing, D.P.; Gough, N.M.; King, J.A.; Hilton, D.J.; Nicola, N.A.; Simpson, R.J.; Nice, E.C.; Kelso, A.; Metcalf, D. Molecular cloning and expression of cDNA encoding a murine myeloid leukaemia inhibitory factor (LIF). EMBO J. 1987, 6, 3995–4002. [Google Scholar] [CrossRef]
- Gough, N.M.; Gearing, D.P.; King, J.A.; Willson, T.A.; Hilton, D.J.; Nicola, N.A.; Metcalf, D. Molecular cloning and expression of the human homologue of the murine gene encoding myeloid leukemia-inhibitory factor. Proc. Natl. Acad. Sci. USA 1988, 85, 2623–2627. [Google Scholar] [CrossRef] [PubMed]
- Hilton, D.J.; Nicola, N.A.; Gough, N.M.; Metcalf, D. Resolution and purification of three distinct factors produced by Krebs ascites cells which have differentiation-inducing activity on murine myeloid leukemic cell lines. J. Biol. Chem. 1988, 263, 9238–9243. [Google Scholar] [CrossRef]
- Williams, R.L.; Hilton, D.J.; Pease, S.; Willson, T.A.; Stewart, C.L.; Gearing, D.P.; Wagner, E.F.; Metcalf, D.; Nicola, N.A.; Gough, N.M. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 1988, 336, 684–687. [Google Scholar] [CrossRef]
- Smith, A.G.; Heath, J.K.; Donaldson, D.D.; Wong, G.G.; Moreau, J.; Stahl, M.; Rogers, D. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature 1988, 336, 688–690. [Google Scholar] [CrossRef]
- Pease, S.; Williams, R.L. Formation of germ-line chimeras from embryonic stem cells maintained with recombinant leukemia inhibitory factor. Exp. Cell Res. 1990, 190, 209–211. [Google Scholar] [CrossRef]
- Ancey, C.; Corbi, P.; Froger, J.; Delwail, A.; Wijdenes, J.; Gascan, H.; Potreau, D.; Lecron, J.C. Secretion of IL-6, IL-11 and LIF by human cardiomyocytes in primary culture. Cytokine 2002, 18, 199–205. [Google Scholar] [CrossRef]
- Morel, D.S.; Taupin, J.L.; Potier, M.; Deminière, C.; Potaux, L.; Gualde, N.; Moreau, J.F. Renal synthesis of leukaemia inhibitory factor (LIF), under normal and inflammatory conditions. Cytokine 2000, 12, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Yu, H.; Zhao, Y.; Zhang, C.; Wang, J.; Yue, X.; Yang, Q.; Hu, W. HIF-2α mediates hypoxia-induced LIF expression in human colorectal cancer cells. Oncotarget 2015, 6, 4406–4417. [Google Scholar] [CrossRef] [PubMed]
- Felling, R.J.; Covey, M.V.; Wolujewicz, P.; Batish, M.; Levison, S.W. Astrocyte-produced leukemia inhibitory factor expands the neural stem/progenitor pool following perinatal hypoxia-ischemia. J. Neurosci. Res. 2016, 94, 1531–1545. [Google Scholar] [CrossRef]
- Ulich, T.R.; Fann, M.J.; Patterson, P.H.; Williams, J.H.; Samal, B.; Del Castillo, J.; Yin, S.; Guo, K.; Remick, D.G. Intratracheal injection of LPS and cytokines. V. LPS induces expression of LIF and LIF inhibits acute inflammation. Am. J. Physiol. 1994, 267, L442–L446. [Google Scholar] [CrossRef]
- Foronjy, R.F.; Dabo, A.J.; Cummins, N.; Geraghty, P. Leukemia inhibitory factor protects the lung during respiratory syncytial viral infection. BMC Immunol. 2014, 15, 41. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, Q.; Corne, J.; Zhu, Z.; Lee, C.G.; Bhandari, V.; Homer, R.J.; Elias, J.A. Pulmonary expression of leukemia inhibitory factor induces B cell hyperplasia and confers protection in hyperoxia. J. Biol. Chem. 2003, 278, 31226–31232. [Google Scholar] [CrossRef]
- Quinton, L.J.; Mizgerd, J.P.; Hilliard, K.L.; Jones, M.R.; Kwon, C.Y.; Allen, E. Leukemia inhibitory factor signaling is required for lung protection during pneumonia. J. Immunol. 2012, 188, 6300–6308. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, P.; Wyman, A.E.; Garcia-Arcos, I.; Dabo, A.J.; Gadhvi, S.; Foronjy, R. STAT3 modulates cigarette smoke-induced inflammation and protease expression. Front. Physiol. 2013, 4, 267. [Google Scholar] [CrossRef] [PubMed]
- Nogueira-Silva, C.; Piairo, P.; Carvalho-Dias, E.; Peixoto, F.O.; Moura, R.S.; Correia-Pinto, J. Leukemia inhibitory factor in rat fetal lung development: Expression and functional studies. PLoS ONE 2012, 7, e30517. [Google Scholar] [CrossRef]
- Gough, N.M.; Wilson, T.A.; Stahl, J.; Brown, M.A. Molecular biology of the leukaemia inhibitory factor gene. Ciba Found. Symp. 1992, 167, 24–38; discussion 38–46. [Google Scholar]
- Taupin, J.L.; Pitard, V.; Dechanet, J.; Miossec, V.; Gualde, N.; Moreau, J.F. Leukemia inhibitory factor: Part of a large ingathering family. Int. Rev. Immunol. 1998, 16, 397–426. [Google Scholar] [CrossRef]
- Rose, T.M.; Lagrou, M.J.; Fransson, I.; Werelius, B.; Delattre, O.; Thomas, G.; de Jong, P.J.; Todaro, G.J.; Dumanski, J.P. The genes for oncostatin M (OSM) and leukemia inhibitory factor (LIF) are tightly linked on human chromosome 22. Genomics 1993, 17, 136–140. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. In Epigenetics; Allis, C.D., Jenuwein, T., Reinberg, D., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2007; pp. 457–476. [Google Scholar]
- Shin, J.E.; Park, S.H.; Jang, Y.K. Epigenetic up-regulation of leukemia inhibitory factor (LIF) gene during the progression to breast cancer. Mol. Cells 2011, 31, 181–189. [Google Scholar] [CrossRef]
- Voyle, R.B.; Haines, B.P.; Pera, M.F.; Forrest, R.; Rathjen, P.D. Human germ cell tumor cell lines express novel leukemia inhibitory factor transcripts encoding differentially localized proteins. Exp. Cell Res. 1999, 249, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Haines, B.P.; Voyle, R.B.; Pelton, T.A.; Forrest, R.; Rathjen, P.D. Complex conserved organization of the mammalian leukemia inhibitory factor gene: Regulated expression of intracellular and extracellular cytokines. J. Immunol. 1999, 162, 4637–4646. [Google Scholar] [CrossRef] [PubMed]
- Haines, B.P.; Voyle, R.B.; Rathjen, P.D. Intracellular and extracellular leukemia inhibitory factor proteins have different cellular activities that are mediated by distinct protein motifs. Mol. Biol. Cell 2000, 11, 1369–1383. [Google Scholar] [CrossRef]
- Albrengues, J.; Bourget, I.; Pons, C.; Butet, V.; Hofman, P.; Tartare-Deckert, S.; Feral, C.C.; Meneguzzi, G.; Gaggioli, C. LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell Rep. 2014, 7, 1664–1678. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, J.R.; Freeman, T.C.; Stephens, R.J.; Kimber, S.; Smith, A.G.; Chambers, I.; Smith, S.K.; Sharkey, A.M. Identification of genes regulated by leukemia-inhibitory factor in the mouse uterus at the time of implantation. Mol. Endocrinol. 2004, 18, 2185–2195. [Google Scholar] [CrossRef]
- Nicola, N.A.; Babon, J.J. Leukemia inhibitory factor (LIF). Cytokine Growth Factor Rev. 2015, 26, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.C.; Grey, L.M.; Staunton, D.; Vankelecom, H.; Vernallis, A.B.; Moreau, J.F.; Stuart, D.I.; Heath, J.K.; Jones, E.Y. The crystal structure and biological function of leukemia inhibitory factor: Implications for receptor binding. Cell 1994, 77, 1101–1116. [Google Scholar] [CrossRef]
- Hinds, M.G.; Maurer, T.; Zhang, J.G.; Nicola, N.A.; Norton, R.S. Solution structure of leukemia inhibitory factor. J. Biol. Chem. 1998, 273, 13738–13745. [Google Scholar] [CrossRef]
- Boulanger, M.J.; Bankovich, A.J.; Kortemme, T.; Baker, D.; Garcia, K.C. Convergent mechanisms for recognition of divergent cytokines by the shared signaling receptor gp130. Mol. Cell 2003, 12, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Skiniotis, G.; Lupardus, P.J.; Martick, M.; Walz, T.; Garcia, K.C. Structural organization of a full-length gp130/LIF-R cytokine receptor transmembrane complex. Mol. Cell 2008, 31, 737–748. [Google Scholar] [CrossRef]
- Stahl, N.; Boulton, T.G.; Farruggella, T.; Ip, N.Y.; Davis, S.; Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Barbieri, G.; Pellegrini, S.; et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science 1994, 263, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Hilton, D.J.; Nicola, N.A. Kinetic analyses of the binding of leukemia inhibitory factor to receptor on cells and membranes and in detergent solution. J. Biol. Chem. 1992, 267, 10238–10247. [Google Scholar] [CrossRef] [PubMed]
- Hilton, D.J. LIF: Lots of interesting functions. Trends Biochem. Sci. 1992, 17, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Thoma, B.; Bird, T.A.; Friend, D.J.; Gearing, D.P.; Dower, S.K. Oncostatin M and leukemia inhibitory factor trigger overlapping and different signals through partially shared receptor complexes. J. Biol. Chem. 1994, 269, 6215–6222. [Google Scholar] [CrossRef]
- Oh, H.; Fujio, Y.; Kunisada, K.; Hirota, H.; Matsui, H.; Kishimoto, T.; Yamauchi-Takihara, K. Activation of phosphatidylinositol 3-kinase through glycoprotein 130 induces protein kinase B and p70 S6 kinase phosphorylation in cardiac myocytes. J. Biol. Chem. 1998, 273, 9703–9710. [Google Scholar] [CrossRef]
- Stahl, N.; Farruggella, T.J.; Boulton, T.G.; Zhong, Z.; Darnell, J.E., Jr.; Yancopoulos, G.D. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science 1995, 267, 1349–1353. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Raz, R.; Lee, C.K.; Cannizzaro, L.A.; d’Eustachio, P.; Levy, D.E. Essential role of STAT3 for embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 1999, 96, 2846–2851. [Google Scholar] [CrossRef]
- Niwa, H.; Burdon, T.; Chambers, I.; Smith, A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998, 12, 2048–2060. [Google Scholar] [CrossRef]
- Haan, S.; Kortylewski, M.; Behrmann, I.; Müller-Esterl, W.; Heinrich, P.C.; Schaper, F. Cytoplasmic STAT proteins associate prior to activation. Biochem. J. 2000, 345 Pt 3, 417–421. [Google Scholar] [CrossRef]
- Hilton, D.J.; Richardson, R.T.; Alexander, W.S.; Viney, E.M.; Willson, T.A.; Sprigg, N.S.; Starr, R.; Nicholson, S.E.; Metcalf, D.; Nicola, N.A. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc. Natl. Acad. Sci. USA 1998, 95, 114–119. [Google Scholar] [CrossRef]
- Starr, R.; Willson, T.A.; Viney, E.M.; Murray, L.J.; Rayner, J.R.; Jenkins, B.J.; Gonda, T.J.; Alexander, W.S.; Metcalf, D.; Nicola, N.A.; et al. A family of cytokine-inducible inhibitors of signalling. Nature 1997, 387, 917–921. [Google Scholar] [CrossRef]
- Naka, T.; Narazaki, M.; Hirata, M.; Matsumoto, T.; Minamoto, S.; Aono, A.; Nishimoto, N.; Kajita, T.; Taga, T.; Yoshizaki, K.; et al. Structure and function of a new STAT-induced STAT inhibitor. Nature 1997, 387, 924–929. [Google Scholar] [CrossRef]
- Kershaw, N.J.; Laktyushin, A.; Nicola, N.A.; Babon, J.J. Reconstruction of an active SOCS3-based E3 ubiquitin ligase complex in vitro: Identification of the active components and JAK2 and gp130 as substrates. Growth Factors 2014, 32, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Weissenbach, M.; Haan, S.; Heinrich, P.C.; Schaper, F. SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J. Biol. Chem. 2000, 275, 12848–12856. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Nakamura, T.; Nakao, K.; Arai, T.; Katsuki, M.; Heike, T.; Yokota, T. STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J. 1999, 18, 4261–4269. [Google Scholar] [CrossRef]
- Poon, J.; Campos, M.; Foronjy, R.F.; Nath, S.; Gupta, G.; Railwah, C.; Dabo, A.J.; Baumlin, N.; Salathe, M.; Geraghty, P. Cigarette smoke exposure reduces leukemia inhibitory factor levels during respiratory syncytial viral infection. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 1305–1315. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Paling, N.R.; Wheadon, H.; Bone, H.K.; Welham, M.J. Regulation of embryonic stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J. Biol. Chem. 2004, 279, 48063–48070. [Google Scholar] [CrossRef]
- Aros, C.J.; Pantoja, C.J.; Gomperts, B.N. Wnt signaling in lung development, regeneration, and disease progression. Commun. Biol. 2021, 4, 601. [Google Scholar] [CrossRef]
- Plate, M.; Guillotin, D.; Chambers, R.C. The promise of mTOR as a therapeutic target pathway in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2020, 29, 200269. [Google Scholar] [CrossRef]
- Moradi, S.; Jarrahi, E.; Ahmadi, A.; Salimian, J.; Karimi, M.; Zarei, A.; Azimzadeh Jamalkandi, S.; Ghanei, M. PI3K signalling in chronic obstructive pulmonary disease and opportunities for therapy. J. Pathol. 2021, 254, 505–518. [Google Scholar] [CrossRef]
- Schiemann, W.P.; Bartoe, J.L.; Nathanson, N.M. Box 3-independent signaling mechanisms are involved in leukemia inhibitory factor receptor alpha- and gp130-mediated stimulation of mitogen-activated protein kinase. Evidence for participation of multiple signaling pathways which converge at Ras. J. Biol. Chem. 1997, 272, 16631–16636. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.G.; Gu, H.; Pao, L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. [Google Scholar] [CrossRef]
- Meloche, S.; Vella, F.D.; Voisin, L.; Ang, S.L.; Saba-El-Leil, M. Erk2 signaling and early embryo stem cell self-renewal. Cell Cycle 2004, 3, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Burdon, T.; Stracey, C.; Chambers, I.; Nichols, J.; Smith, A. Suppression of SHP-2 and ERK signalling promotes self-renewal of mouse embryonic stem cells. Dev. Biol. 1999, 210, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Robb, L.; Rakar, S.; Hartley, L.; Cluse, L.; Nicola, N.A.; Metcalf, D.; Hilton, D.J.; Alexander, W.S. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc. Natl. Acad. Sci. USA 2001, 98, 9324–9329. [Google Scholar] [CrossRef]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.A.; Lydell, C.P.; Zhou, D.; Weir, T.D.; Robert Schellenberg, R.; Bai, T.R. Leukemia inhibitory factor (LIF) and LIF receptor in human lung. Distribution and regulation of LIF release. Am. J. Respir. Cell Mol. Biol. 1999, 20, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, I.; Nakane, A.; Kurihara, K. Induction of LIF-mRNA by TGF-beta 1 in Schwann cells. Brain Res. 1997, 776, 170–180. [Google Scholar] [CrossRef]
- Peñuelas, S.; Anido, J.; Prieto-Sánchez, R.M.; Folch, G.; Barba, I.; Cuartas, I.; García-Dorado, D.; Poca, M.A.; Sahuquillo, J.; Baselga, J.; et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell 2009, 15, 315–327. [Google Scholar] [CrossRef]
- Wetzler, M.; Talpaz, M.; Lowe, D.G.; Baiocchi, G.; Gutterman, J.U.; Kurzrock, R. Constitutive expression of leukemia inhibitory factor RNA by human bone marrow stromal cells and modulation by IL-1, TNF-alpha, and TGF-beta. Exp. Hematol. 1991, 19, 347–351. [Google Scholar]
- Carlson, C.D.; Bai, Y.; Jonakait, G.M.; Hart, R.P. Interleukin-1 beta increases leukemia inhibitory factor mRNA levels through transient stimulation of transcription rate. Glia 1996, 18, 141–151. [Google Scholar] [CrossRef]
- Jorens, P.G.; De Jongh, R.; Bossaert, L.L.; De Backer, W.; Herman, A.G.; Pollet, H.; Bosmans, E.; Taupin, J.L.; Moreau, J.F. High levels of leukaemia inhibitory factor in ARDS. Cytokine 1996, 8, 873–876. [Google Scholar] [CrossRef]
- Traber, K.E.; Symer, E.M.; Allen, E.; Kim, Y.; Hilliard, K.L.; Wasserman, G.A.; Stewart, C.L.; Jones, M.R.; Mizgerd, J.P.; Quinton, L.J. Myeloid-epithelial cross talk coordinates synthesis of the tissue-protective cytokine leukemia inhibitory factor during pneumonia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L548–L558. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ferrari, J.D.; Cao, Y.; Ramirez, M.I.; Jones, M.R.; Quinton, L.J.; Mizgerd, J.P. Type I alveolar epithelial cells mount innate immune responses during pneumococcal pneumonia. J. Immunol. 2012, 189, 2450–2459. [Google Scholar] [CrossRef]
- Hu, W.; Feng, Z.; Teresky, A.K.; Levine, A.J. p53 regulates maternal reproduction through LIF. Nature 2007, 450, 721–724. [Google Scholar] [CrossRef]
- Cheng, X.; Liu, J.; Shan, H.; Sun, L.; Huang, C.; Yan, Q.; Jiang, R.; Ding, L.; Jiang, Y.; Zhou, J.; et al. Activating transcription factor 3 promotes embryo attachment via up-regulation of leukemia inhibitory factor in vitro. Reprod. Biol. Endocrinol. 2017, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Terawaki, K.; Kashiwase, Y.; Uzu, M.; Nonaka, M.; Sawada, Y.; Miyano, K.; Higami, Y.; Yanagihara, K.; Yamamoto, M.; Uezono, Y. Leukemia inhibitory factor via the Toll-like receptor 5 signaling pathway involves aggravation of cachexia induced by human gastric cancer-derived 85As2 cells in rats. Oncotarget 2018, 9, 34748–34764. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Galvez, R.; Fleurot, I.; Chamero, P.; Trapp, S.; Olivier, M.; Chevaleyre, C.; Barc, C.; Riou, M.; Rossignol, C.; Guillon, A.; et al. Airway Administration of Flagellin Regulates the Inflammatory Response to Pseudomonas aeruginosa. Am. J. Respir. Cell Mol. Biol. 2021, 65, 378–389. [Google Scholar] [CrossRef]
- Wang, H.; Si, S.; Jiang, M.; Chen, L.; Huang, K.; Yu, W. Leukemia inhibitory factor is involved in the pathogenesis of NSCLC through activation of the STAT3 signaling pathway. Oncol. Lett. 2021, 22, 663. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gandler, H.I.; Tosic, I.; Ye, D.Q.; Giaccone, Z.T.; Frank, D.A. Mutant KRAS Downregulates the Receptor for Leukemia Inhibitory Factor (LIF) to Enhance a Signature of Glycolysis in Pancreatic Cancer and Lung Cancer. Mol. Cancer Res. 2021, 19, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Haura, E.B.; Zheng, Z.; Song, L.; Cantor, A.; Bepler, G. Activated epidermal growth factor receptor-Stat-3 signaling promotes tumor survival in vivo in non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 8288–8294. [Google Scholar] [CrossRef]
- Sanchez-Ceja, S.G.; Reyes-Maldonado, E.; Vazquez-Manriquez, M.E.; Lopez-Luna, J.J.; Belmont, A.; Gutierrez-Castellanos, S. Differential expression of STAT5 and Bcl-xL, and high expression of Neu and STAT3 in non-small-cell lung carcinoma. Lung Cancer 2006, 54, 163–168. [Google Scholar] [CrossRef]
- Borazanci, E.; Schram, A.M.; Garralda, E.; Brana, I.; Vieito Villar, M.; Spreafico, A.; Oliva, M.; Lakhani, N.J.; Hoffman, K.; Hallett, R.M.; et al. Phase I, first-in-human study of MSC-1 (AZD0171), a humanized anti-leukemia inhibitory factor monoclonal antibody, for advanced solid tumors. ESMO Open 2022, 7, 100530. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.M.; Collier, L.A.; Winford, E.D.; Leonardo, C.C.; Ajmo, C.T., Jr.; Foran, E.A.; Kopper, T.J.; Gensel, J.C.; Pennypacker, K.R. Leukemia inhibitory factor modulates the peripheral immune response in a rat model of emergent large vessel occlusion. J. Neuroinflammation 2018, 15, 288. [Google Scholar] [CrossRef]
- Jeannin, P.; Duluc, D.; Delneste, Y. IL-6 and leukemia-inhibitory factor are involved in the generation of tumor-associated macrophage: Regulation by IFN-gamma. Immunotherapy 2011, 3, 23–26. [Google Scholar] [CrossRef]
- Yue, X.; Zhao, Y.; Zhang, C.; Li, J.; Liu, Z.; Liu, J.; Hu, W. Leukemia inhibitory factor promotes EMT through STAT3-dependent miR-21 induction. Oncotarget 2016, 7, 3777–3790. [Google Scholar] [CrossRef]
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef]
- Foronjy, R.F.; Dabo, A.J.; Taggart, C.C.; Weldon, S.; Geraghty, P. Respiratory syncytial virus infections enhance cigarette smoke induced COPD in mice. PLoS ONE 2014, 9, e90567. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.L.; Kaspar, P.; Brunet, L.J.; Bhatt, H.; Gadi, I.; Kontgen, F.; Abbondanzo, S.J. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature 1992, 359, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Gruson, D.; Hilbert, G.; Juzan, M.; Taupin, J.L.; Coulon, V.; Moreau, J.F.; Gualde, N.; Gbikpi-Benissan, G. Sequential production of leukaemia inhibitory factor by blood cell culture in patients with ARDS. Intensive Care Med. 1998, 24, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Na, E.; Allen, E.; Baird, L.A.; Odom, C.V.; Korkmaz, F.T.; Shenoy, A.T.; Matschulat, A.M.; Jones, M.R.; Kotton, D.N.; Mizgerd, J.P.; et al. Epithelial LIF signaling limits apoptosis and lung injury during bacterial pneumonia. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2022, 322, L550–L563. [Google Scholar] [CrossRef]
- Chen, X.; Qi, D.; Fan, S.; He, Y.; Jing, H.; Wang, D. Interferon regulatory factor 1 (IRF1) inhibits lung endothelial regeneration following inflammation-induced acute lung injury. Clin. Sci. 2023, 137, 367–383. [Google Scholar] [CrossRef]
- Xu, S.; Yang, X.; Chen, Q.; Liu, Z.; Chen, Y.; Yao, X.; Xiao, A.; Tian, J.; Xie, L.; Zhou, M.; et al. Leukemia inhibitory factor is a therapeutic target for renal interstitial fibrosis. EBioMedicine 2022, 86, 104312. [Google Scholar] [CrossRef]
- Leach, H.G.; Chrobak, I.; Han, R.; Trojanowska, M. Endothelial cells recruit macrophages and contribute to a fibrotic milieu in bleomycin lung injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ncbi.nlm.nih.gov/gene/442898 (accessed on 10 May 2023).
- Li, R.; Wang, Y.; Song, X.; Sun, W.; Zhang, J.; Liu, Y.; Li, H.; Meng, C.; Zhang, J.; Zheng, Q.; et al. Potential regulatory role of circular RNA in idiopathic pulmonary fibrosis. Int. J. Mol. Med. 2018, 42, 3256–3268. [Google Scholar] [CrossRef]
- Zheng, X.; Knight, D.A.; Zhou, D.; Weir, T.; Peacock, C.; Schellenberg, R.R.; Bai, T.R. Leukemia inhibitory factor is synthesized and released by human eosinophils and modulates activation state and chemotaxis. J. Allergy Clin. Immunol. 1999, 104, 136–144. [Google Scholar] [CrossRef]
- Bjorkander, S.; Klevebro, S.; Hernandez-Pacheco, N.; Kere, M.; Ekstrom, S.; Sparreman Mikus, M.; van Hage, M.; James, A.; Kull, I.; Bergstrom, A.; et al. Obese asthma phenotypes display distinct plasma biomarker profiles. Clin. Transl. Allergy 2023, 13, e12238. [Google Scholar] [CrossRef]
- Kurz, T.; Hoffjan, S.; Hayes, M.G.; Schneider, D.; Nicolae, R.; Heinzmann, A.; Jerkic, S.P.; Parry, R.; Cox, N.J.; Deichmann, K.A.; et al. Fine mapping and positional candidate studies on chromosome 5p13 identify multiple asthma susceptibility loci. J. Allergy Clin. Immunol. 2006, 118, 396–402. [Google Scholar] [CrossRef]
- Xiong, W.; Zeng, D.; Xu, Y.; Xiong, S.; Fang, H.; Cao, Y.; Song, Q.; Cao, C. Expression of leukemia inhibitory factor in airway epithelial tissue of asthmatic rats. J. Huazhong Univ. Sci. Technol. 2007, 27, 372–374. [Google Scholar] [CrossRef]
- Lin, M.J.; Lao, X.J.; Liu, S.M.; Xu, Z.H.; Zou, W.F. Leukemia inhibitory factor in the neuroimmune communication pathways in allergic asthma. Neurosci. Lett. 2014, 563, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Chetty, A.; Sharda, A.; Warburton, R.; Weinberg, E.O.; Dong, J.; Fang, M.; Sahagian, G.G.; Chen, T.; Xue, C.; Castellot, J.J.; et al. A purinergic P2Y6 receptor agonist prodrug modulates airway inflammation, remodeling, and hyperreactivity in a mouse model of asthma. J. Asthma Allergy 2018, 11, 159–171. [Google Scholar] [CrossRef]
- Abdoli, Z.; Assarehzadegan, M.A.; Pipelzadeh, M.H.; Iranparast, S.; Dashti Gerdabi, N.; Parsanahad, M.; Khodadadi, A. Leukemia Inhibitory Factor Suppresses NKG2D mRNA Expression and Presentation on Human Natural Killer Cells. Iran. J. Allergy Asthma Immunol. 2021, 20, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Fayon, M.; Rebola, M.; Berger, P.; Daburon, S.; Ousova, O.; Lavrand, F.; Moukaila, B.; Pujol, W.; Taupin, J.L.; Labbe, A.; et al. Increased secretion of leukemia inhibitory factor by immature airway smooth muscle cells enhances intracellular signaling and airway contractility. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L244–L251. [Google Scholar] [CrossRef] [PubMed]
- Veldhuizen, R.A.; McCaig, L.A.; Akino, T.; Lewis, J.F. Pulmonary surfactant subfractions in patients with the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1995, 152, 1867–1871. [Google Scholar] [CrossRef] [PubMed]
- Hokuto, I.; Ikegami, M.; Yoshida, M.; Takeda, K.; Akira, S.; Perl, A.K.; Hull, W.M.; Wert, S.E.; Whitsett, J.A. Stat-3 is required for pulmonary homeostasis during hyperoxia. J. Clin. Investig. 2004, 113, 28–37. [Google Scholar] [CrossRef]
- Yan, C.; Naltner, A.; Martin, M.; Naltner, M.; Fangman, J.M.; Gurel, O. Transcriptional stimulation of the surfactant protein B gene by STAT3 in respiratory epithelial cells. J. Biol. Chem. 2002, 277, 10967–10972. [Google Scholar] [CrossRef]
- Ladenburger, A.; Seehase, M.; Kramer, B.W.; Thomas, W.; Wirbelauer, J.; Speer, C.P.; Kunzmann, S. Glucocorticoids potentiate IL-6-induced SP-B expression in H441 cells by enhancing the JAK-STAT signaling pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L578–L584. [Google Scholar] [CrossRef]
- Ikegami, M.; Falcone, A.; Whitsett, J.A. STAT-3 regulates surfactant phospholipid homeostasis in normal lung and during endotoxin-mediated lung injury. J. Appl. Physiol. 2008, 104, 1753–1760. [Google Scholar] [CrossRef]
- Matsuzaki, Y.; Xu, Y.; Ikegami, M.; Besnard, V.; Park, K.S.; Hull, W.M.; Wert, S.E.; Whitsett, J.A. Stat3 is required for cytoprotection of the respiratory epithelium during adenoviral infection. J. Immunol. 2006, 177, 527–537. [Google Scholar] [CrossRef]
- Lian, X.; Qin, Y.; Hossain, S.A.; Yang, L.; White, A.; Xu, H.; Shipley, J.M.; Li, T.; Senior, R.M.; Du, H.; et al. Overexpression of Stat3C in pulmonary epithelium protects against hyperoxic lung injury. J. Immunol. 2005, 174, 7250–7256. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Chakravarty, S.D.; Ivashkiv, L.B. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol. Rev. 2008, 226, 41–56. [Google Scholar] [CrossRef]
- Dallagi, A.; Girouard, J.; Hamelin-Morrissette, J.; Dadzie, R.; Laurent, L.; Vaillancourt, C.; Lafond, J.; Carrier, C.; Reyes-Moreno, C. The activating effect of IFN-γ on monocytes/macrophages is regulated by the LIF-trophoblast-IL-10 axis via Stat1 inhibition and Stat3 activation. Cell. Mol. Immunol. 2015, 12, 326–341. [Google Scholar] [CrossRef]
- Lee, J.W.; Chun, W.; Lee, H.J.; Min, J.H.; Kim, S.M.; Seo, J.Y.; Ahn, K.S.; Oh, S.R. The Role of Macrophages in the Development of Acute and Chronic Inflammatory Lung Diseases. Cells 2021, 10, 897. [Google Scholar] [CrossRef]
- Shen, M.M.; Skoda, R.C.; Cardiff, R.D.; Campos-Torres, J.; Leder, P.; Ornitz, D.M. Expression of LIF in transgenic mice results in altered thymic epithelium and apparent interconversion of thymic and lymph node morphologies. EMBO J. 1994, 13, 1375–1385. [Google Scholar] [CrossRef]
- Gao, W.; Thompson, L.; Zhou, Q.; Putheti, P.; Fahmy, T.M.; Strom, T.B.; Metcalfe, S.M. Treg versus Th17 lymphocyte lineages are cross-regulated by LIF versus IL-6. Cell Cycle 2009, 8, 1444–1450. [Google Scholar] [CrossRef]
- Hu, Y.; Long, H.; Cao, Y.; Guo, Y. Prognostic value of lymphocyte count for in-hospital mortality in patients with severe AECOPD. BMC Pulm. Med. 2022, 22, 376. [Google Scholar] [CrossRef]
- Moon, S.W.; Leem, A.Y.; Kim, Y.S.; Lee, J.H.; Kim, T.H.; Oh, Y.M.; Shin, H.; Chang, J.; Jung, J.Y.; Group, K.S. Low serum lymphocyte level is associated with poor exercise capacity and quality of life in chronic obstructive pulmonary disease. Sci. Rep. 2020, 10, 11700. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Todd, I.; Fairclough, L.C. The role of CD8 + T lymphocytes in chronic obstructive pulmonary disease: A systematic review. Inflamm. Res. 2021, 70, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Ai, X.; Liao, Z.; You, C.; Cheng, Y. The prognostic values of neutrophil to lymphocyte ratio for outcomes in chronic obstructive pulmonary disease. Medicine 2019, 98, e16371. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, J.; Matthay, M.A. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu. Rev. Physiol. 2013, 75, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Tapia, A.; Salamonsen, L.A.; Manuelpillai, U.; Dimitriadis, E. Leukemia inhibitory factor promotes human first trimester extravillous trophoblast adhesion to extracellular matrix and secretion of tissue inhibitor of metalloproteinases-1 and -2. Hum. Reprod. 2008, 23, 1724–1732. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Zaveri, S.; Pillai, M.; Taluru, H.; Schaible, M.; Chaddha, S.; Ahmed, A.; Tfaili, S.; Geraghty, P. The Role of Leukemia Inhibitory Factor in Counteracting the Immunopathology of Acute and Chronic Lung Inflammatory Diseases. J. Respir. 2023, 3, 86-100. https://doi.org/10.3390/jor3020009
Yu H, Zaveri S, Pillai M, Taluru H, Schaible M, Chaddha S, Ahmed A, Tfaili S, Geraghty P. The Role of Leukemia Inhibitory Factor in Counteracting the Immunopathology of Acute and Chronic Lung Inflammatory Diseases. Journal of Respiration. 2023; 3(2):86-100. https://doi.org/10.3390/jor3020009
Chicago/Turabian StyleYu, Howard, Sahil Zaveri, Meshach Pillai, Harsha Taluru, Michael Schaible, Sahil Chaddha, Asad Ahmed, Said Tfaili, and Patrick Geraghty. 2023. "The Role of Leukemia Inhibitory Factor in Counteracting the Immunopathology of Acute and Chronic Lung Inflammatory Diseases" Journal of Respiration 3, no. 2: 86-100. https://doi.org/10.3390/jor3020009
APA StyleYu, H., Zaveri, S., Pillai, M., Taluru, H., Schaible, M., Chaddha, S., Ahmed, A., Tfaili, S., & Geraghty, P. (2023). The Role of Leukemia Inhibitory Factor in Counteracting the Immunopathology of Acute and Chronic Lung Inflammatory Diseases. Journal of Respiration, 3(2), 86-100. https://doi.org/10.3390/jor3020009