
Ferroptosis in Cancer Immunotherapy—Implications for Hepatocellular Carcinoma



Abstract
:1. Introduction
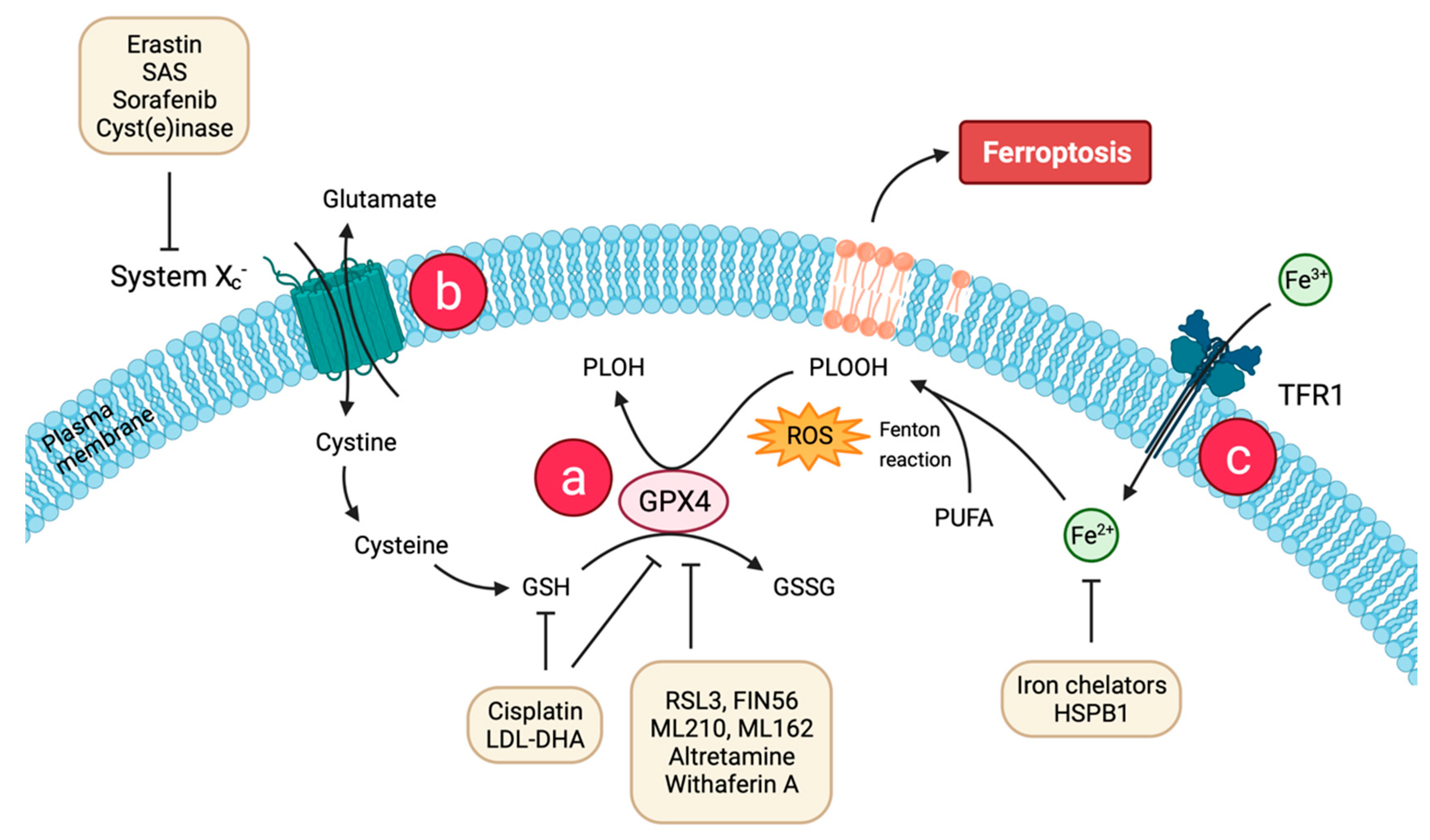
2. Molecular Basis of Ferroptosis
2.1. Lipid Peroxidation and Antioxidant Defense
2.2. Iron Accumulation
2.3. Regulatory Pathways
2.4. Immunogenic Features of Ferroptosis (in Cancer Cells)
2.5. Immune Response to DAMPs Released by Ferroptotic Cells
3. Ferroptosis in Cancer Therapy
3.1. System xc− Regulators
3.2. GPX4 Regulators
3.3. Targeting SLC7A11
3.4. Targeting NRF
3.5. Nanoparticles
3.6. Other Classes
3.7. Ferroptosis in Cancer Immunotherapy
3.8. Clinical Trials in HCC Treatment
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features (Review). World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J. Ferroptosis: Bug or feature? Immunol. Rev. 2017, 277, 150–157. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. 1), 4–13. [Google Scholar] [CrossRef]
- Dasgupta, P.; Henshaw, C.; Youlden, D.R.; Clark, P.J.; Aitken, J.F.; Baade, P.D. Global Trends in Incidence Rates of Primary Adult Liver Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 171. [Google Scholar] [CrossRef] [Green Version]
- Lurje, I.; Czigany, Z.; Bednarsch, J.; Roderburg, C.; Isfort, P.; Neumann, U.P.; Lurje, G. Treatment Strategies for Hepatocellular Carcinoma (-) a Multidisciplinary Approach. Int. J. Mol. Sci. 2019, 20, 1465. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, R.; Sahu, M.K.; Tripathy, A.; Uthansingh, K.; Behera, M. Hepatocellular carcinoma treatment: Hurdles, advances and prospects. Hepat. Oncol. 2018, 5, HEP08. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, M.; Okusaka, T.; Mitsunaga, S.; Ueno, H.; Tamai, T.; Suzuki, T.; Hayato, S.; Kadowaki, T.; Okita, K.; Kumada, H. Safety and Pharmacokinetics of Lenvatinib in Patients with Advanced Hepatocellular Carcinoma. Clin. Cancer Res. 2016, 22, 1385–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou-Alfa, G.K.; Schwartz, L.; Ricci, S.; Amadori, D.; Santoro, A.; Figer, A.; De Greve, J.; Douillard, J.Y.; Lathia, C.; Schwartz, B.; et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2006, 24, 4293–4300. [Google Scholar] [CrossRef] [Green Version]
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef] [Green Version]
- Makarova-Rusher, O.V.; Medina-Echeverz, J.; Duffy, A.G.; Greten, T.F. The yin and yang of evasion and immune activation in HCC. J. Hepatol. 2015, 62, 1420–1429. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, N.; Flecken, T.; Thimme, R. Tumor-associated antigen specific CD8(+) T cells in hepatocellular carcinoma—a promising target for immunotherapy. Oncoimmunology 2014, 3, e954919. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2021, 23, 77–90. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewerenz, J.; Ates, G.; Methner, A.; Conrad, M.; Maher, P. Oxytosis/Ferroptosis-(Re-) Emerging Roles for Oxidative Stress-Dependent Non-apoptotic Cell Death in Diseases of the Central Nervous System. Front. Neurosci. 2018, 12, 214. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, X.; Yan, C. Ferroptosis: An emerging therapeutic opportunity for cancer. Genes Dis. 2020, 9, 334–346. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [Green Version]
- Dierge, E.; Debock, E.; Guilbaud, C.; Corbet, C.; Mignolet, E.; Mignard, L.; Bastien, E.; Dessy, C.; Larondelle, Y.; Feron, O. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab. 2021, 33, 1701–1715. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Forster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Radmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422. [Google Scholar] [CrossRef] [Green Version]
- Xiaoxuan, W.; Zicheng, L.; Lijuan, M.; Haijie, Y. Ferroptosis and its emerging role in tumor. Biophys. Rep. 2021, 7, 280. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torti, S.V.; Manz, D.H.; Paul, B.T.; Blanchette-Farra, N.; Torti, F.M. Iron and Cancer. Annu. Rev. Nutr. 2018, 38, 97–125. [Google Scholar] [CrossRef] [PubMed]
- Fleming, M.D.; Romano, M.A.; Su, M.A.; Garrick, L.M.; Garrick, M.D.; Andrews, N.C. Nramp2 is mutated in the anemic Belgrade (b) rat: Evidence of a role for Nramp2 in endosomal iron transport. Proc. Natl. Acad. Sci. USA 1998, 95, 1148–1153. [Google Scholar] [CrossRef] [Green Version]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Lan, H.; Gao, Y.; Zhao, Z.; Mei, Z.; Wang, F. Ferroptosis: Redox Imbalance and Hematological Tumorigenesis. Front. Oncol. 2022, 12. [Google Scholar] [CrossRef]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef]
- Nie, Q.; Hu, Y.; Yu, X.; Li, X.; Fang, X. Induction and application of ferroptosis in cancer therapy. Cancer Cell Int. 2022, 22, 12. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Roumenina, L.T.; Rayes, J.; Lacroix-Desmazes, S.; Dimitrov, J.D. Heme: Modulator of Plasma Systems in Hemolytic Diseases. Trends Mol. Med. 2016, 22, 200–213. [Google Scholar] [CrossRef] [PubMed]
- NaveenKumar, S.K.; SharathBabu, B.N.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S.; Mugesh, G. The Role of Reactive Oxygen Species and Ferroptosis in Heme-Mediated Activation of Human Platelets. ACS Chem. Biol. 2018, 13, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef] [Green Version]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Flora, S.J.S. Positive and Negative Regulation of Ferroptosis and Its Role in Maintaining Metabolic and Redox Homeostasis. Oxid. Med. Cell Longev. 2021, 2021, 9074206. [Google Scholar] [CrossRef]
- Efimova, I.; Catanzaro, E.; Van der Meeren, L.; Turubanova, V.D.; Hammad, H.; Mishchenko, T.A.; Vedunova, M.V.; Fimognari, C.; Bachert, C.; Coppieters, F.; et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Wen, Q.; Liu, J.; Kang, R.; Zhou, B.; Tang, D. The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 2019, 510, 278–283. [Google Scholar] [CrossRef]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release mechanisms of major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Zeh, H.J., 3rd; Lotze, M.T. High-mobility group box 1 and cancer. Biochim. Biophys Acta 2010, 1799, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demuynck, R.; Efimova, I.; Naessens, F.; Krysko, D.V. Immunogenic ferroptosis and where to find it? J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Ye, F.; Chai, W.; Xie, M.; Yang, M.; Yu, Y.; Cao, L.; Yang, L. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRAS(Q61L) cells. Am. J. Cancer Res. 2019, 9, 730–739. [Google Scholar] [PubMed]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, W.; Yang, H.; Shao, D.; Zhao, X.; Zhang, G. Intraperitoneal injection of 4-hydroxynonenal (4-HNE), a lipid peroxidation product, exacerbates colonic inflammation through activation of Toll-like receptor 4 signaling. Free Radic. Biol. Med. 2019, 131, 237–242. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Luo, X.; Gong, H.B.; Gao, H.Y.; Wu, Y.P.; Sun, W.Y.; Li, Z.Q.; Wang, G.; Liu, B.; Liang, L.; Kurihara, H.; et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ. 2021, 28, 1971–1989. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Dai, E.; Han, L.; Liu, J.; Xie, Y.; Kroemer, G.; Klionsky, D.J.; Zeh, H.J.; Kang, R.; Wang, J.; Tang, D. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 2020, 16, 2069–2083. [Google Scholar] [CrossRef]
- Luo, L.; Wang, H.; Tian, W.; Zeng, J.; Huang, Y.; Luo, H. Targeting ferroptosis for cancer therapy: Iron metabolism and anticancer immunity. Am. J. Cancer Res. 2021, 11, 5508–5525. [Google Scholar] [PubMed]
- Matsushita, M.; Freigang, S.; Schneider, C.; Conrad, M.; Bornkamm, G.W.; Kopf, M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 2015, 212, 555–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drijvers, J.M.; Gillis, J.E.; Muijlwijk, T.; Nguyen, T.H.; Gaudiano, E.F.; Harris, I.S.; LaFleur, M.W.; Ringel, A.E.; Yao, C.H.; Kurmi, K.; et al. Pharmacologic Screening Identifies Metabolic Vulnerabilities of CD8(+) T Cells. Cancer Immunol. Res. 2021, 9, 184–199. [Google Scholar] [CrossRef]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloditz, K.; Fadeel, B. Three cell deaths and a funeral: Macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discov. 2019, 5, 65. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Yan, R.; Zhu, J.; Cheng, S.; Kong, C.; Chen, W.; Fang, S.; Wang, Y.; Yang, Y.; Qiu, R.; et al. Integrative analysis of the molecular mechanisms, immunological features and immunotherapy response of ferroptosis regulators across 33 cancer types. Int. J. Biol. Sci. 2022, 18, 180–198. [Google Scholar] [CrossRef]
- Lang, X.; Green, M.D.; Wang, W.; Yu, J.; Choi, J.E.; Jiang, L.; Liao, P.; Zhou, J.; Zhang, Q.; Dow, A.; et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019, 9, 1673–1685. [Google Scholar] [CrossRef] [Green Version]
- Fonseca-Nunes, A.; Jakszyn, P.; Agudo, A. Iron and cancer risk--a systematic review and meta-analysis of the epidemiological evidence. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 12–31. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Hatcher, H.C.; Singh, R.N.; Torti, F.M.; Torti, S.V. Synthetic and natural iron chelators: Therapeutic potential and clinical use. Future Med. Chem. 2009, 1, 1643–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grignano, E.; Birsen, R.; Chapuis, N.; Bouscary, D. From Iron Chelation to Overload as a Therapeutic Strategy to Induce Ferroptosis in Leukemic Cells. Front. Oncol. 2020, 10, 586530. [Google Scholar] [CrossRef] [PubMed]
- Capece, T.; Walling, B.L.; Lim, K.; Kim, K.D.; Bae, S.; Chung, H.L.; Topham, D.J.; Kim, M. A novel intracellular pool of LFA-1 is critical for asymmetric CD8(+) T cell activation and differentiation. J. Cell Biol. 2017, 216, 3817–3829. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.; Xue, X. Targeting iron metabolism in cancer therapy. Theranostics 2021, 11, 8412–8429. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Han, X.; Lan, X.; Gao, Y.; Wan, J.; Durham, F.; Cheng, T.; Yang, J.; Wang, Z.; Jiang, C.; et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017, 2, e90777. [Google Scholar] [CrossRef] [Green Version]
- Dey, S.; Ashrafi, A.; Vidal, C.; Jain, N.; Kalainayakan, S.P.; Ghosh, P.; Alemi, P.S.; Salamat, N.; Konduri, P.C.; Kim, J.W.; et al. Heme Sequestration Effectively Suppresses the Development and Progression of Both Lung Adenocarcinoma and Squamous Cell Carcinoma. Mol. Cancer Res. 2022, 20, 139–149. [Google Scholar] [CrossRef]
- Ghosh, P.; Guo, Y.; Ashrafi, A.; Chen, J.; Dey, S.; Zhong, S.; Liu, J.; Campbell, J.; Konduri, P.C.; Gerberich, J.; et al. Oxygen-Enhanced Optoacoustic Tomography Reveals the Effectiveness of Targeting Heme and Oxidative Phosphorylation at Normalizing Tumor Vascular Oxygenation. Cancer Res. 2020, 80, 3542–3555. [Google Scholar] [CrossRef]
- Wang, T.; Ashrafi, A.; Konduri, P.C.; Ghosh, P.; Dey, S.; Modareszadeh, P.; Salamat, N.; Alemi, P.S.; Berisha, E.; Zhang, L. Heme Sequestration as an Effective Strategy for the Suppression of Tumor Growth and Progression. Mol. Cancer Ther. 2021, 20, 2506–2518. [Google Scholar] [CrossRef]
- Kong, R.; Wang, N.; Han, W.; Bao, W.; Lu, J. IFNgamma-mediated repression of system xc(-) drives vulnerability to induced ferroptosis in hepatocellular carcinoma cells. J. Leukoc. Biol. 2021, 110, 301–314. [Google Scholar] [CrossRef]
- Li, X.; Wang, T.X.; Huang, X.; Li, Y.; Sun, T.; Zang, S.; Guan, K.L.; Xiong, Y.; Liu, J.; Yuan, H.X. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int. 2020, 40, 1378–1394. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, X.; Ge, C.; Min, J.; Wang, F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 2022. [Google Scholar] [CrossRef] [PubMed]
- Devisscher, L.; Van Coillie, S.; Hofmans, S.; Van Rompaey, D.; Goossens, K.; Meul, E.; Maes, L.; De Winter, H.; Van Der Veken, P.; Vandenabeele, P.; et al. Discovery of Novel, Drug-Like Ferroptosis Inhibitors with in Vivo Efficacy. J. Med. Chem. 2018, 61, 10126–10140. [Google Scholar] [CrossRef] [PubMed]
- Codenotti, S.; Poli, M.; Asperti, M.; Zizioli, D.; Marampon, F.; Fanzani, A. Cell growth potential drives ferroptosis susceptibility in rhabdomyosarcoma and myoblast cell lines. J. Cancer Res. Clin. Oncol. 2018, 144, 1717–1730. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell Oncol. 2015, 2, e1054549. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Gai, C.; Li, Z.; Ding, D.; Zheng, J.; Zhang, W.; Lv, S.; Li, W. Targeted exosome-encapsulated erastin induced ferroptosis in triple negative breast cancer cells. Cancer Sci. 2019, 110, 3173–3182. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623–633. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. OncoTargets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef]
- Zheng, J.; Sato, M.; Mishima, E.; Sato, H.; Proneth, B.; Conrad, M. Sorafenib fails to trigger ferroptosis across a wide range of cancer cell lines. Cell Death Dis. 2021, 12, 698. [Google Scholar] [CrossRef]
- Sun, Y.; Deng, R.; Zhang, C. Erastin induces apoptotic and ferroptotic cell death by inducing ROS accumulation by causing mitochondrial dysfunction in gastric cancer cell HGC27. Mol. Med. Rep. 2020, 22, 2826–2832. [Google Scholar] [CrossRef]
- Basuli, D.; Tesfay, L.; Deng, Z.; Paul, B.; Yamamoto, Y.; Ning, G.; Xian, W.; McKeon, F.; Lynch, M.; Crum, C.P.; et al. Iron addiction: A novel therapeutic target in ovarian cancer. Oncogene 2017, 36, 4089–4099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, N.; Shi, B.J.; Li, S.L.; Zhong, Z.Y.; Li, Y.C.; Xua, W.L.; Zhou, H.; Cai, J.H. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3826–3836. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yan, H.; Xu, X.; Liu, H.; Wu, C.; Zhao, L. Erastin/sorafenib induces cisplatin-resistant non-small cell lung cancer cell ferroptosis through inhibition of the Nrf2/xCT pathway. Oncol. Lett. 2020, 19, 323–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhao, Y.; Zhang, Z.; Tan, N.; Zhao, F.; Ge, C.; Liang, L.; Jia, D.; Chen, T.; Yao, M.; et al. Disruption of xCT inhibits cell growth via the ROS/autophagy pathway in hepatocellular carcinoma. Cancer Lett. 2011, 312, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Mimura, J.; Okada, T.; Sato, H.; Liu, T.; Maruyama, A.; Ohyama, C.; Itoh, K. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol. Cell Biol. 2014, 34, 3421–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, M.Z.; Chen, G.; Wang, P.; Lu, W.H.; Zhu, C.F.; Song, M.; Yang, J.; Wen, S.; Xu, R.H.; Hu, Y.; et al. Xc- inhibitor sulfasalazine sensitizes colorectal cancer to cisplatin by a GSH-dependent mechanism. Cancer Lett. 2015, 368, 88–96. [Google Scholar] [CrossRef]
- Chung, W.J.; Lyons, S.A.; Nelson, G.M.; Hamza, H.; Gladson, C.L.; Gillespie, G.Y.; Sontheimer, H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J. Neurosci. 2005, 25, 7101–7110. [Google Scholar] [CrossRef]
- Yu, H.; Yang, C.; Jian, L.; Guo, S.; Chen, R.; Li, K.; Qu, F.; Tao, K.; Fu, Y.; Luo, F.; et al. Sulfasalazineinduced ferroptosis in breast cancer cells is reduced by the inhibitory effect of estrogen receptor on the transferrin receptor. Oncol. Rep. 2019, 42, 826–838. [Google Scholar] [CrossRef]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.C.; Maziere, J.C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014, 34, 6417–6422. [Google Scholar] [PubMed]
- Chang, Y.S.; Adnane, J.; Trail, P.A.; Levy, J.; Henderson, A.; Xue, D.; Bortolon, E.; Ichetovkin, M.; Chen, C.; McNabola, A.; et al. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother. Pharmacol. 2007, 59, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Krainz, T.; Gaschler, M.M.; Lim, C.; Sacher, J.R.; Stockwell, B.R.; Wipf, P. A Mitochondrial-Targeted Nitroxide Is a Potent Inhibitor of Ferroptosis. ACS Cent. Sci. 2016, 2, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Kissel, M.; Berndt, S.; Fiebig, L.; Kling, S.; Ji, Q.; Gu, Q.; Lang, T.; Hafner, F.T.; Teufel, M.; Zopf, D. Antitumor effects of regorafenib and sorafenib in preclinical models of hepatocellular carcinoma. Oncotarget 2017, 8, 107096–107108. [Google Scholar] [CrossRef]
- Zhang, W.; Konopleva, M.; Shi, Y.X.; McQueen, T.; Harris, D.; Ling, X.; Estrov, Z.; Quintas-Cardama, A.; Small, D.; Cortes, J.; et al. Mutant FLT3: A direct target of sorafenib in acute myelogenous leukemia. J. Natl. Cancer Inst. 2008, 100, 184–198. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Wang, W.; Liu, B.; Yang, G.; Xu, Z.; Li, J.; Liu, Z. RNA binding protein DAZAP1 promotes HCC progression and regulates ferroptosis by interacting with SLC7A11 mRNA. Exp. Cell Res. 2021, 399, 112453. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Kroemer, G.; Tang, D. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021, 28, 1135–1148. [Google Scholar] [CrossRef]
- Hu, F.; Li, G.; Huang, C.; Hou, Z.; Yang, X.; Luo, X.; Feng, Y.; Wang, G.; Hu, J.; Cao, Z. The autophagy-independent role of BECN1 in colorectal cancer metastasis through regulating STAT3 signaling pathway activation. Cell Death Dis. 2020, 11, 304. [Google Scholar] [CrossRef]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc(−) Activity. Curr. Biol. 2018, 28, 2388–2399. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Cai, Y.; Xu, K.; Ren, X.; Sun, J.; Lu, S.; Chen, J.; Xu, P. Beclin1 overexpression suppresses tumor cell proliferation and survival via an autophagydependent pathway in human synovial sarcoma cells. Oncol. Rep. 2018, 40, 1927–1936. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Zhang, C.; Yi, H.; Wu, C.; Wang, J.; Liu, Y.; Tan, J.; Wen, J. Downregulation of Beclin 1 and impairment of autophagy in a small population of colorectal cancer. Dig. Dis. Sci. 2013, 58, 2887–2894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Z.; Hu, T.; Han, L.; Yu, S.; Yao, Y.; Ruan, Z.; Tian, T.; Huang, T.; Wang, M.; et al. Nrf2 promotes progression of non-small cell lung cancer through activating autophagy. Cell Cycle 2017, 16, 1053–1062. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, C.; Zhang, L.; Yang, Q.; Zhou, S.; Wen, Q.; Wang, J. Nrf2 is a potential prognostic marker and promotes proliferation and invasion in human hepatocellular carcinoma. BMC Cancer 2015, 15, 531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Zhou, C.; Zhao, Y.; Zhang, X.; Chen, W.; Zhou, Q.; Hu, B.; Gao, D.; Raatz, L.; Wang, Z.; et al. Quiescin sulfhydryl oxidase 1 promotes sorafenib-induced ferroptosis in hepatocellular carcinoma by driving EGFR endosomal trafficking and inhibiting NRF2 activation. Redox Biol. 2021, 41, 101942. [Google Scholar] [CrossRef]
- Zhang, X.F.; Wang, J.; Jia, H.L.; Zhu, W.W.; Lu, L.; Ye, Q.H.; Nelson, P.J.; Qin, Y.; Gao, D.M.; Zhou, H.J.; et al. Core fucosylated glycan-dependent inhibitory effect of QSOX1-S on invasion and metastasis of hepatocellular carcinoma. Cell Death Discov. 2019, 5, 84. [Google Scholar] [CrossRef] [Green Version]
- de Andrade, C.R.; Stolf, B.S.; Debbas, V.; Rosa, D.S.; Kalil, J.; Coelho, V.; Laurindo, F.R. Quiescin sulfhydryl oxidase (QSOX) is expressed in the human atheroma core: Possible role in apoptosis. In Vitro Cell Dev. Biol. Anim. 2011, 47, 716–727. [Google Scholar] [CrossRef]
- Katchman, B.A.; Ocal, I.T.; Cunliffe, H.E.; Chang, Y.H.; Hostetter, G.; Watanabe, A.; LoBello, J.; Lake, D.F. Expression of quiescin sulfhydryl oxidase 1 is associated with a highly invasive phenotype and correlates with a poor prognosis in Luminal B breast cancer. Breast Cancer Res. 2013, 15, R28. [Google Scholar] [CrossRef] [Green Version]
- Fifield, A.L.; Hanavan, P.D.; Faigel, D.O.; Sergienko, E.; Bobkov, A.; Meurice, N.; Petit, J.L.; Polito, A.; Caulfield, T.R.; Castle, E.P.; et al. Molecular Inhibitor of QSOX1 Suppresses Tumor Growth In Vivo. Mol. Cancer Ther. 2020, 19, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Pernodet, N.; Hermetet, F.; Adami, P.; Vejux, A.; Descotes, F.; Borg, C.; Adams, M.; Pallandre, J.R.; Viennet, G.; Esnard, F.; et al. High expression of QSOX1 reduces tumorogenesis, and is associated with a better outcome for breast cancer patients. Breast Cancer Res. 2012, 14, R136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Zhang, Z.; Tsai, H.I.; Liu, Y.; Gao, J.; Wang, M.; Song, L.; Cao, X.; Xu, Z.; Chen, H.; et al. Branched-chain amino acid aminotransferase 2 regulates ferroptotic cell death in cancer cells. Cell Death Differ. 2021, 28, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Antanaviciute, I.; Mikalayeva, V.; Cesleviciene, I.; Milasiute, G.; Skeberdis, V.A.; Bordel, S. Transcriptional hallmarks of cancer cell lines reveal an emerging role of branched chain amino acid catabolism. Sci. Rep. 2017, 7, 7820. [Google Scholar] [CrossRef] [Green Version]
- Pankevičiūtė, M.; Mikalayeva, V.; Raškevičius, V.; Sarapinienė, I. A Novel chemical compound inhibits BCAT2 in cancer cells. In Proceedings of the FEBS Open Bio: Supplement: 45th FEBS Congress, Molecules of Life: Towards New Horizons, Ljubljana, Slovenia, 3–8 July 2021; Federation of European Biochemical Societies (FEBS). John Wiley & Sons Ltd.: West Sussex, UK, 2021; Volume 11. [Google Scholar]
- Lei, M.Z.; Li, X.X.; Zhang, Y.; Li, J.T.; Zhang, F.; Wang, Y.P.; Yin, M.; Qu, J.; Lei, Q.Y. Acetylation promotes BCAT2 degradation to suppress BCAA catabolism and pancreatic cancer growth. Signal. Transduct. Target. Ther. 2020, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Cho, Y.R.; Kim, J.H.; Kim, J.; Nam, H.Y.; Kim, S.W.; Son, J. Branched-chain amino acids sustain pancreatic cancer growth by regulating lipid metabolism. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef]
- Khalil, M.I.; Ibrahim, M.M.; El-Gaaly, G.A.; Sultan, A.S. Trigonella foenum (Fenugreek) Induced Apoptosis in Hepatocellular Carcinoma Cell Line, HepG2, Mediated by Upregulation of p53 and Proliferating Cell Nuclear Antigen. Biomed. Res. Int. 2015, 2015, 914645. [Google Scholar] [CrossRef] [Green Version]
- Al-Oqail, M.M.; Farshori, N.N.; Al-Sheddi, E.S.; Musarrat, J.; Al-Khedhairy, A.A.; Siddiqui, M.A. In Vitro Cytotoxic Activity of Seed Oil of Fenugreek AgainstVarious Cancer Cell Lines. Asian Pac. J. Cancer Prev. 2013, 14, 1829–1832. [Google Scholar] [CrossRef] [Green Version]
- Bai, T.; Lei, P.; Zhou, H.; Liang, R.; Zhu, R.; Wang, W.; Zhou, L.; Sun, Y. Sigma-1 receptor protects against ferroptosis in hepatocellular carcinoma cells. J. Cell Mol. Med. 2019, 23, 7349–7359. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Bai, X.; Li, X.; Zhu, C.; Wu, Z.P. Overexpression of sigma—1 receptor in MCF—7 cells enhances proliferation via the classic protein kinase C subtype signaling pathway. Oncol Lett. 2018, 16, 6763–6769. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.; Kim, E.H.; Lee, J.; Roh, J.L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 2018, 129, 454–462. [Google Scholar] [CrossRef]
- Yang, H.; Zhao, L.; Gao, Y.; Yao, F.; Marti, T.M.; Schmid, R.A.; Peng, R.W. Pharmacotranscriptomic Analysis Reveals Novel Drugs and Gene Networks Regulating Ferroptosis in Cancer. Cancers 2020, 12, 3273. [Google Scholar] [CrossRef] [PubMed]
- Ghoochani, A.; Hsu, E.C.; Aslan, M.; Rice, M.A.; Nguyen, H.M.; Brooks, J.D.; Corey, E.; Paulmurugan, R.; Stoyanova, T. Ferroptosis Inducers Are a Novel Therapeutic Approach for Advanced Prostate Cancer. Cancer Res. 2021, 81, 1583–1594. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Probst, L.; Dachert, J.; Schenk, B.; Fulda, S. Lipoxygenase inhibitors protect acute lymphoblastic leukemia cells from ferroptotic cell death. Biochem. Pharmacol. 2017, 140, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, S.; He, C.; Wang, C.; Wang, L.; Piao, M.; Chi, G.; Luo, Y.; Ge, P. RSL3 induced autophagic death in glioma cells via causing glycolysis dysfunction. Biochem. Biophys. Res. Commun. 2019, 518, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef]
- Woo, J.H.; Shimoni, Y.; Yang, W.S.; Subramaniam, P.; Iyer, A.; Nicoletti, P.; Rodriguez Martinez, M.; Lopez, G.; Mattioli, M.; Realubit, R.; et al. Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell 2015, 162, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Kusumi, R.; Hamashima, S.; Kobayashi, S.; Sasaki, S.; Komiyama, Y.; Izumikawa, T.; Conrad, M.; Bannai, S.; Sato, H. The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 2018, 8, 968. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A Novel Anti-tumor Action for Cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Lin, D.; Yu, Q.; Li, Z.; Lenahan, C.; Dong, Y.; Wei, Q.; Shao, A. A Promising Future of Ferroptosis in Tumor Therapy. Front. Cell Dev. Biol. 2021, 9, 629150. [Google Scholar] [CrossRef] [PubMed]
- Cobler, L.; Zhang, H.; Suri, P.; Park, C.; Timmerman, L.A. xCT inhibition sensitizes tumors to gamma-radiation via glutathione reduction. Oncotarget 2018, 9, 32280–32297. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Lin, Z.; Jiang, D.; Yu, Y.; Yang, D.; Zhou, H.; Zhan, D.; Liu, S.; Peng, G.; Chen, Z.; et al. Erastin decreases radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis. Oncol Lett. 2019, 17, 3001–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020, 30, 146–162. [Google Scholar] [CrossRef]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wang, Y.; Jiang, R.; Xue, R.; Yin, X.; Wu, M.; Meng, Q. Ferroptosis in liver disease: New insights into disease mechanisms. Cell Death Discov. 2021, 7, 276. [Google Scholar] [CrossRef]
- Shen, Z.; Liu, T.; Li, Y.; Lau, J.; Yang, Z.; Fan, W.; Zhou, Z.; Shi, C.; Ke, C.; Bregadze, V.I.; et al. Fenton-Reaction-Acceleratable Magnetic Nanoparticles for Ferroptosis Therapy of Orthotopic Brain Tumors. ACS Nano 2018, 12, 11355–11365. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.; Shi, J.; Wen, K.; Lin, J.; Liu, Q.; Shi, B.; Yan, Y.; Xiao, Z. Molecular Targets of Ferroptosis in Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2021, 8, 985–996. [Google Scholar] [CrossRef]
- Sun, X.; Niu, X.; Chen, R.; He, W.; Chen, D.; Kang, R.; Tang, D. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 2016, 64, 488–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahnson, R.R.; Basu, A.; Lazo, J.S. The role of metallothioneins in anticancer drug resistance. Cancer Treat. Res. 1991, 57, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhu, J.Y.; Zang, X.; Zhai, Y.Z. The Emerging Role of Ferroptosis in Liver Diseases. Front. Cell Dev. Biol. 2021, 9, 801365. [Google Scholar] [CrossRef] [PubMed]
- Louandre, C.; Marcq, I.; Bouhlal, H.; Lachaier, E.; Godin, C.; Saidak, Z.; Francois, C.; Chatelain, D.; Debuysscher, V.; Barbare, J.C.; et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015, 356, 971–977. [Google Scholar] [CrossRef]
- Harding, J.J.; Abou-Alfa, G.K.; Shi, Y.; Whang-Peng, J.; Yuen, M.F.; Saif, W.M.; Tian, A.; Gu, S.; Lam, W.; Liu, S.-H.; et al. A phase II randomized placebo-controlled study investigating the combination of yiv-906 and sorafenib (SORA) in HBV (+) patients (Pts) with advanced hepatocellular carcinoma (HCC). J. Clin. Oncol. 2020, 38, TPS601. [Google Scholar] [CrossRef]
- Wang, Q.; Bin, C.; Xue, Q.; Gao, Q.; Huang, A.; Wang, K.; Tang, N. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021, 12, 426. [Google Scholar] [CrossRef]
- He, Q.; Liu, Y.; Zou, Q.; Guan, Y.S. Transarterial injection of H101 in combination with chemoembolization overcomes recurrent hepatocellular carcinoma. World J. Gastroenterol. 2011, 17, 2353–2355. [Google Scholar] [CrossRef]
- Cramer, S.L.; Saha, A.; Liu, J.; Tadi, S.; Tiziani, S.; Yan, W.; Triplett, K.; Lamb, C.; Alters, S.E.; Rowlinson, S.; et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 2017, 23, 120–127. [Google Scholar] [CrossRef]
- Chaiswing, L.; Zhong, W.; Liang, Y.; Jones, D.P.; Oberley, T.D. Regulation of prostate cancer cell invasion by modulation of extra- and intracellular redox balance. Free Radic. Biol. Med. 2012, 52, 452–461. [Google Scholar] [CrossRef] [Green Version]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Bebber, C.M.; Muller, F.; Prieto Clemente, L.; Weber, J.; von Karstedt, S. Ferroptosis in Cancer Cell Biology. Cancers 2020, 12, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol 2015, 10, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Damia, G.; D’Incalci, M. Clinical pharmacokinetics of altretamine. Clin. Pharmacokinet. 1995, 28, 439–448. [Google Scholar] [CrossRef]
- Alberts, D.S.; Jiang, C.; Liu, P.Y.; Wilczynski, S.; Markman, M.; Rothenberg, M.L. Long-term follow-up of a phase II trial of oral altretamine for consolidation of clinical complete remission in women with stage III epithelial ovarian cancer in the Southwest Oncology Group. Int J. Gynecol. Cancer 2004, 14, 224–228. [Google Scholar] [CrossRef]
- Mendiola, C.; Valdiviezo, N.; Vega, E.; Munoz, A.S.; Ciruelos, E.M.; Manso, L.; Ghanem, I.; Dorta, M.; Manneh, R.; Flores, C.J.; et al. Altretamine for recurrent ovarian cancer or as maintenance after response to second-line therapy. J. Clin. Oncol. 2011, 29, e15589. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Mello, S.S.; Attardi, L.D. Deciphering p53 signaling in tumor suppression. Curr. Opin. Cell Biol. 2018, 51, 65–72. [Google Scholar] [CrossRef]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef]
- Wang, J.; Shanmugam, A.; Markand, S.; Zorrilla, E.; Ganapathy, V.; Smith, S.B. Sigma 1 receptor regulates the oxidative stress response in primary retinal Muller glial cells via NRF2 signaling and system xc(-), the Na(+)-independent glutamate-cystine exchanger. Free Radic. Biol. Med. 2015, 86, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.K.; Patel, A.K.; Shah, N.; Chaudhary, A.K.; Jha, U.K.; Yadav, U.C.; Gupta, P.K.; Pakuwal, U. Oxidative stress and antioxidants in disease and cancer: A review. Asian Pac. J. Cancer Prev. 2014, 15, 4405–4409. [Google Scholar] [CrossRef] [Green Version]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayir, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef]
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef]
- Yu, J.; Wang, J.Q. Research mechanisms of and pharmaceutical treatments for ferroptosis in liver diseases. Biochimie 2021, 180, 149–157. [Google Scholar] [CrossRef]
- Cabral, H.; Makino, J.; Matsumoto, Y.; Mi, P.; Wu, H.; Nomoto, T.; Toh, K.; Yamada, N.; Higuchi, Y.; Konishi, S.; et al. Systemic Targeting of Lymph Node Metastasis through the Blood Vascular System by Using Size-Controlled Nanocarriers. ACS Nano 2015, 9, 4957–4967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Jiang, J.; Xu, S.; Liu, W.; Xie, Q.; Cai, X.; Zhang, J.; Liu, S.; Li, R. Nanoparticle-induced ferroptosis: Detection methods, mechanisms and applications. Nanoscale 2021, 13, 2266–2285. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.; Yan, J.; Liu, Y.; Yang, R.; Pan, D.; Wang, L.; Xu, Y.; Li, X.; Yang, M. Nanoparticle ferritin-bound erastin and rapamycin: A nanodrug combining autophagy and ferroptosis for anticancer therapy. Biomater. Sci. 2019, 7, 3779–3787. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Zhang, L.; Ma, K.; Riegman, M.; Chen, F.; Ingold, I.; Conrad, M.; Turker, M.Z.; Gao, M.; Jiang, X.; et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 2016, 11, 977–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, W.; Mulik, R.S.; Anwar, A.; McDonald, J.G.; He, X.; Corbin, I.R. Low-density lipoprotein docosahexaenoic acid nanoparticles induce ferroptotic cell death in hepatocellular carcinoma. Free Radic. Biol. Med. 2017, 112, 597–607. [Google Scholar] [CrossRef]
- Lim, K.; Han, C.; Dai, Y.; Shen, M.; Wu, T. Omega-3 polyunsaturated fatty acids inhibit hepatocellular carcinoma cell growth through blocking beta-catenin and cyclooxygenase-2. Mol. Cancer Ther. 2009, 8, 3046–3055. [Google Scholar] [CrossRef] [Green Version]
- Weylandt, K.H.; Krause, L.F.; Gomolka, B.; Chiu, C.Y.; Bilal, S.; Nadolny, A.; Waechter, S.F.; Fischer, A.; Rothe, M.; Kang, J.X. Suppressed liver tumorigenesis in fat-1 mice with elevated omega-3 fatty acids is associated with increased omega-3 derived lipid mediators and reduced TNF-alpha. Carcinogenesis 2011, 32, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Zaffaroni, N.; Beretta, G. Nanoparticles for Ferroptosis Therapy in Cancer. Pharmaceutics 2021, 13, 1785. [Google Scholar] [CrossRef]
- Ye, Z.; Hu, Q.; Zhuo, Q.; Zhu, Y.; Fan, G.; Liu, M.; Sun, Q.; Zhang, Z.; Liu, W.; Xu, W.; et al. Abrogation of ARF6 promotes RSL3-induced ferroptosis and mitigates gemcitabine resistance in pancreatic cancer cells. Am. J. Cancer Res. 2020, 10, 1182–1193. [Google Scholar]
- Scheck, A.C.; Perry, K.; Hank, N.C.; Clark, W.D. Anticancer activity of extracts derived from the mature roots of Scutellaria baicalensis on human malignant brain tumor cells. BMC Complement. Altern. Med. 2006, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.; Shi, C.; Li, T.; Wu, Y.; Hu, C.; Huang, G. Solasonine promotes ferroptosis of hepatoma carcinoma cells via glutathione peroxidase 4-induced destruction of the glutathione redox system. Biomed. Pharmacother. 2020, 129, 110282. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sun, T.; Zhong, R.; You, C.; Tian, M. PEGylation of Deferoxamine for Improving the Stability, Cytotoxicity, and Iron-Overload in an Experimental Stroke Model in Rats. Front. Bioeng. Biotechnol. 2020, 8, 592294. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, M.; Liu, Y.; Qiao, Z.; Bai, T.; Yang, L.; Liu, B. Dihydroartemisinin triggers ferroptosis in primary liver cancer cells by promoting and unfolded protein responseinduced upregulation of CHAC1 expression. Oncol. Rep. 2021, 46. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.D.; Tan, S.H.; Ng, S.; Shi, Y.; Zhou, J.; Tan, K.S.; Wong, W.S.; Shen, H.M. Artesunate induces cell death in human cancer cells via enhancing lysosomal function and lysosomal degradation of ferritin. J. Biol. Chem. 2014, 289, 33425–33441. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Zhao, D.; Jin, C.; Li, Z.; Sun, S.; Xia, S.; Zhang, Y.; Zhang, Z.; Zhang, F.; Xu, X.; et al. Dihydroartemisinin Induces Ferroptosis in HCC by Promoting the Formation of PEBP1/15-LO. Oxidative Med. Cell. Longev. 2021, 2021, 3456725. [Google Scholar] [CrossRef]
- Li, Z.J.; Dai, H.Q.; Huang, X.W.; Feng, J.; Deng, J.H.; Wang, Z.X.; Yang, X.M.; Liu, Y.J.; Wu, Y.; Chen, P.H.; et al. Artesunate synergizes with sorafenib to induce ferroptosis in hepatocellular carcinoma. Acta Pharmacol. Sin. 2021, 42, 301–310. [Google Scholar] [CrossRef]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Lim, S.O.; Yan, M.; Hsu, J.L.; Yao, J.; Wei, Y.; Chang, S.S.; Yamaguchi, H.; Lee, H.H.; Ke, B.; et al. TYRO3 induces anti-PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis. J. Clin. Investig. 2021, 131, e139434. [Google Scholar] [CrossRef]
- Sun, L.L.; Linghu, D.L.; Hung, M.C. Ferroptosis: A promising target for cancer immunotherapy. Am. J. Cancer Res. 2021, 11, 5856–5863. [Google Scholar]
- Talty, R.; Bosenberg, M. The role of ferroptosis in melanoma. Pigment Cell Melanoma Res. 2022, 35, 18–25. [Google Scholar] [CrossRef]
- Watson, M.J.; Vignali, P.D.A.; Mullett, S.J.; Overacre-Delgoffe, A.E.; Peralta, R.M.; Grebinoski, S.; Menk, A.V.; Rittenhouse, N.L.; DePeaux, K.; Whetstone, R.D.; et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 2021, 591, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; DuBois, R.N. The Role of Prostaglandin E(2) in Tumor-Associated Immunosuppression. Trends Mol. Med. 2016, 22, 1257–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; Rathmell, J.C. Metabolic Barriers to T Cell Function in Tumors. J. Immunol. 2018, 200, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Muri, J.; Kopf, M. Redox regulation of immunometabolism. Nat. Rev. Immunol. 2021, 21, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Kishton, R.J.; Sukumar, M.; Restifo, N.P. Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab. 2017, 26, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Xiao, L.; Liu, L.; Ye, L.; Su, P.; Bi, E.; Wang, Q.; Yang, M.; Qian, J.; Yi, Q. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021, 33, 1001–1012. [Google Scholar] [CrossRef]
- Xu, S.; Chaudhary, O.; Rodriguez-Morales, P.; Sun, X.; Chen, D.; Zappasodi, R.; Xu, Z.; Pinto, A.F.M.; Williams, A.; Schulze, I.; et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 2021, 54, 1561–1577. [Google Scholar] [CrossRef]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Zhu, S.; Luo, Z.; Li, X.; Han, X.; Shi, S.; Zhang, T. Tumor-associated macrophages: Role in tumorigenesis and immunotherapy implications. J. Cancer 2021, 12, 54–64. [Google Scholar] [CrossRef]
- Sacco, A.; Battaglia, A.M.; Botta, C.; Aversa, I.; Mancuso, S.; Costanzo, F.; Biamonte, F. Iron Metabolism in the Tumor Microenvironment-Implications for Anti-Cancer Immune Response. Cells 2021, 10, 303. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, H.; Evstatiev, R.; Kornek, G.; Aapro, M.; Bauernhofer, T.; Buxhofer-Ausch, V.; Fridrik, M.; Geissler, D.; Geissler, K.; Gisslinger, H.; et al. Iron metabolism and iron supplementation in cancer patients. Wien. Klin. Wochenschr. 2015, 127, 907–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifhofer-Obermair, C.; Tymoszuk, P.; Petzer, V.; Weiss, G.; Nairz, M. Iron in the Tumor Microenvironment-Connecting the Dots. Front. Oncol. 2018, 8, 549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Luo, M.; Yao, F.; Wang, S.; Yuan, Z.; Yang, Y. Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell Signal. 2020, 72, 109633. [Google Scholar] [CrossRef]
- Ruiz-de-Angulo, A.; Bilbao-Asensio, M.; Cronin, J.; Evans, S.J.; Clift, M.J.D.; Llop, J.; Feiner, I.V.J.; Beadman, R.; Bascaran, K.Z.; Mareque-Rivas, J.C. Chemically Programmed Vaccines: Iron Catalysis in Nanoparticles Enhances Combination Immunotherapy and Immunotherapy-Promoted Tumor Ferroptosis. iScience 2020, 23, 101499. [Google Scholar] [CrossRef]
- Irvine, D.J.; Swartz, M.A.; Szeto, G.L. Engineering synthetic vaccines using cues from natural immunity. Nat. Mater. 2013, 12, 978–990. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.-L.; Wu, M.-Y.; Zhang, T.-N.; Wang, C.-G. Ferroptosis Regulator Modification Patterns and Tumor Microenvironment Immune Infiltration Characterization in Hepatocellular Carcinoma. Front. Mol. Biosci. 2022, 9. [Google Scholar] [CrossRef]
- Gao, L.; Xue, J.; Liu, X.; Cao, L.; Wang, R.; Lei, L. A scoring model based on ferroptosis genes for prognosis and immunotherapy response prediction and tumor microenvironment evaluation in liver hepatocellular carcinoma. Aging 2021, 13, 24866–24881. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.H.; Shen, Y.C.; Shao, Y.Y.; Hsu, C.; Cheng, A.L. Sorafenib in advanced hepatocellular carcinoma: Current status and future perspectives. J. Hepatocell. Carcinoma 2014, 1, 85–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacco, R.; Bargellini, I.; Ginanni, B.; Bertini, M.; Faggioni, L.; Federici, G.; Romano, A.; Bertoni, M.; Metrangolo, S.; Altomare, E.; et al. Long-term results of sorafenib in advanced-stage hepatocellular carcinoma: What can we learn from routine clinical practice? Expert. Rev. Anticancer Ther. 2012, 12, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Shingina, A.; Hashim, A.M.; Haque, M.; Suen, M.; Yoshida, E.M.; Gill, S.; Donnellan, F.; Weiss, A.A. In a ‘real-world’, clinic-based community setting, sorafenib dose of 400 mg/day is as effective as standard dose of 800 mg/day in patients with advanced hepatocellular carcimona, with better tolerance and similar survival. Can. J. Gastroenterol. 2013, 27, 393–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Zeng, L.; Zhu, S.; Xie, Y.; Liu, J.; Wen, Q.; Cao, L.; Xie, M.; Ran, Q.; Kroemer, G.; et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 2018, 24, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Montal, R.; Villanueva, A. Randomized trials and endpoints in advanced HCC: Role of PFS as a surrogate of survival. J. Hepatol. 2019, 70, 1262–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.L.; Kang, Y.K.; Lin, D.Y.; Park, J.W.; Kudo, M.; Qin, S.; Chung, H.C.; Song, X.; Xu, J.; Poggi, G.; et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: Results of a randomized phase III trial. J. Clin. Oncol. 2013, 31, 4067–4075. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Llovet, J.M. Targeted therapies for hepatocellular carcinoma. Gastroenterology 2011, 140, 1410–1426. [Google Scholar] [CrossRef] [Green Version]
- Yao, F.; Deng, Y.; Zhao, Y.; Mei, Y.; Zhang, Y.; Liu, X.; Martinez, C.; Su, X.; Rosato, R.R.; Teng, H.; et al. A targetable LIFR-NF-kappaB-LCN2 axis controls liver tumorigenesis and vulnerability to ferroptosis. Nat. Commun. 2021, 12, 7333. [Google Scholar] [CrossRef]
- Walters, R.; Mousa, S.A. Modulations of ferroptosis in lung cancer therapy. Expert. Opin. Ther. Targets 2022. [Google Scholar] [CrossRef]
- Zuo, S.; Yu, J.; Pan, H.; Lu, L. Novel insights on targeting ferroptosis in cancer therapy. Biomark. Res. 2020, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Mbah, N.E.; Lyssiotis, C.A. Metabolic regulation of ferroptosis in the tumor microenvironment. J. Biol. Chem. 2022, 101617. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Kalathur, R.K.R.; Coto-Llerena, M.; Ercan, C.; Buechel, D.; Shuang, S.; Piscuoglio, S.; Dill, M.T.; Camargo, F.D.; Christofori, G.; et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol. Med. 2021, 13, e14351. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Schulte, N.; Eastman, A. Multiple mechanisms of resistance to cis-diamminedichloroplatinum(II) in murine leukemia L1210 cells. Cancer Res. 1987, 47, 2056–2061. [Google Scholar] [PubMed]
- Shen, Z.; Song, J.; Yung, B.C.; Zhou, Z.; Wu, A.; Chen, X. Emerging Strategies of Cancer Therapy Based on Ferroptosis. Adv. Mater. 2018, 30, e1704007. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheung, Y.K.; Ng, D.K.P.; Fong, W.P. Immunogenic necroptosis in the anti-tumor photodynamic action of BAM-SiPc, a silicon(IV) phthalocyanine-based photosensitizer. Cancer Immunol. Immunother. 2021, 70, 485–495. [Google Scholar] [CrossRef]
- Yang, Y.; Lin, J.; Guo, S.; Xue, X.; Wang, Y.; Qiu, S.; Cui, J.; Ma, L.; Zhang, X.; Wang, J. RRM2 protects against ferroptosis and is a tumor biomarker for liver cancer. Cancer Cell Int. 2020, 20, 587. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Category | Drug/ Compound | Mechanism | Cancer Research | |
|---|---|---|---|---|
| Tumor Type | Effect on Ferroptosis | |||
| System xc− inhibition | Erastin | Inhibition of system xc− and VDAC2/3 [2] | human lung cancer cell line (A549, Calu-1) [98,99] human liver cancer cell line (HepG2) [100] human leukemia cell line (HL60) [95] human B cell lymphoma cell line (SU-DHL-8, WSU-DLCL-2) [50] human fibrosarcoma cell line (HT1080) [99] human kidney carcinoma cell line (Caki-1, 786-O) [99] human ovarian epithelial-endometroid carcinoma cell line (COV362) [101] human neuroblastoma cell line (SH-SY5Y) [102] | Induction |
| lung cancer xenograft mice (N5CP) [103] high grade serous ovarian carcinoma (HGSOC) xenograft mice [101] | ||||
| Sulfasalazine | A prodrug of 5-aminosalicylic acid that inhibits cystine uptake [104] | human liver cancer cell lines (Huh-7, SK-Hep-1, HepG2, PLC/ PRF/5, Hep3B) [105] human bladder cancer cell line (T24) [106] human cervix carcinoma cell line (HeLa) [98] human astrocytoma cell line (U373MG) [98] human glioblastoma cell line (T98G) [98] human embryonic kidney cell line (293T) [98] human colorectal cancer cell lines (HCT116, HT29, LOVO, DLD1) [107] human glioma cell line (D54-MG, STTG-1, U251-MG, U87-MG) [108] human breast cancer cell lines (MDA-MB-231, T47D, BT549 and MCF7) [109] | Induction | |
| human glioma xenograft mice [108] | ||||
| Sorafenib | Inhibits system xc−-mediated cystine import, leading to GSH depletion and iron-dependent accumulation of lipid-ROS [104,110] | human hepatocellular carcinoma cell line (Huh7, PLC/PRF5, Hep3B, HepG2) [111] human kidney carcinoma cell line (Caki-1, ACHN, 786-O) [111,112] human melanoma cell line (SK-MEL-3) [111] human lung carcinoma cell line (NCI-H460, N5CP, A549) [103] human pancreatic carcinoma cell lines (PANC-1) [111] human colon carcinoma cell lines (HCT116, HT-29) [111] human fibrosarcoma cell line (HT-1080) [113] | Induction | |
| patient-derived human hepatocellular carcinoma xenograft mice (HCC-PDX) [114] thyroid cancer xenograft mice acute myelogenous leukemia xenograft mice [115] | ||||
| DAZAP1 | Maintains SLC7A11 mRNA stability [116] | human hepatocellular carcinoma cell lines (HepG2, SMMC-7721, Hep3B, Bel-7402, Huh7, L02) [116] | Inhibition | |
| BECN1 | Blocks system xc− activity via binding of SLC7A11 [117] | human colorectal cancer cell lines (HCT116, CX-1, LoVo, SW48) [118] Human pancreatic cancer cell line (PANC1) [119] human fibrosarcoma cell line (HT1080) [119] human non-small-cell lung cancer cell line (Calu-1) [119] human synovial sarcoma cell line (SW982) [120] | Induction | |
| human colon cancer xenograft mice (HT-29) [121] human fibrosarcoma xenograft mice (HT1080) [119] | ||||
| NRF2 | Key regulator of antioxidant response including system xc−-expression [122] | human lung cell lines (H460, H1975, H1299,95D, A549) [123] human hepatocellular carcinoma cell lines (Hep3B, Bel-7402, and HepG2) [124] human breast carcinoma cell line (MDA-MB-231) [125] human neuroblastoma cell line (SH-SY5Y) [125] | Inhibition | |
| human lung cancer xenograft mice [123] | ||||
| QSOX1 | Inhibition of NRF2 activation [126] | human hepatocellular carcinoma cell lines (HCC-LM3, MHCC97L, MHCC97H, SMMC7721, SMMC-7402, HuH7, PLC, Hep3B, HepG2) [127] cervical carcinoma cell line (HeLa) [128] human breast carcinoma cell line (BT549, BT474) [129] human kidney carcinoma cell line (786-O) [130] | Induction | |
| human primary breast tumor clinical study [131] human kidney carcinoma xenograft mice [130] | ||||
| BCAT2 | Regulates intracellular glutamate concentration [132] | murine hepatocellular carcinoma cell line (H22) [132] murine pancreatic carcinoma cell line (Panc02) [132] human pancreas carcinoma cell line (AsPC-1) [132] human hepatocellular carcinoma cell line (HepG2) [132] human fibrosarcoma cell line (HT1080) [132] human colon cancer cell line (SW-480) [132] patient-derived human breast carcinoma cell line (BCC, MCF-7) [133] human ovarian cancer cell line (HeLa) [134] human colon cancer cell line (HCT116) [134] | Inhibition | |
| hepatocellular carcinoma xenograft mice (H22) pancreatic carcinoma xenograft mice (SW1990) [135] pancreatic ductal carcinoma xenograft mice [136] | ||||
| Fenugreek (trigonelline) | Blocks NRF2 | human hepatocellular carcinoma cell line (HepG2) [137] human epidermoid cancer cell line (HEp2) [138] human breast carcinoma cell line (MCF-7) [138] | Induction | |
| σ1R | Regulates ROS accumulation via NRF2 [139] | human hepatocellular carcinoma cell line (HepG2, Huh-7, SMMC-7721, PLC/PRF/5) hepatocellular carcinoma xenograft mice (Huh-7) [139] human breast cancer cell lines (MCF-7, MCF-41) [140] | Inhibition | |
| GPX4 Inhibition | RSL3 | Inhibition of GPX4 phospholipid peroxidase activity [50] | human head and neck cancer cell line (AMC-HN2-11) [141] human lung cancer cell lines (H1650, HCC827, PC9, NCI-H1975) [142] human prostate cancer cell lines (DU145, PC-3, 22Rv1, LNCaP, NCI-H660) [143] human colon cancer cell lines (HCT116, LoVo, HT29) [144] human acute lymphoblastic leukemia cell line [145] rat glioma cell line (C6) [146] human glioma cell line (U373, U87, U251 cell lines) [146] human pancreatic carcinoma cell lines (PANC-1) [147] human ovarian carcinoma cell line (IGROV-1) [147] human bone rhabdomyosarcoma cell line (A-673) [147] human breast carcinoma cell line (MCF7) [147] human prostate carcinoma cell line (PC-3) [147] | Induction |
| head and neck cancer xenograft mice [141] prostate cancer xenograft mice [143] | ||||
| FIN56 | Degradation of GPX4 protein [147] | human lung carcinoma cell line (HT-1080, BJeLR, Calu-1) [147] human osteosarcoma cell line (143B) [147] human kidney carcinoma cell line (CAKI-1, UO-31, 786-O) [148] human bone rhabdomyosarcoma cell line (A-673) [147] human breast adenocarcinoma cell line (MCF7) [147] human prostate carcinoma cell line (PC-3) [147] human non-small cell lung cancer cell line (NCI-H1975) [147] human colorectal carcinoma cell line (LS411N) [147] human pancreatic adenocarcinoma cell lines (PANC-1) [147] human ovarian carcinoma cell line (IGROV-1) [147] | Induction | |
| lung carcinoma xenograft mice [50] | ||||
| DPIs | Inhibition of GPX4 via GSH depletion [50] | human lung carcinoma cell line (BJeLR, Calu-1) [2,50] human renal cell carcinoma cell lines (A498, TK-10, SN12C, CAKI-1, RXF-393, ACHN, 786-0, UO-31) [50] human fibrosarcoma cell line (HT1080) [50] | Inhibition | |
| lung carcinoma xenograft mice [50] | ||||
| Altretamine | Anticancer agent inhibiting GPX4 without depleting cells of GSH [149] | human B-cell lymphoma cell line (OCI-LY3, OCI-LY7, U-2932 DLBCL) [149] human osteosarcoma cell line U-2-OS [149] human breast cancer cell lines (MCF-7, MDA-MB-231) [149] | Induction | |
| Trial Identifier | Phase | Drug | Target | N | Study Start | Estimated Study Completion |
|---|---|---|---|---|---|---|
| NCT03518502 | IV | Sorafenib monotherapy vs TACE + sorafenib | VEGFR | 130 | 1 March 2012 | 28 February 2022 |
| NCT01624285 | II | Sorafenib tosylate | VEGFR | 356 | 16 July 2012 | July 2023 |
| NCT01730937 | III | SBRT + sorafenib tosylate | radiation, VEGFR | 193 | April 2013 | June 2025 |
| NCT01840592 | II | Sorafenib + doxorubicin (TACE) | cytostatica, VEGFR | 30 | April 2013 | April 2022 |
| NCT02733809 | IV | Sorafenib | VEGFR | 40 | January 2014 | December 2024 |
| NCT02143401 | I | Navitoclax + sorafenib tosylate | Bcl-2, VEGFR | 44 | 7 November 2014 | 7 May 2022 |
| NCT02576509 | III | Nivolumab vs. sorafenib | PD-1, VEGFR | 743 | 7 December 2015 | 30 June 2022 |
| NCT02716012 | I | MTL-CEBPA + sorafenib | VEGFR | 51 | 1 March 2016 | 1 January 2022 |
| NCT03037437 | II | Sorafenib + HCQ | autophagy inhibition, VEGFR | 68 | 16 February 2017 | April 2023 |
| NCT03298451 | III | Durvalumab + tremelimumab vs. sorafenib or lenvatinib | PD-1 + CTLA-4, VEGFR | 1504 | 11 October 2017 | 30 April 2023 |
| NCT03211416 | II | Sorafenib tosylate + pembrolizumab | PD-1, VEGFR | 41 | 7 December 2017 | 7 December 2022 |
| NCT03412773 | III | Tislelizumab vs. sorafenib | PD-1, VEGFR | 674 | 28 December 2017 | May 2022 |
| NCT03434379 | III | Atezolizumab + bevacizumab vs. sorafenib | PD-L1 + VEGFR | 558 | 15 March 2018 | 30 June 2022 |
| NCT03439891 | II | Nivolumab + sorafenib | PD-1, VEGFR | 24 | 16 April 2018 | 30 September 2023 |
| NCT03652467 | I | Deferoxamine + conventional TACE | Ferric ions | 100 | 1 September 2018 | 31 December 2023 |
| NCT03645980 | II | DKN-01 + sorafenib | DKK-1, VEGFR | 70 | 10 October 2018 | 31 August 2022 |
| NCT03755791 | III | Cabozantinib + atezolizumab vs. sorafenib | VEGFR | 740 | 10 December 2018 | 1 December 2021 |
| NCT03775395 | III | HAIC + sorafenib | chemotherapy, VEGFR | 250 | 12 December 2018 | 1 December 2021 |
| NCT02436902 | III | TACE + sorafenib vs. monotherapy | VEGFR | 240 | 1 February 2019 | 30 August 2022 |
| NCT04926532 | II | Toripalimab + sorafenib | PD-1, VEGFR | 30 | 1 August 2019 | 31 December 2021 |
| NCT03606590 | II | NovoTTF-100L(P) + sorafenib | electrical field + VEGFR | 25 | 15 February 2019 | September 2021 |
| NCT03965546 | I | Sorafenib + ET140202-T cell | AFP/MHC complex, VEGFR | 27 | 30 May 2019 | June 2022 |
| NCT04926532 | II | Toripalimab + sorafenib | PD-1, VEGFR | 30 | 1 August 2019 | 31 December 2021 |
| NCT03971201 | II | Surgery + sorafenib vs. sorafenib | VEGFR | 200 | 6 September 2019 | 30 June 2023 |
| NCT04143191 | III | Sorafenib + TACE | VEGFR | 158 | 15 September 2019 | 15 September 2023 |
| NCT04039607 | III | Nivolumab + ipilimumab vs. sorafenib or lenvatinib | PD-1 + CTLA-4, VEGFR | 634 | 30 September 2019 | 4 January 2025 |
| NCT04232722 | II | Sorafenib + arsenical | VEGFR + apoptosis | 43 | 1 January 2020 | 30 June 2022 |
| NCT04000737 | II | YIV-906 + sorafenib | VEGFR | 125 | 10 January 2020 | 19 May 2023 |
| NCT04387695 | III | SBRT + TACE + sorafenib | radiation, VEGFR | 54 | 30 April 2020 | 1 June 2023 |
| NCT04518852 | II | TACE + sorafenib and PD-1 mAb | PD-1, VEGFR | 60 | 14 September 2020 | 31 January 2023 |
| NCT04599777 | II | TACE + sorafenib and tislelizumab | PD-1, VEGFR | 30 | 1 October 2020 | December 2022 |
| NCT04687163 | III | Sorafenib or lenvatinib + HAIC | chemotherapy, VEGFR | 400 | 30 December 2020 | 1 December 2022 |
| NCT04229355 | III | DEB-TACE + sorafenib | VEGFR | 90 | 2 February 2021 | 30 December 2022 |
| NCT04763408 | IV | Sorafenib, lenvatinib | VEGFR | 1000 | 9 April 2021 | 31 July 2029 |
| NCT04770896 | III | Atezolizumab + lenvatinib or sorafenib | PD-L1 + VEGFR | 554 | 26 April 2021 | 8 October 2024 |
| NCT04967495 | not applicable | TACE + sorafenib or lenvatinib + iodion-125 seeds brachytherapy (TACE-MKI-I) | radiotherapy, VEGFR | 171 | 9 July 2021 | 8 January 2025 |
| NCT04992143 | II | TACE + tilelizumab and sorafenib | PD-1, VEGFR | 20 | 20 August 2021 | 30 June 2023 |
| NCT04465734 | III | HLX10 mAb + HLX04 mAb vs. sorafenib | PD-1, VEGFR | 477 | 15 November 2021 | 15 March 2024 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kusnick, J.; Bruneau, A.; Tacke, F.; Hammerich, L. Ferroptosis in Cancer Immunotherapy—Implications for Hepatocellular Carcinoma. Immuno 2022, 2, 185-217. https://doi.org/10.3390/immuno2010014
Kusnick J, Bruneau A, Tacke F, Hammerich L. Ferroptosis in Cancer Immunotherapy—Implications for Hepatocellular Carcinoma. Immuno. 2022; 2(1):185-217. https://doi.org/10.3390/immuno2010014
Chicago/Turabian StyleKusnick, Johanna, Alix Bruneau, Frank Tacke, and Linda Hammerich. 2022. "Ferroptosis in Cancer Immunotherapy—Implications for Hepatocellular Carcinoma" Immuno 2, no. 1: 185-217. https://doi.org/10.3390/immuno2010014