Abstract
RAGE (receptor for advanced glycation end-products) represents a class of multi-ligand pattern recognition receptors highly expressed in the vertebrate lung. Our previous work demonstrated unique patterns of RAGE expression in the developing murine lung and regulation by key transcription factors including NKX2.1 and FoxA2. The current investigation employed conditional lung-specific upregulation via a TetOn transgenic mouse model (RAGE TG) and nontransgenic controls. RAGE expression was induced in RAGE TG mice throughout gestation (embryonic day, E0-E18.5) or from E15.5-E18.5 and compared to age-matched controls. High-resolution respirometry was used to assess mitochondrial respiration and context was provided by quantifying ATP and reactive oxygen species (ROS) generation. Lung lysates were also screened by immunoblotting for MAPK/PI3K signaling intermediates. RAGE upregulation increased mitochondrial oxygen consumption in the E0-E18.5 and E15.5-E18.5 groups compared to controls. RAGE TG mice also had increased ATP concentrations, which persisted even after controlling for oxygen consumption. In contrast, ROS generation was diminished in RAGE TG animals compared to controls. Lastly, in both RAGE TG groups, pERK and pp38 were significantly decreased, whereas pAKT was significantly elevated, suggesting that RAGE signaling is likely perpetuated via pAKT pathways. Together, these data demonstrate that despite lung hypoplasia in RAGE TG mice, the remaining tissue experiences a favorable shift in mitochondrial bioenergetics without excessive redox assault and a preference for AKT signaling over ERK or p38.
1. Introduction
The development of pulmonary tissue is governed by a highly coordinated system that relies upon a genetically controlled environment. Classically, pulmonary development is organized into five major stages: embryonic, embryonic day 9.5 (E9.5) to E11.5; pseudoglandular, E11.5-E16.6; canalicular, E16.6-E17.4; saccular, E17.4-postnatal day 5 (PN)5; and alveolar PN5-PN30 [1]. The embryonic period encompasses the initiation of lung tissue and the creation of lung buds that branch to form lobar bronchi. The pseudoglandular period encompasses comprehensive branching and the additional generation of the peripheral airways. The canalicular phase of development is a period of pronounced distal airway cell differentiation and the culmination of pulmonary branching morphogenesis. The saccular phase coincides with the expansion of airspaces, enabling proper gas exchange. When developmental stages are altered due to developmental errors stemming from genetic abnormalities or exposure to exogenous stimuli, impaired branching may lead to pulmonary simplification and cell type dysplasia coincident with numerous disease states.
The receptor for advanced glycation end products (RAGE) is a multiligand cell surface receptor primarily expressed within lung tissue. As a member of the immunoglobulin superfamily, RAGE is expressed by smooth muscle cells, endothelial cells, fibroblasts, macrophages, and epithelial cells [2,3]. RAGE is composed of an extracellular V region-like domain which interacts with endogenous ligands including high-mobility group box 1 protein (HMGB-1), S100 calgranulins, and advanced glycation end products (AGEs) that initiate intracellular signaling [4]. As a pattern recognition receptor, RAGE also perpetuates signaling following interaction with patterned external ligands. While RAGE normally functions during healthy inflammatory responses and cell signaling that controls cell turnover and differentiation, RAGE overexpression, either genetically and in response to exposure to noxious external stimulation, leads to pathologic inflammation, hindered morphogenesis, and deleterious cell signaling cascades [5,6]. It is estimated that abnormal RAGE expression could participate in a host of compromising lung diseases including bronchopulmonary dysplasia (BPD), cystic fibrosis (CF), or interstitial lung disease (ILD) [5]
Previous work in our lab has histologically demonstrated that RAGE TG mice that genetically upregulate RAGE in the lung during development culminates in marked pulmonary simplification [4,7]. Subsequent research found that lung hypoplasia in RAGE TG mice was not primarily due to aberrant proliferation, rather the induction of apoptosis in part due to increased levels of FasL signaling through the NF-κB pathway [8]. It was also found that RAGE overexpression coincided with a significant decrease in the expression of essential transcription factors including NKX2.1 and FoxA2, which caused a significant inhibition of surfactant protein C expression necessary for postnatal gas exchange [7]. The current undertaking builds upon these discoveries and sought to further clarify cellular compromise in RAGE TG mice, with specific attention to mitochondrial activity and signaling intermediates that orchestrate NF-κB-mediated responses.
Mitochondria have a central role in the liberation of cellular energy. In a developmental context, aberrant mitochondrial function has been implicated in embryological processes such as cellular differentiation, the epithelial-to-mesenchymal transition (EMT), and in particular, the essential differentiation of alveolar type 1 and alveolar type 2 cells [9,10]. When lung tissue is compromised, data suggest a role for immune cells that enhance mitochondria activity and increased oxygen consumption observed during an inflammatory cytokine response. These outcomes may also occur in tandem with the generation of reactive oxygen species (ROS) created by increased mitochondrial respiration [11,12]. In fact, roles for enhanced ROS formation have been proposed in cases of increased apoptosis, DNA damage, and altered cell signaling, leading to the development of diverse pre- and postnatal lung diseases [11]. Increased ROS formation has also been tied to the NF-κB pathway through p38 and/or ERK intermediaries [13], and the related compromise of ATP accrual may also underpin inflammation and apoptosis [14,15].
The current investigation sought to elucidate mitochondrial bioenergetics in the developing lung that overexpresses RAGE. We discovered that RAGE TG mice had increased mitochondrial oxygen consumption, more ATP, and decreased ROS accrual. We also demonstrated that the NF-κB pathway, specifically its intermediates p38 (MAPK pathway), ERK (MAPK pathway), and AKT (PI3K pathway), were differentially regulated. The controlled expression of ERKs is required for proper lung formation and can affect mechanisms of proliferation, apoptosis, and inflammation [16,17,18]. AKT signaling within the PI3K cascade in particular leads to the significant simplification of lung tissue and increased apoptosis when misregulated [19,20].
2. Materials and Methods
2.1. Mice
All mice were in a C57Bl/6 background. RAGE overexpression (RAGE TG) was accomplished by mating two transgenic lines to create mice that upregulate RAGE via the doxycycline (dox)-inducible mechanism as described previously [4,8]. PCR genotyping confirmed the existence of transgenes as noted previously [4,8]. Time-mated 12–18-week-old pregnant mice (RAGE TG and nontransgenic littermate controls) were fed dox food (625 mg/kg; Invigo Teklad, St. Louis, MO, USA) from before conception until E18.5 or from E15.5 to E18.5 (during the canalicular/saccular periods of development). Dams (at least 3 pregnant dams per group) were sacrificed on day E18.5 of pregnancy. Lungs were removed from pups (n = 8 per group) on day E18.5 and total homogenates were obtained. Lungs from RAGE TG and nontransgenic littermate controls (n = 8 per group) were also procured at E18.5 and mitochondrial respiration was assessed as detailed previously [21]. All mice were housed and used in accordance with an animal use protocol approved by the Institutional Animal Care and Use Committee at Brigham Young University (Protocol number 21-0203) with the approval period being 16 March 2021 through 15 March 2024.
2.2. Mitochondrial Respiration
Immediately following euthanasia, lungs from pups (n = 8) in each animal group were removed and oxygen consumption was measured. Tissues were permeabilized in saponin and added into an OROBOS FluoRespirometer (Oroboros Instruments, Innsbruck, Austria). We measured the effects of RAGE upregulation on the electron transport system (ETS) using five substrates added in the following order: glutamate and malate (GM), adenine diphosphate (ADP), succinate (S), and carbonyl cyanide (trifluoromethoxy)phenylhydrazone (FCCP). Malate (8 μL of 500 mM) and glutamate (40 μL of 500 mM) were added together to support the creation of NADH, via the malate aspartate transporter, which feeds into complex 1 of the ETS. The introduction of ADP (10 μL of 500 mM) allows for the hydrogen from the added GM to flow through the ATP synthase. Next, succinate (40 μL of 500 mM) was added in order to measure the activation of complex 2. Lastly, FCCP (2 μL of 1 mM) was used as an uncoupler to allow hydrogen to flow through the inner membrane of the mitochondria without the need for ATP synthase. Following these procedures, tissues were homogenized in a radioimmunoprecipitation assay (RIPA) (Thermo Fisher Scientific REF 89901, Rockford, IL, USA) and their protein concentration was calculated using a Bicinchoninic assay (BCA; REF 23225, Thermo Fisher Scientific, Pittsburg, PA, USA) for the normalization of total protein measured in each well.
2.3. Protein Expression
Whole lung lysates from RAGE TG and nontransgenic controls (n = 8 for each group) were homogenized in RIPA (Thermo Fisher Scientific) supplemented with protease and phosphatase inhibitors (Thermo Fisher Scientific). Protein concentrations of lysed tissue were obtained using a BCA Protein Assay Kit (Thermo Fisher Scientific). Protein samples (30 μg each) were separated on a 4–20% SDS-PAGE gel and then transferred to a nitrocellulose membrane that was blocked with 5% BSA and separately incubated with the following primary antibodies: pAKT anti-Rabbit at 1:250 (Cat# 9271L, Cell Signaling Technology, Danvers, MA, USA), pERK anti-Rabbit at 1:250 (Cat# 5683T, Cell Signaling Technology), and pp38 anti-Rabbit at 1:250 (cat# 4511S, Cell signaling Technology). We used mouse anti-beta Actin (Cat# sc-81178, Santa Cruz Biotechnology, Santa Cruz, CA, USA) as a loading control at 1:1000. Membranes were incubated overnight at 4 °C with the corresponding secondary antibodies: IRDye® 680RD Donkey anti-Rabbit IgG at 1:2500 (LiCor 926-68074, Lincoln, NE, USA) and IRDye® 800CW donkey anti-Mouse IgG at 1:2500 (LiCor 926-32212). They were then imaged using a Li-Cor Odyssey machine and quantified using image studio software (Version 1.1, Licor). The figures presented are representative of three different immunoblotting experiments performed in quadruplicate.
2.4. ROS Quantification
General ROS levels (n = 8 for each group) were quantified using a 2′,7′-dichlorofluorescin diacetate assay (DCFDA) (Sigma-Aldrich D6883, Saint Louis, MO, USA). Snap-frozen lung tissues were homogenized in PBS and then diluted at a 1:5 ratio in ddH2O. An amount of 90 μL of the diluted homogenates was loaded into black, opaque, clear-bottom 96-well plates, and then 10 μL of DCFDA (500 uM) was subsequently added to each well. The 96-well plate was covered in aluminum foil and incubated at 37 °C for 30 min. The reaction was terminated on ice for 2 min. Fluorescence was measured at 480/530 nm using a Victor Nivo Multimode Plate Reader (Perkin Elmer, Waltham, MA, USA). Values were normalized to protein concentration using the BCA Protein Assay Kit (Thermo Fisher Scientific).
2.5. ATP Quantification
ATP concentration (n = 8 for each group) was measured using an ATPlite Luminescence Assay kit (Perkin Elmer). Tissues were homogenized in ATP stabilization buffer (three volumes of ice-cold PBS containing 20 mM glycine, 50 mM MgSO4, and 4 mM EDTA). Homogenates were then diluted at a 1-to-5 ratio in ddH2O. Amounts of 100 μL of ATP-stabilizing buffer and 50 μL of mammalian cell lysis buffer were added to an opaque, white, 96-well plate in wells used for standards and were then placed on a shaker for 5 min at 25 °C at 700 RPM. Next, 100 μL of diluted samples was added into open wells and 10 μL of standard was added to the aforementioned ATP-stabilizing buffer. Then, the dish was put on the shaker for 5 min at 25 °C at 700 RPM. Next, 50 μL of substrate was added into each well (in the dark) and was placed on a shaker for 5 min at 25 °C at 700 RPM. Solutions in wells were allowed to dark adapt for 10 min. Luminescence was subsequently measured with a Victor Nivo Multimode Plate Reader (Perkin Elmer), and the ATP concentration was normalized to the protein concentration measured by the BCA protein assay (Thermo Fisher Scientific).
2.6. Statistical Analysis
Mean values ± S.D. from at least eight animals per group were assessed by Mann–Whitney tests. The results are representative and those with p values < 0.05 were considered significant. Statistical analysis was performed with GraphPad Prism 7.0.
3. Results
3.1. Mitochondrial Respiration
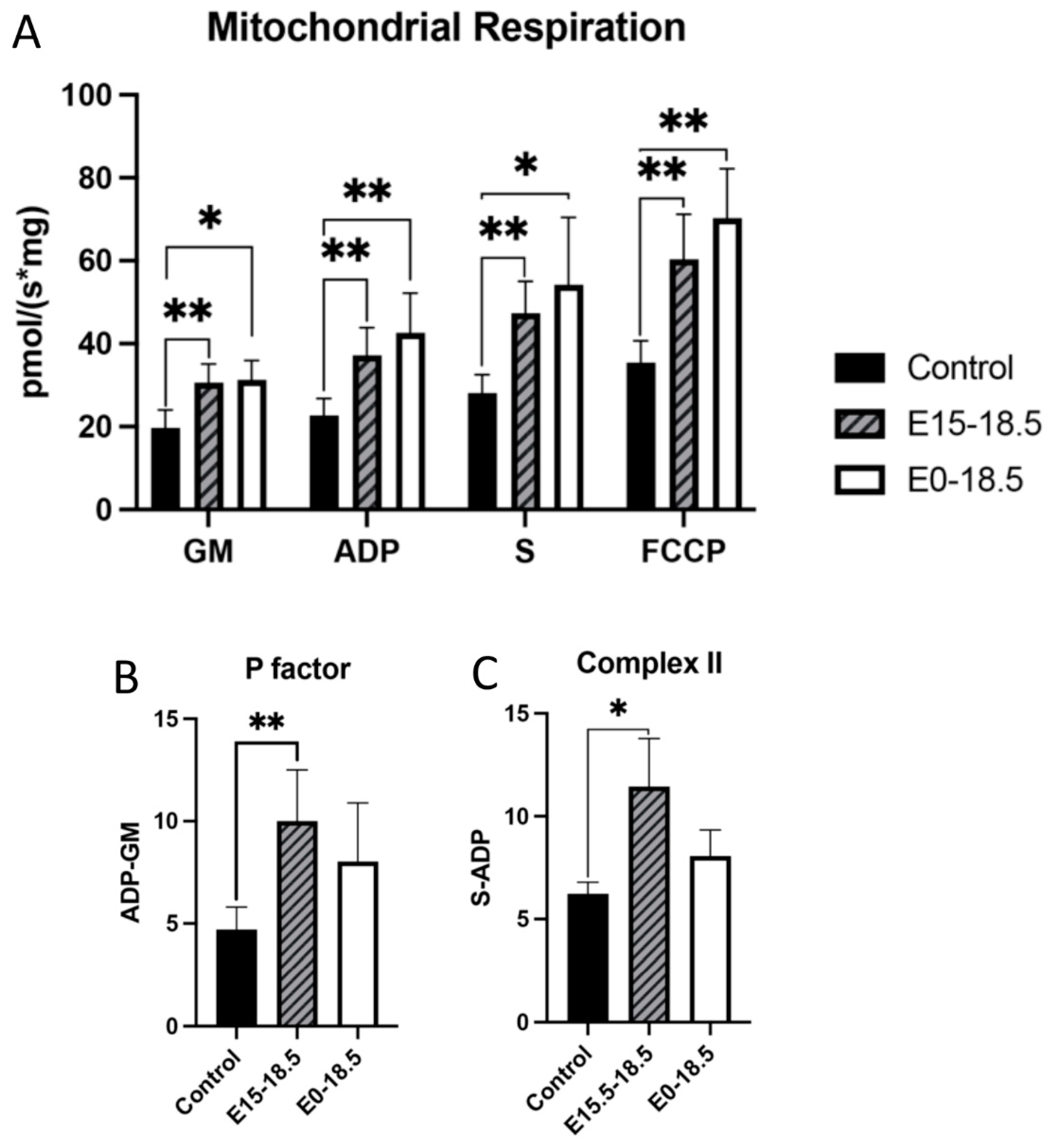
To understand how RAGE upregulation affects mitochondrial respiration, we measured oxygen consumption in response to different substrates. When RAGE was upregulated in lung tissue from pups in both treatment groups (E15-E18.5 and E0-E18.5), there was a significant increase in maximal oxygen consumption in complex 1 activation (GM), complex 2 activation (S), ATP synthase activation, and with FCCP exposure (Figure 1A). To assess whether mitochondria functioned at healthy levels, ATP synthase activation was normalized to complex 1 activation (GM) to evaluate if hydrogen ions produced through complex 1 (and therefore 3 and 4) can be used by ATP synthase at the same rate in controls as in RAGE TG samples. No significant difference was found between groups, suggesting that the mitochondria evaluated from each animal group were of similar capacity in their response to ADP addition (Figure 1B). We measured complex 2 activation alone by excluding ADP levels from succinate inclusion and found that RAGE TG lungs in the E15-E18.5 group had significant increases in complex 2 activation compared to controls (Figure 1C), which is notable given its implication in metabolic control, inflammation, and the determination of cell fate.
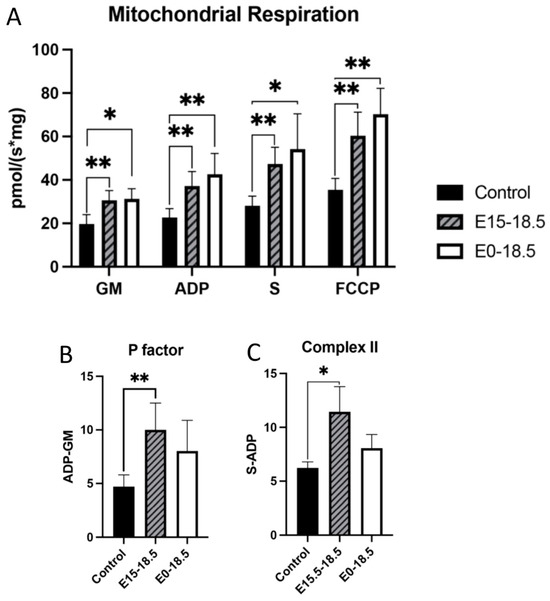
Figure 1.
Altered mitochondrial activity in response to increased expression of RAGE in lung tissue. RAGE upregulation alters mitochondrial bioenergetics in developing lungs. Upregulation of RAGE was induced in RAGE TG pup lung tissue from E15-18.5 and E0-18.5 and compared to dox-fed nontransgenic controls (n = 8 mice per group). Mice were sacrificed on day E18.5 and mitochondrial respiration (A), respiratory P factor (B), and Complex II activity (C) were evaluated. Permeabilized lung tissue samples were sequentially treated with glutamate (10 mM) and malate (GM; 2 mM); +ADP (2.5 mM); +succinate (S; 10 mM); + FCCP (2 mM). Mann–Whitney tests were used, resulting in differences noted as * p ≤ 0.05 or ** p ≤ 0.01.
3.2. ATP and ROS
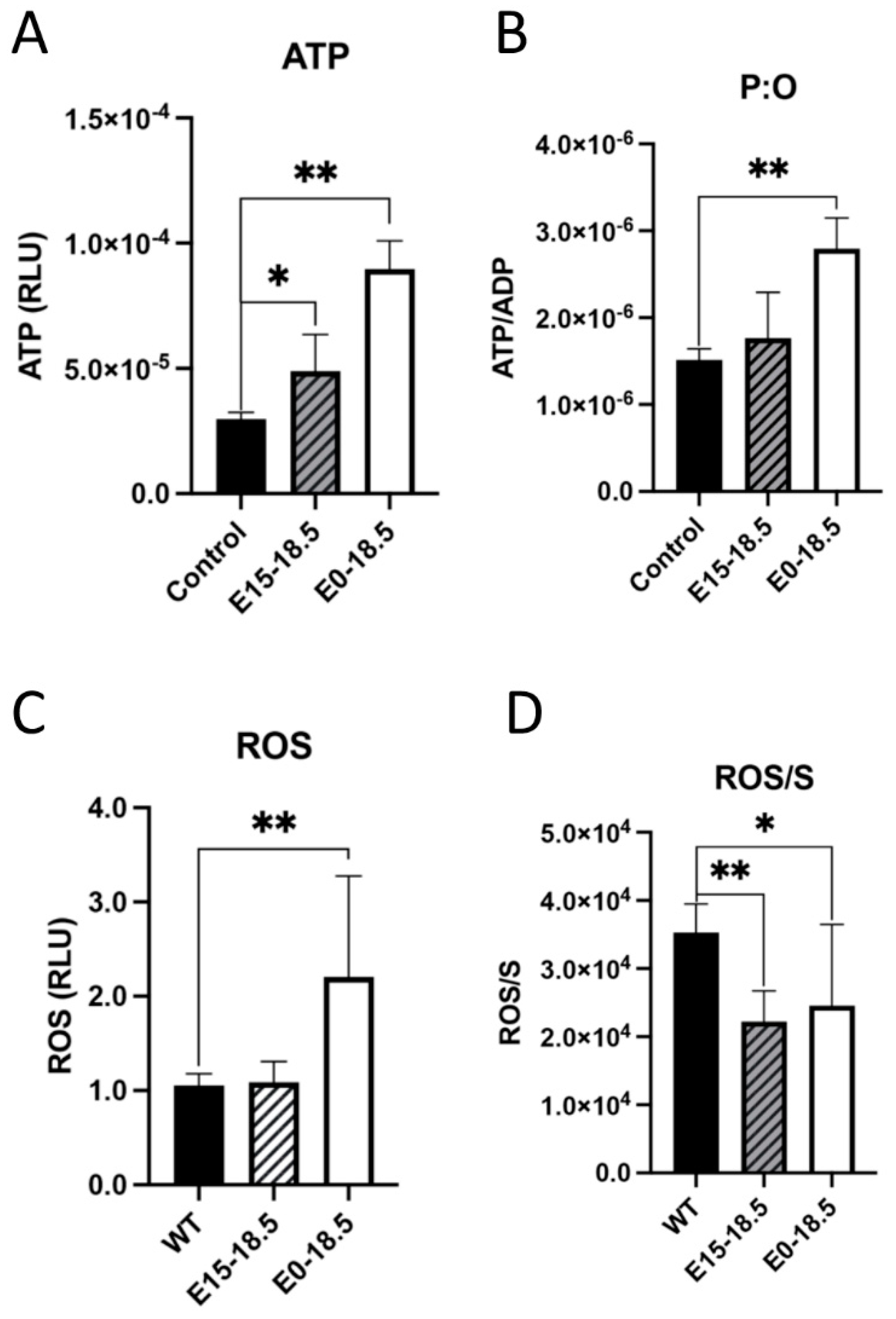
One approach to understanding the effects of increased oxygen consumption in response to RAGE overexpression was to measure ATP levels. It was found that when RAGE is overexpressed, there was a significant increase in ATP detected in both experimental groups (Figure 2A), and this trend of ATP synthesis continued when controlling for oxygen consumed during respiration (Figure 2B). Levels of reactive oxygen species (ROS) were also measured based on our discovery that RAGE overexpression led to increased oxygen consumption and ATP generation. We found that levels of ROS were significantly increased in mice with RAGE upregulation from day E0-E18.5 (Figure 2C). We also found that ROS production was not significantly elevated when indexed to oxygen respiration (Figure 2D).
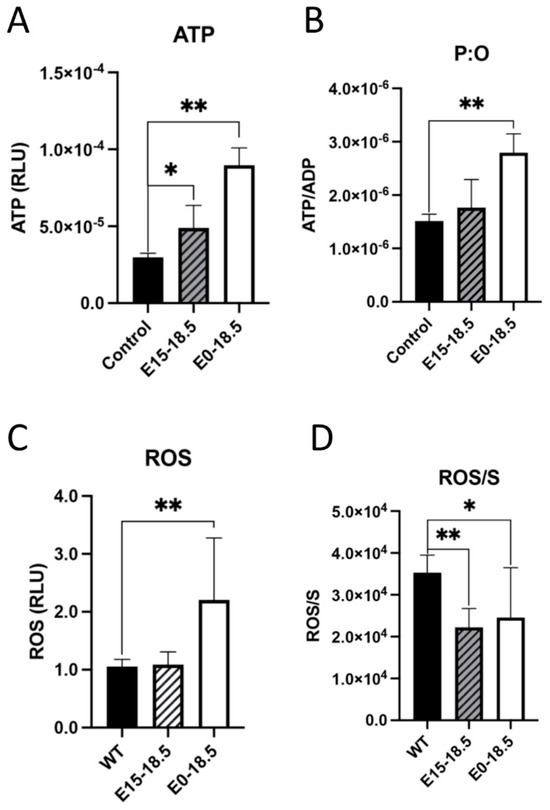
Figure 2.
Altered levels of ATP or reactive oxygen species (ROS) in response to upregulated expression of RAGE in lung tissue. Following RAGE upregulation from E15-18.5 or E0-18.5, ATP levels were measured in RAGE TG and nontransgenic controls. Levels of ATP were significantly increased in both groups of RAGE TG mice (A). When normalized to ATP synthase activity (ADP), ATP was only significantly increased in E0-18.5 treatment mice compared to controls (B). The DCFDA assay was used to quantify changes in ROS in RAGE TG mice with RAGE upregulation from E15-18.5 or E0-18.5 and compared to nontransgenic controls. ROS was significantly increased in tissue treated from E0-18.5 compared to controls (C). When normalized to complex 2 activity (S), there was a significant decrease in production of ROS in both RAGE TG groups compared to controls (D). Mann–Whitney tests were used, resulting in differences noted as * p ≤ 0.05 or ** p ≤ 0.01.
3.3. Inflammatory Signaling Intermediates
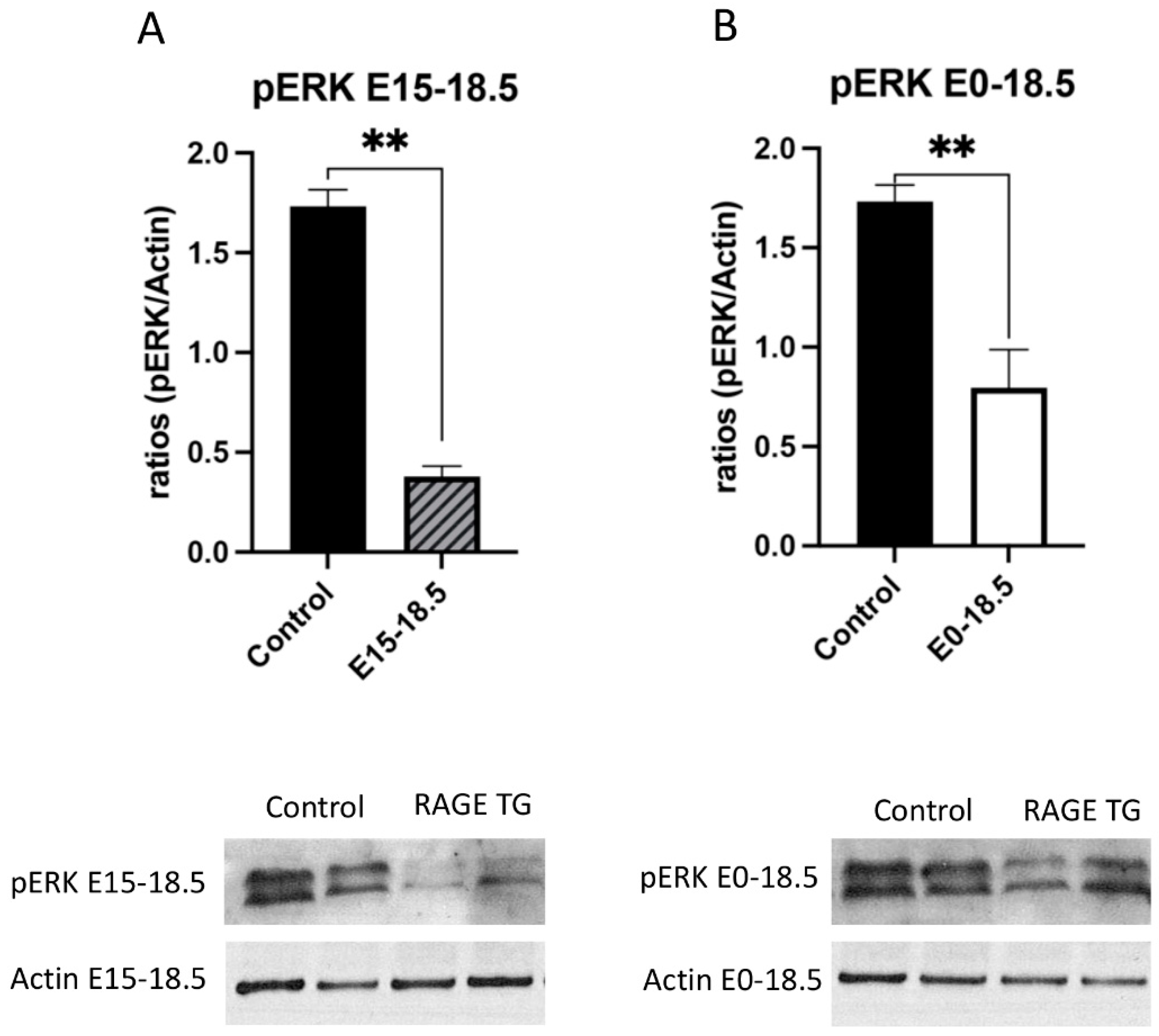
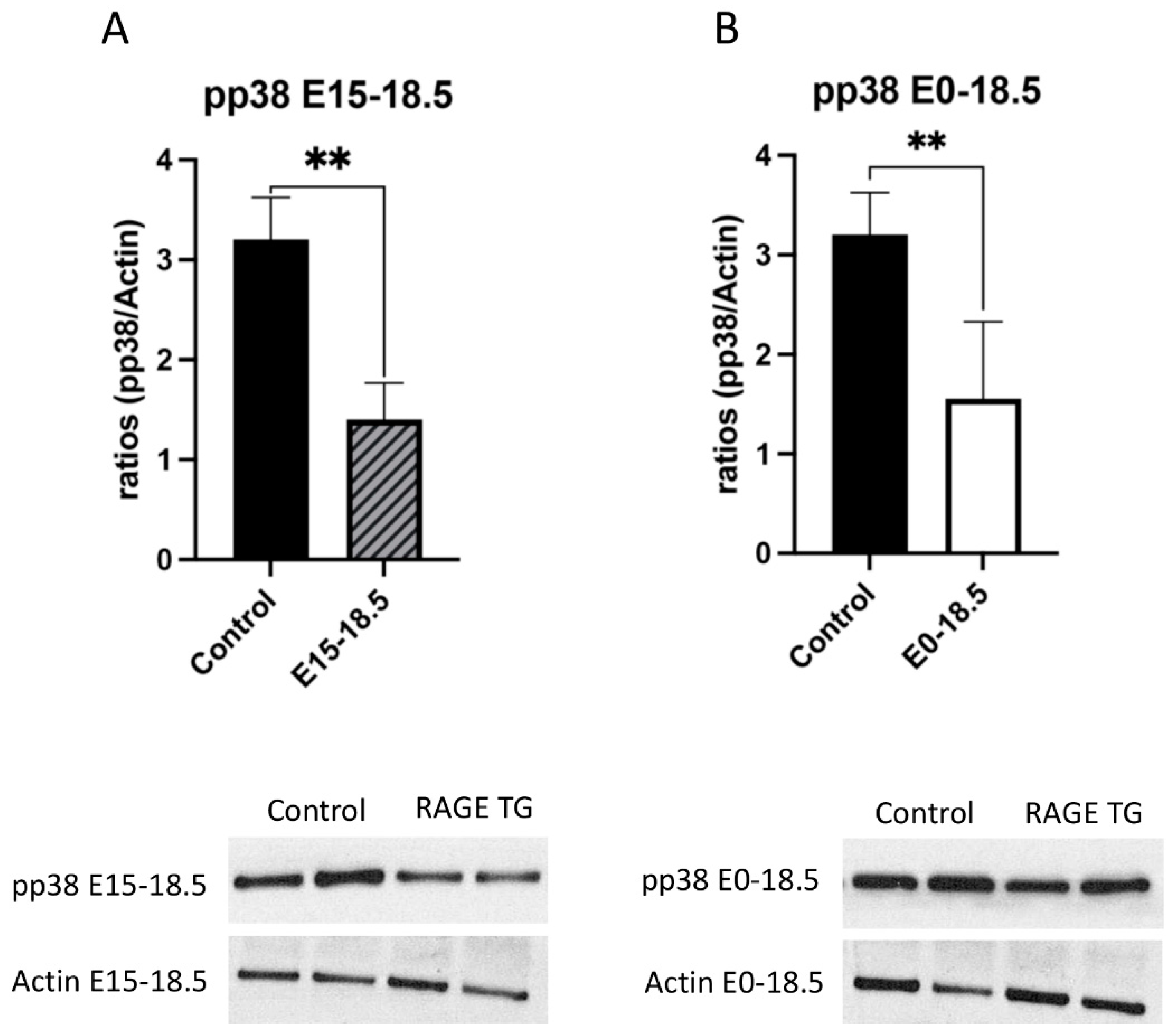
Due to its effects on mitochondrial respiration and a key role in managing inflammation, we next sought to dissect signaling factors potentially altered in RAGE TG mice. It was previously shown that NF-κB, a transcriptional regulator centrally involved in RAGE-mediated inflammation, was significantly upregulated within lung tissue in response to alveolar-cell-specific RAGE expression [22,23]. We found that RAGE TG mice expressed significantly increased levels of phosphorylated AKT (pAKT), a key PI3K perpetuator that feeds toward NF-κB activation (Figure 3). P38 and ERK activation also culminate in NF-κB activation, and the phosphorylation of these MAPKs enhance NF-κB signaling [24]. We discovered that activated p38 and ERK were significantly decreased in both RAGE TG groups compared to controls (Figure 4 and Figure 5).
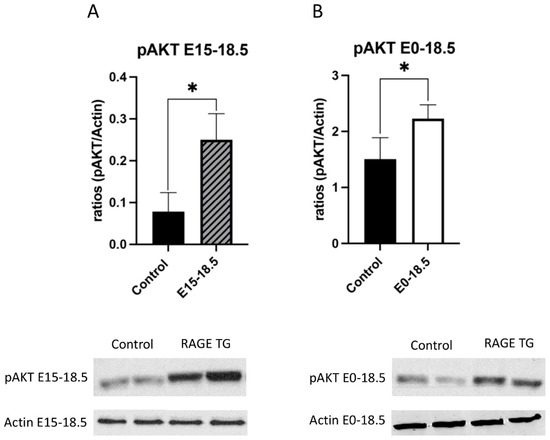
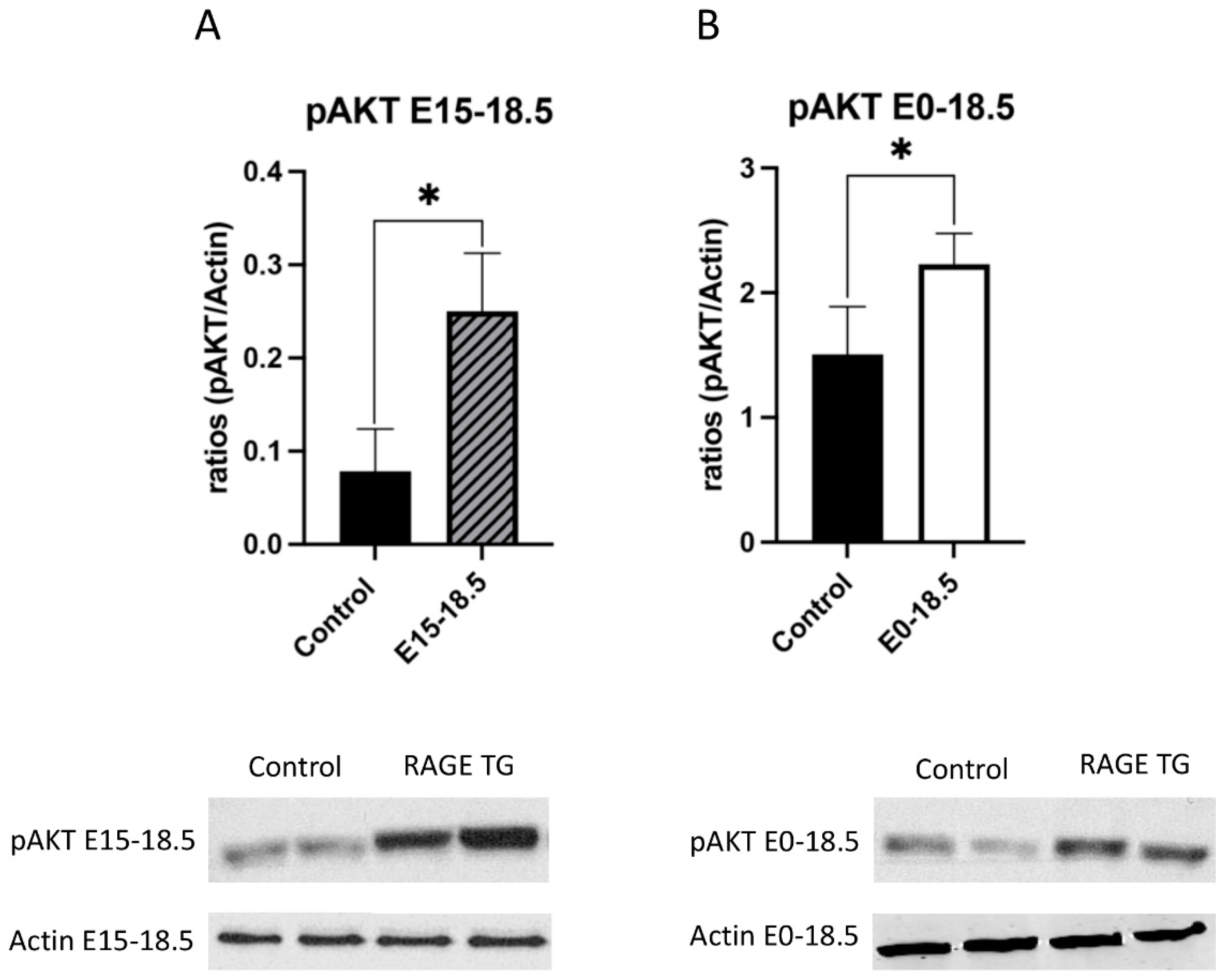
Figure 3.
Increased pAKT in response to RAGE upregulation in lung tissue. Levels of phosphorylated AKT (pAKT) were screened in lungs from both groups of RAGE TG mice and compared to nontransgenic controls. Active pAKT was significantly increased in RAGE TG lungs treated from E15-18.5 (A) or E0-18.5 (B) compared to controls (n = 4 per group). Representative blots for pAKT and actin are shown and Mann–Whitney tests revealed differences noted as * p ≤ 0.05.
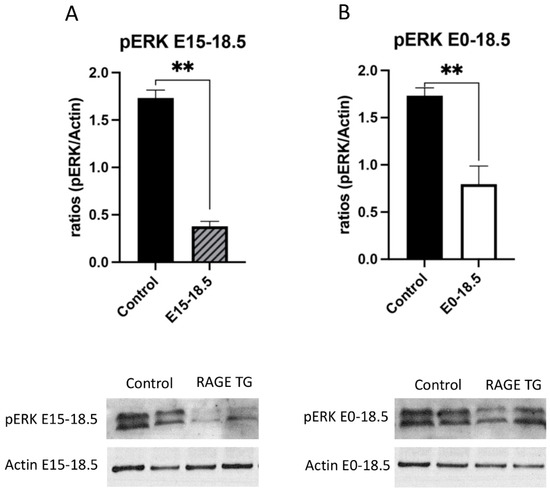
Figure 4.
Decreased pERK in response to RAGE upregulation in lung tissue. Levels of phosphorylated ERK (pERK) were screened in lungs from both groups of RAGE TG mice and compared to nontransgenic controls. Active pERK was significantly decreased in RAGE TG lungs treated from E15-18.5 (A) or E0-18.5 (B) compared to controls (n = 4 per group). Representative blots for pERK and actin are shown and Mann–Whitney tests revealed differences noted as ** p ≤ 0.01.
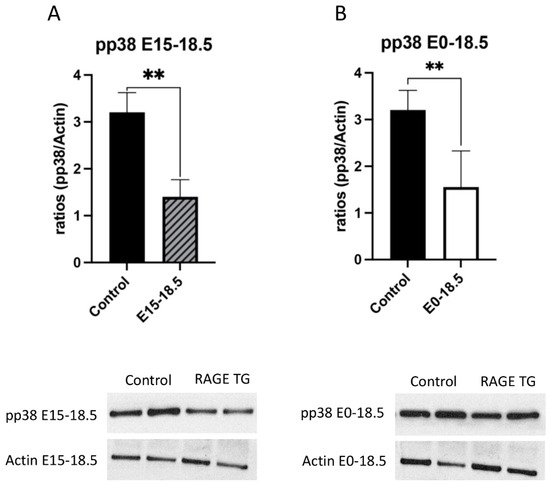
Figure 5.
Decreased pp38 in response to RAGE upregulation in lung tissue. Levels of phosphorylated p38 (pp38) were screened in lungs from both groups of RAGE TG mice and compared to nontransgenic controls. Active pp38 was significantly decreased in RAGE TG lungs treated from E15-18.5 (A) or E0-18.5 (B) compared to controls (n = 4 per group). Representative blots for pp38 and actin are shown and Mann–Whitney tests revealed differences noted as ** p ≤ 0.01.
4. Discussion
The current investigation provides additional insight into the effects of genetic RAGE upregulation in the prenatal lung and identifies candidate mechanisms that culminate in RAGE-mediated lung simplification. We discovered that RAGE upregulation coincided with a significant increase in mitochondrial respiration, coincident with elevated ATP levels, and when controlling for respiratory rate, no additional ROS burden. We also clarified differential expression profiles for mediators that function in the NF-κB signaling pathway, specifically elevated pAKT and diminished pERK and pp38. It is noteworthy that these collective observations were consistent whether RAGE upregulation was induced throughout gestation (E0-E18.5) or limited to the final three days of development (E15.5-E18.5). This undertaking provides a better understanding of how RAGE overexpression may alter mitochondrial activity as a means of tissue loss compensation and to what extent preferred signaling pathways are modulated in the aberrant lung morphogenesis phenotype. Future evaluation of these outcomes should include RAGE null mice in order to contextualize the effects or normal RAGE expression (nontransgenic controls) in comparison to both overexpression (RAGE TG) and absent expression (nulls).
The upregulation of RAGE enhanced mitochondrial respiration in both RAGE TG groups compared to controls. Despite marked tissue reduction in the RAGE TG lung [7], a greater respiratory efficiency denotes a likelihood that the remaining tissue attempts to meet energy demands via greater mitochondrial output. Such was proposed by Huang et al. describing tissue compromise and the resulting shift in mitochondrial morphology and output [25]. Depending on the disease type, mitochondrial morphology may or may not be affected and the resulting respiration outcomes may not be as anticipated. Furthermore, although mitochondrial bioenergetics are complex and intertwined, metabolic flexibility may underpin the observation that diseased tissues can experience enhanced mitochondrial output [26]. One of the main differences we discovered was altered complex 2, which has been identified as a player in metabolic pathways, inflammation, and cell fate determination. This discovery highlighted the possibility that complex 2 differences may be due in part to an increased electron flow that stimulates increases in ROS generation [27,28]. While not a focus of the current investigation, it remains plausible that mitochondrial biogenesis is also affected by RAGE overexpression. Such a concept is supported by recent research that revealed a connection between AKT and NRF1, a key transcription factor implicated in mitochondrial biogenesis [29].
A more efficiently working mitochondrion led to the perplexing notion that while more ATP per mass of tissue may result, the unfortunate accumulation of ROS was likely as well. We indeed observed higher levels of ATP in both groups of RAGE TG mice. This observation is the probable result of the compensation of unmet energy demands by tissue that remained after the course of impaired branching morphogenesis and/or excessive remodeling via enhanced apoptosis. In fact, such an observation supports the concept of maintaining apoptotic trajectories and the avoidance of shifts toward necrosis [30]. Because ATP levels were elevated, a natural concern emerged related to the potential accumulation of ROS in the developing RAGE TG lung. Decades of research has supported the conclusion that enhanced mitochondrial bioenergetics is balanced by the accumulation of deleterious ROS [31,32]. While we did observe elevated ROS levels in the E0-E18.5 RAGE TG lung, ROS abundance when controlled for oxygen consumption during respiration was intriguingly decreased in both RAGE TG groups compared to controls. While additional research involving the RAGE TG mouse lung specific to ROS accrual during oxidative stress is needed, our initial discovery of dampened ROS abundance may foreshadow future endeavors that focus on cells in the innate immune system. Immune cells produce ROS such as superoxide and hydrogen peroxide, which are common in long-term inflammatory responses. For example, inflammation mediated by Th-1 and Th-17 phenotypes may be active when tissue remodeling occurs, and regulatory T cells (Tregs) may be induced to release immunosuppressive mediators that reduce oxidative stress [33]. These potential mechanisms should be pursued, particularly as ROS signaling has also been linked to diseases of perinatal lung simplification like bronchopulmonary dysplasia (BPD) and other chronic lung diseases [34].
Due to relationships between mitochondrial bioenergetics and inflammation [35,36], we also evaluated inflammatory intermediaries of the NF-κB pathway during RAGE augmentation in the developing lung. Abundant previous research has firmly established links between RAGE signaling and inflammation modulated by NF-κB [2,37,38]. Additionally, our own work has shown that the NF-κB pathway stimulates apoptosis via FasL [8], a likely component of the simplified lung resulting from RAGE upregulation observed here. We discovered that activated p38 and ERK were both significantly decreased while levels of active AKT were increased in both RAGE TG groups compared to controls. While p38 and ERK are MAPKs and AKT is considered a component of the PI3K cascade, both culminate in the activation of NF-κB outcomes [39,40]. Our data clearly show that RAGE upregulation in RAGE TG mouse lungs preferentially signal via PI3K/AKT pathways over ERK or p38 MAPKs. These discoveries open the door to additional research into the mechanisms of signaling axes that precisely coordinate inflammation, apoptosis, and deleterious tissue remodeling. For instance, studies have highlighted the involvement of specific pathways and factors in mediating the effects of RAGE and TGF-β, such as the activation of STAT5 and p21 by AGEs and TGF-β, suggesting a conversion point in their signaling mechanisms that regulate cell cycle progression [41]. Moreover, the interaction between RAGE and TGF-β has been shown to regulate extracellular matrix turnover and cytokine synthesis in fibroblast cells exposed to AGEs, underlining the cross-talk between RAGE and TGF-β1 signaling in the lung simplification we observe in RAGE TG mice [42]. A future extension of this additional research may be the identification of these and other targets in RAGE/NF-κB signaling, potentially helpful in managing developmental disorders of pulmonary simplification such as BPD [20,43,44,45,46,47].
In summary, RAGE upregulation by the alveolar epithelium during development causes significant pulmonary malformation via altered mitochondrial bioenergetics and impaired NF-κB signaling. Further research is still needed that identifies and contextualizes other potential pathways of apoptosis and inflammatory signaling. Such research may employ the screening of single-cell mitochondrial bioenergetics and a full characterization of redox status and inflammatory signaling programs in the hopes of identifying possible treatment modalities for impaired pulmonary development.
Author Contributions
Conceptualization, P.R.R., Methodology, D.M.C., K.L.C., B.T.B., J.A.A. and P.R.R.; Formal analysis, D.M.C., K.L.C., B.T.B., J.A.A. and P.R.R.; Investigation, D.M.C., K.L.C., K.H., J.S., B.M.S., M.N.K., E.T.C., P.R., E.C. and C.E.W.; D.M.C., J.A.A., B.T.B. and P.R.R. contributed to the experimental design; D.M.C., B.M.S., M.N.K., E.T.C. and P.R. maintained animals and assisted with surgical procedures; D.M.C. and C.E.W. performed and interpreted mitochondrial, ATP, and ROS assays; D.M.C., K.H. and J.S. conducted immunoblotting; D.M.C., J.A.A. and P.R.R. assisted with data collection and interpretation. The work was conceived, funded, and supervised by J.A.A. and P.R.R. The manuscript was written primarily by D.M.C. and P.R.R. with the assistance of J.A.A. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the National Institutes of Health (1R15HL152257 and 1R15HD108743; PRR and JAA).
Institutional Review Board Statement
All experimental animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at Brigham Young University and all methods were carried out in accordance with relevant animal guidelines and regulations. The reporting of animal methods was in accordance with ARRIVE guidelines for the reporting of animal experiments.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data are presented within the article. Data and other materials are available from the corresponding author on reasonable request.
Acknowledgments
Much appreciation is extended to a team of exceptional undergraduate students in the Lung and Placenta Laboratory at Brigham Young University for vital assistance with various experiments.
Conflicts of Interest
The authors declare that they have no financial or non-financial conflicts of interest to declare.
References
- Warburton, D.; El-Hashash, A.; Carraro, G.; Tiozzo, C.; Sala, F.; Rogers, O.; De Langhe, S.; Kemp, P.J.; Riccardi, D.; Torday, J.; et al. Lung organogenesis. Curr. Top Dev. Biol. 2010, 90, 73–158. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Downs, C.A.; Johnson, N.M.; Tsaprailis, G.; Helms, M.N. RAGE-induced changes in the proteome of alveolar epithelial cells. J. Proteom. 2018, 177, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.R.; Stogsdill, J.A.; Stogsdill, M.P.; Heimann, N.B. Up-Regulation of Receptors for Advanced Glycation End-Products by Alveolar Epithelium Influences Cytodifferentiation and Causes Severe Lung Hypoplasia. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Oczypok, E.A.; Perkins, T.N.; Oury, T.D. All the “RAGE” in lung disease: The receptor for advanced glycation endproducts (RAGE) is a major mediator of pulmonary inflammatory responses. Paediatr. Respir. Rev. 2017, 23, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.Y.F.; Tullis, B.; Breithaupt, K.L.; Fowers, R.; Jones, N.; Grajeda, S.; Reynolds, P.R.; Arroyo, J.A. A Role for RAGE in DNA Double Strand Breaks (DSBs) Detected in Pathological Placentas and Trophoblast Cells. Cells 2021, 10, 857. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.M.; Curtis, K.L.; Wendt, R.A.; Stapley, B.M.; Clark, E.T.; Beckett, N.; Campbell, K.M.; Arroyo, J.A.; Reynolds, P.R. Decreased Expression of Pulmonary Homeobox NKX2.1 and Surfactant Protein C in Developing Lungs That Over-Express Receptors for Advanced Glycation End-Products (RAGE). J. Dev. Biol. 2023, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Stogsdill, J.A.; Stogsdill, M.P.; Porter, J.L.; Hancock, J.M.; Robinson, A.B.; Reynolds, P.R. Embryonic overexpression of receptors for advanced glycation end-products by alveolar epithelium induces an imbalance between proliferation and apoptosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 60–66. [Google Scholar] [CrossRef]
- Zhang, K.; Yao, E.; Chen, B.; Chuang, E.; Wong, J.; Seed, R.I.; Nishimura, S.L.; Wolters, P.J.; Chuang, P.-T. Acquisition of cellular properties during alveolar formation requires differential activity and distribution of mitochondria. eLife 2022, 11, e68598. [Google Scholar] [CrossRef]
- Madan, S.; Uttekar, B.; Chowdhary, S.; Rikhy, R. Mitochondria Lead the Way: Mitochondrial Dynamics and Function in Cellular Movements in Development and Disease. Front. Cell Dev. Biol. 2021, 9, 781933. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Suliman, H.B. Mitochondrial Dysfunction in Lung Pathogenesis. Annu. Rev. Physiol. 2017, 79, 495–515. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, S.M.; Choi, A.M. Mitochondria in lung disease. J. Clin. Investig. 2016, 126, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Keshari, R.S.; Verma, A.; Barthwal, M.K.; Dikshit, M. Reactive oxygen species-induced activation of ERK and p38 MAPK mediates PMA-induced NETs release from human neutrophils. J. Cell Biochem. 2013, 114, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-A.; Chen, J.-C.; Wu, C.-C.; Hsu, C.-W.; Ko, A.M.-S.; Chen, L.-C.; Kuo, M.-L. Reducing Lung ATP Levels and Alleviating Asthmatic Airway Inflammation through Adeno-Associated Viral Vector-Mediated CD39 Expression. Biomedicines 2021, 9, 656. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Berg, N.K.; Harting, M.T.; Li, X.; Eltzschig, H.K.; Yuan, X. Purinergic Signaling in Pulmonary Inflammation. Front. Immunol. 2019, 10, 1633. [Google Scholar] [CrossRef] [PubMed]
- Boucherat, O.; Landry-Truchon, K.; Aoidi, R.; Houde, N.; Nadeau, V.; Charron, J.; Jeannotte, L. Lung development requires an active ERK/MAPK pathway in the lung mesenchyme. Dev. Dyn. 2017, 246, 72–82. [Google Scholar] [CrossRef]
- Mercer, B.A.; D’Armiento, J.M. Emerging role of MAP kinase pathways as therapeutic targets in COPD. Int. J. Chron. Obs. Pulmon. Dis. 2006, 1, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Cagnol, S.; Chambard, J.-C. ERK and cell death: Mechanisms of ERK-induced cell death—Apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Wang, J.; Ito, T.; Udaka, N.; Okudela, K.; Yazawa, T.; Kitamura, H. PI3K–AKT pathway mediates growth and survival signals during development of fetal mouse lung. Tissue Cell 2005, 37, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Los, M.; Maddika, S.; Erb, B.; Schulze-Osthoff, K. Switching Akt: From survival signaling to deadly response. Bioessays 2009, 31, 492–495. [Google Scholar] [CrossRef]
- Walton, C.M.; Jacobsen, S.M.; Dallon, B.W.; Saito, E.R.; Bennett, S.L.H.; Davidson, L.E.; Thomson, D.M.; Hyldahl, R.D.; Bikman, B.T. Ketones Elicit Distinct Alterations in Adipose Mitochondrial Bioenergetics. Int. J. Mol. Sci. 2020, 21, 6255. [Google Scholar] [CrossRef] [PubMed]
- Curtis, K.L.; Homer, K.M.; Wendt, R.A.; Stapley, B.M.; Clark, E.T.; Harward, K.; Chang, A.; Clarke, D.M.; Arroyo, J.A.; Reynolds, P.R. Inflammatory Cytokine Elaboration Following Secondhand Smoke (SHS) Exposure Is Mediated in Part by RAGE Signaling. Int. J. Mol. Sci. 2023, 24, 15645. [Google Scholar] [CrossRef] [PubMed]
- Hirschi-Budge, K.M.; Tsai, K.Y.F.; Curtis, K.L.; Davis, G.S.; Theurer, B.K.; Kruyer, A.M.M.; Homer, K.W.; Chang, A.; Van Ry, P.M.; Arroyo, J.A.; et al. RAGE signaling during tobacco smoke-induced lung inflammation and potential therapeutic utility of SAGEs. BMC Pulm. Med. 2022, 22, 160. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Marwick, J.; Kirkham, P. Redox modulation of chromatin remodeling: Impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expression. Biochem. Pharmacol. 2004, 68, 1255–1267. [Google Scholar] [CrossRef]
- Huang, C.; Deng, K.; Wu, M. Mitochondrial cristae in health and disease. Int. J. Biol. Macromol. 2023, 235, 123755. [Google Scholar] [CrossRef] [PubMed]
- San-Millan, I. The Key Role of Mitochondrial Function in Health and Disease. Antioxidants 2023, 12, 782. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2016, 167, 457–470.e413. [Google Scholar] [CrossRef]
- Vohwinkel, C.U.; Coit, E.J.; Burns, N.; Elajaili, H.; Hernandez-Saavedra, D.; Yuan, X.; Eckle, T.; Nozik, E.; Tuder, R.M.; Eltzschig, H.K. Targeting alveolar-specific succinate dehydrogenase A attenuates pulmonary inflammation during acute lung injury. FASEB J. 2021, 35, e21468. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Feng, J.; Wang, M.; Wufuer, R.; Liu, K.; Zhang, Z.; Zhang, Y. Nrf1 is an indispensable redox-determining factor for mitochondrial homeostasis by integrating multi-hierarchical regulatory networks. Redox Biol. 2022, 57, 102470. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Doran, E.; Gillespie, J.P.; O’Toole, A. Mitochondria and cell death. Biochem. Soc. Trans. 2000, 28, 170–177. [Google Scholar] [CrossRef]
- Okoye, C.N.; Koren, S.A.; Wojtovich, A.P. Mitochondrial complex I ROS production and redox signaling in hypoxia. Redox Biol. 2023, 67, 102926. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef]
- Agita, A.; Alsagaff, M.T. Inflammation, Immunity, and Hypertension. Acta Med. Indones. 2017, 49, 158–165. [Google Scholar] [PubMed]
- Valencia, A.M.; Abrantes, M.A.; Hasan, J.; Aranda, J.V.; Beharry, K.D. Reactive Oxygen Species, Biomarkers of Microvascular Maturation and Alveolarization, and Antioxidants in Oxidative Lung Injury. React. Oxyg. Species 2018, 6, 373–388. [Google Scholar] [CrossRef]
- Jassim, A.H.; Inman, D.M.; Mitchell, C.H. Crosstalk Between Dysfunctional Mitochondria and Inflammation in Glaucomatous Neurodegeneration. Front. Pharmacol. 2021, 12, 699623. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta 2014, 1842, 446–462. [Google Scholar] [CrossRef]
- Cha, S.R.; Jang, J.; Park, S.M.; Ryu, S.M.; Cho, S.J.; Yang, S.R. Cigarette Smoke-Induced Respiratory Response: Insights into Cellular Processes and Biomarkers. Antioxidants 2023, 12, 1210. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.B.; Stogsdill, J.A.; Lewis, J.B.; Wood, T.T.; Reynolds, P.R. RAGE and tobacco smoke: Insights into modeling chronic obstructive pulmonary disease. Front. Physiol. 2012, 3, 301. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Li, Y.; Deng, B.; Lin, A.; Zhang, G.; Ma, M.; Wang, Y.; Yang, Y.; Kang, X. The PI3K/AKT signalling pathway in inflammation, cell death and glial scar formation after traumatic spinal cord injury: Mechanisms and therapeutic opportunities. Cell Prolif. 2022, 55, e13275. [Google Scholar] [CrossRef]
- Keskinidou, C.; Vassiliou, A.G.; Dimopoulou, I.; Kotanidou, A.; Orfanos, S.E. Mechanistic Understanding of Lung Inflammation: Recent Advances and Emerging Techniques. J. Inflamm. Res. 2022, 15, 3501–3546. [Google Scholar] [CrossRef]
- Brizzi, M.F.; Dentelli, P.; Rosso, A.; Calvi, C.; Gambino, R.; Cassader, M.; Salvidio, G.; Deferrari, G.; Camussi, G.; Pegoraro, L. RAGE-and TGF-β receptor-mediated signals converge on STAT5 and p21waf to control cell-cycle progression of mesangial cells: A possible role in the development and progression of diabetic nephropathy. FASEB J. 2004, 18, 1249–1251. [Google Scholar] [CrossRef] [PubMed]
- Serban, A.I.; Stanca, L.; Geicu, O.I.; Munteanu, M.C.; Dinischiotu, A. RAGE and TGF-β1 cross-talk regulate extracellular matrix turnover and cytokine synthesis in AGEs exposed fibroblast cells. PLoS ONE 2016, 11, e0152376. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ren, D.; Wang, J.; Liu, X.; Zhang, H.; Wu, M.; Yang, G. Bruceine D induces lung cancer cell apoptosis and autophagy via the ROS/MAPK signaling pathway in vitro and in vivo. Cell Death Dis. 2020, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fan, D. Ginsenoside Rg5 induces G2/M phase arrest, apoptosis and autophagy via regulating ROS-mediated MAPK pathways against human gastric cancer. Biochem. Pharmacol. 2019, 168, 285–304. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Zhu, M.; Li, W.; Si, G.; Xing, Y. Activation of PI3K/p110α in the Lung Mesenchyme Affects Branching Morphogenesis and Club Cell Differentiation. Front. Cell Dev. Biol. 2022, 10, 880206. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Luis, G.E.; van Westering-Kroon, E.; Villamor-Martinez, E.; Huizing, M.J.; Kilani, M.A.; Kramer, B.W.; Villamor, E. Tobacco Smoking During Pregnancy Is Associated With Increased Risk of Moderate/Severe Bronchopulmonary Dysplasia: A Systematic Review and Meta-Analysis. Front. Pediatr. 2020, 8, 160. [Google Scholar] [CrossRef]
- Luo, H.R.; Hattori, H.; Hossain, M.A.; Hester, L.; Huang, Y.; Lee-Kwon, W.; Donowitz, M.; Nagata, E.; Snyder, S.H. Akt as a mediator of cell death. Proc. Natl. Acad. Sci. USA 2003, 100, 11712–11717. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).