1. Introduction
This review shall focus on the critical intra-cellular signalling dysregulation caused by SARS-CoV-2, which triggers the chronic inflammation that deteriorates into COVID-19 [
1,
2]. Two effective therapeutic solutions to COVID-19, 28-amino-acid Vasoactive Intestinal Peptide and 14-amino-acid Human Ezrin Peptides, seem to have related mechanisms of action, which may be based on TIRAP inhibition, PKA activation, and increased transcription-factor CREB-mediated expression. These therapeutic peptides not only stop the virus-triggered innate-immune-activation, which is the cause of the destructive COVID-19 inflammatory-disease progression, they also amplify adaptive anti-viral B-Cell and T-Cell immune responses that inhibit the SARS-CoV-2 virus life cycle and prevent re-infection.
Regulated innate and adaptive immune responses are essential to limit viral infections and stop inflammation-related tissue damage. Innate immunity is the ancient first line of defence, which is mobilised within minutes against viral, bacterial, or fungal invasion of multicellular organisms. The innate immune system evolved with multicellular animal species to defend against invading microorganisms, mainly by detecting characteristic conserved carbohydrate and lipid polymers that display Pathogen-Associated-Molecular-Patterns (PAMPs), which are recognised by genetically pre-programmed Pattern Recognition Receptors (PRRs), for example the Toll-Like-Receptors (TLRs).
On detection of PAMPs, a defensive inflammatory response is rapidly triggered, which is mediated by the transcription, translation, activation, and secretion of different soluble intra- and inter-cellular signals such as interferons (IFN), interleukins, chemokines, and growth factors. The main cell types of the innate-immune-system are myelo-monocyte cells, natural killer (NK) cells, neutrophils, and eosinophils, but elements of the innate defence mechanism are found in all cell types, particularly in epithelial cells of the mucus membranes, which are constantly exposed to the external environment and pathogens.
In contrast, the adaptive-immune-system evolved later and is based on more flexible biological software that can learn, memorise and recognise polypeptide targets that represent invading microorganisms, during an organism’s lifetime. The adaptive flexible-targeting response is based on the expression of novel antibodies produced by B-cells and novel TCR receptors on T-cells. A few days after infection, B-cells and T-cells, including helper (Th), T regulatory (Treg), and T cytotoxic (Tc) subsets, are mobilized to eliminate invaders.
Effectors of both the innate and adaptive immune responses are usually highly regulated and coordinated. Unfortunately, SARS-CoV-2 has been adapted to knock out and corrupt elements of the innate immune response, leading to shut-down of interferon defence and induction of a wild-fire of inflammation, which is the main cause of COVID-19. Virus-induced signalling-chaos in the immune-system allows SARS-CoV-2 to evade detection and elimination.
3. COVID-19: Mild>Moderate>Severe
3.1. COVID-19 Disease Progression
As the disease progresses in the airways and lungs, viral infection spreads through the patchwork of ciliated cells and mucus secretory cells, penetrating further down the respiratory tract. Eventually, SARS-CoV-2 infects the alveolar compartment of the lungs and the alveolar type II cells, which also express ACE2 and TMPRSS2 on their apical cell-membranes.
Alveolar type II cells secrete the pulmonary-surfactant that is essential for the low-surface-tension air-liquid interface that facilitates gas exchange. Secretion of the pulmonary-surfactant is controlled by the apical protein complex of ezrin+NHERF+NHE+CFTR and viral-interference with pulmonary-surfactant production reduces oxygen saturation of the pulmonary blood. Viscous airway-surfactant is the origin of the characteristic COVID-19 low blood-oxygen saturation and “dry-cough”.
In the peripheral blood of moderate-COVID-19 patients, there is a steady increase in the concentration of pro-inflammatory cytokines; IL-1, IL-6, IL-8, TNFα, plus negative regulators, such as anti-inflammatory interleukin-10 (IL-10). However, the peripheral blood mononuclear cells of these patients have impaired inflammatory cytokine gene-expression responses, compared to healthy controls. High plasma levels of circulating anti-inflammatory IL-10 is the probable cause of this inhibition of inflammatory cytokine expression by PBMCs. However, the origin of the high plasma levels of pro-inflammatory IL-1, IL-6, IL-8, and TNFα, and anti-inflammatory IL-10, is deep in COVID-19-damaged alveoli tissues of the lungs [
18].
In the SARS-CoV-2 infected lungs, the progressive breakdown of cell–cell tight-junctions, immune cell invasion of the airway-lumen, the failure of lung-surfactant production, together with alveolar flooding and collapse, lead to severe COVID-19 that produces the unique “ground glass” appearance of lung-tissue on X-ray images. As patients deteriorate to end-stage COVID-19, an extremely complex life-threatening condition develops, associated with out-of-control inflammatory processes affecting multiple organ systems.
3.2. Severe-COVID-19 Is Controlled by m-RAGE: s-RAGE Inflammation in the Lungs
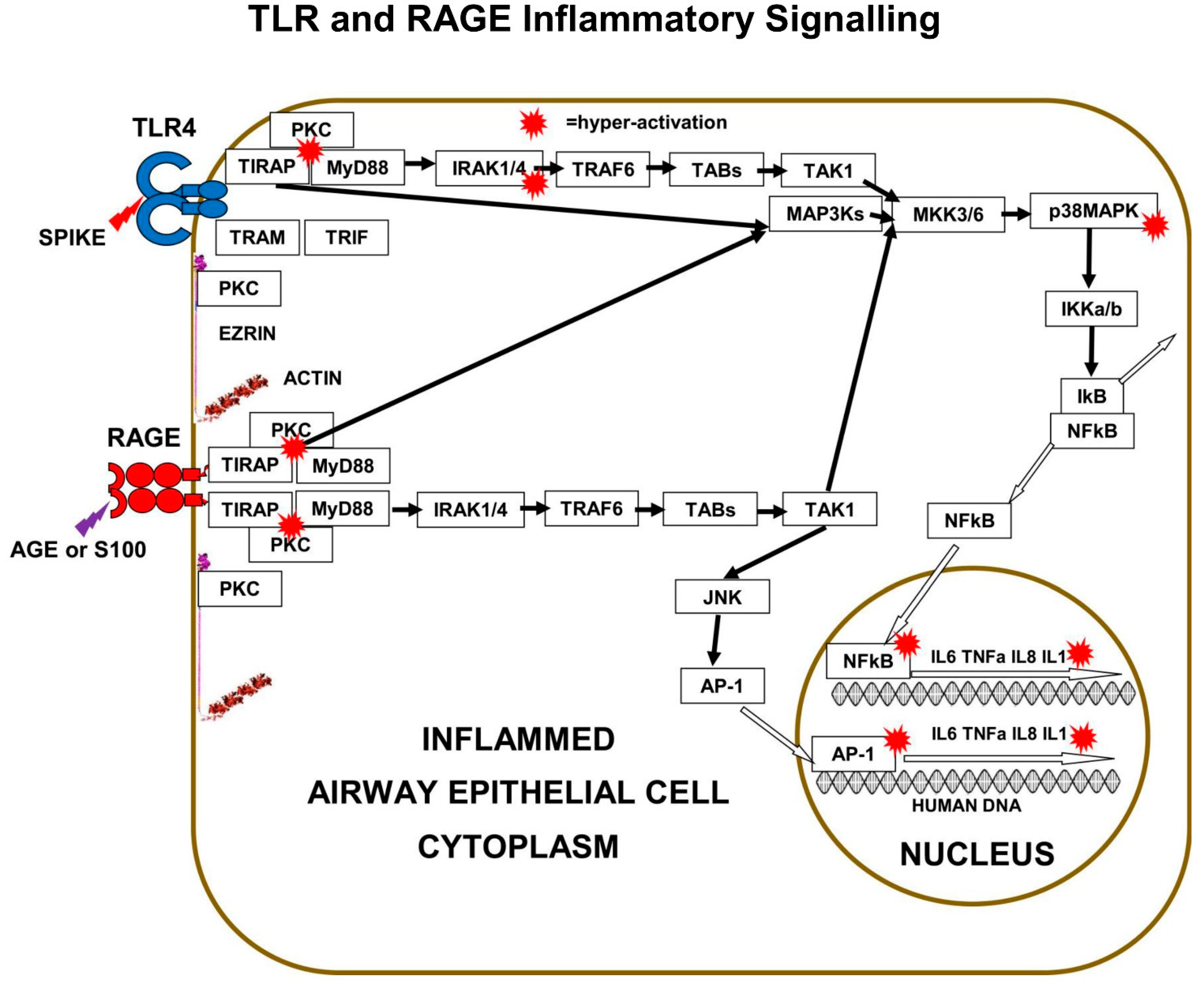
Cell-surface m-RAGE is a pattern recognition receptor of the innate-immune-system, which triggers an inflammation pathway that becomes very significant during the deterioration towards severe COVID-19. m-RAGE binds broad classes of ligands such as Damage Associated Molecular Patterns (DAMPs), and Pathogen-Associated Molecular Patterns (PAMPs). m-RAGE is not only found in the cells of the mucus membranes of the airways, but also in multiple cell types throughout the body, where it is triggered by various ligands associated with diverse chronic inflammatory states.
Cell-membrane expressed m-RAGE is a 35-kDa protein of the immunoglobulin superfamily. The 382-amino-acids of the m-RAGE protein-chain are folded into a tertiary structure with three extra-cellular domains exposed on the cell-surface, in the form of an outer N-terminal variable V-domain, connected to two distinct C1 and C2 immunoglobin-like domains. The V+C1+C2 domains comprise of 314-amino-acids and are attached to a single trans-membrane helix of 27 amino-acids and a short highly charged cytoplasmic-tail of 41 amino-acids.
The variable V-domain of m-RAGE is involved in ligand binding, while its highly charged cytoplasmic tail binds to the FERM-domain of phosphorylated-ezrin and to cell-signalling proteins. The homo-dimerization of two m-RAGE proteins in the cell membrane is an important step for ligand binding, receptor activation, and induction of various inflammatory signalling cascades.
Both TLRs and m-RAGE depend on PKC and the two adaptor proteins, MyD88 and TIRAP, to form a PKC+TIRAP+MyD88 multi-protein complex, needed to signal downstream to induce inflammatory cytokines. However, only TLRs bind to “TRIF-Related-Adaptor-Molecule” (TRAM), which forms a complex with “TIR domain-containing adaptor Inducing-InterFeron-β” (TRIF).
MMP-9 protease digestion of the cytoplasmic tail of m-RAGE simultaneously reduces the cell-surface receptor-density of m-RAGE and causes the external V+C1+C2 domains of m-RAGE (known as s-RAGE) to become soluble in the inter-cellular medium. Plasma-soluble s-RAGE competitively-inhibits m-RAGE signalling, by binding and removing m-RAGE-activating-ligands. Increasing m-RAGE proteolysis and the plasma concentration of s-RAGE inhibits the intensity of m-RAGE inflammatory signalling.
s-RAGE accumulates in plasma and Broncho Alveolar Lavage Fluid (BALF) during Acute Lung Injury (ALI). In a clinical study of SARS-CoV-2 induced COVID-19, 22 healthy volunteers were compared, with 23 asymptomatic infected patients, and with 35 infected patients with COVID-19 lung damage. The plasma-levels of soluble s-RAGE were measured, and it was found that younger and healthier patients generally had high stable plasma-levels of inhibitory s-RAGE. In contrast, elderly patients developing severe-COVID-19 had low but rising s-RAGE levels. Increasing plasma s-RAGE indicated inflammatory processes in the lungs induced by active m-RAGE [
19]. High and rising plasma concentrations of s-RAGE in severe-COVID-19 cases predicts disease-deterioration and death [
20].
3.3. Severe-COVID-19 in the Elderly & the Obese, Due to AGE-Ligands of RAGE
m-RAGE receptors are expressed on the cell surface of many cell types and recognise proteins that have been chemically modified by non-enzymatic reactions in chronic pro-oxidant and hyper-glycaemic disorders. Concentrations of these ligands called “Advanced Glycation End products”, or AGEs, are elevated in old-age and obesity, and result in chronic triggering of m-RAGE.
In addition, the cell-surface expression density of m-RAGE increases with both increasing age and increasing obesity. High expression levels of m-RAGE are associated with various pathologies such as arthritis, arteriosclerosis, diabetes, and cancer. It is significant that knockout of m-RAGE in mice models prevents some age-related diseases such as atherosclerosis, nephron-sclerosis, and Alzheimer’s disease. The absolute and relative m-RAGE: s-RAGE expression levels are part of the mechanism of the underlying co-morbidities that confer the higher risk of severe-COVID-19 in the elderly and the obese. AGE triggering of m-RAGE leads to phosphorylation of both transcription factor NFκB and transcription factor AP-1 that cause pro-inflammatory cytokine expression (
Figure 3) [
21].
3.4. COVID-19 Involves S100A12-Protein Activation of m-RAGE
S100-Calgranulin C, also known as EN-RAGE and S100A12, binds to the C1-domain of m-RAGE, without a significant involvement of either the V or C2 domains. It is also excreted by damaged cells and behaves like a cytokine, amplifying systemic pro-inflammatory signalling. S100A12 activates m-RAGE to induce inflammatory cytokines [
23].
S100A12 is strongly expressed in many inflammatory diseases such as Crohn’s disease, cystic fibrosis, atherosclerosis, rheumatoid arthritis, psoriasis, and the gastric tissue inflammation triggered by Helicobacter pylori. S100A12 protein is a biomarker of pulmonary injury and is implicated in sepsis-induced Acute Respiratory Distress Syndrome (ARDS).
In the plasma of COVID-19 patients, there are pathologically high levels of S100A12 that correlates with deterioration to severe COVID-19 [
24]. Activated macrophages from the peripheral blood of COVID-19 patients also display
S100A12 gene expression that significantly correlates with plasma S100A12 protein concentrations. In type-I alveolar epithelial cells in the lung, S100A12 binds m-RAGE where it maintains chronic inflammation.
Some of the immune dysregulation in COVID-19 can be partly explained by the observation that HLA receptors are down-regulated when
S100A12 gene expression is up-regulated. This strong inverse-correlation between innate-immune-activation and gene-expression of HLA-DPA1, HLA-DPB1, and HLA-DR, part of the adaptive-immunity antigen-presentation system, could be part of the explanation for chronic viral infection [
18,
25].
3.5. m-RAGE Downstream-Signalling via PKC, TIRAP, & MyD88
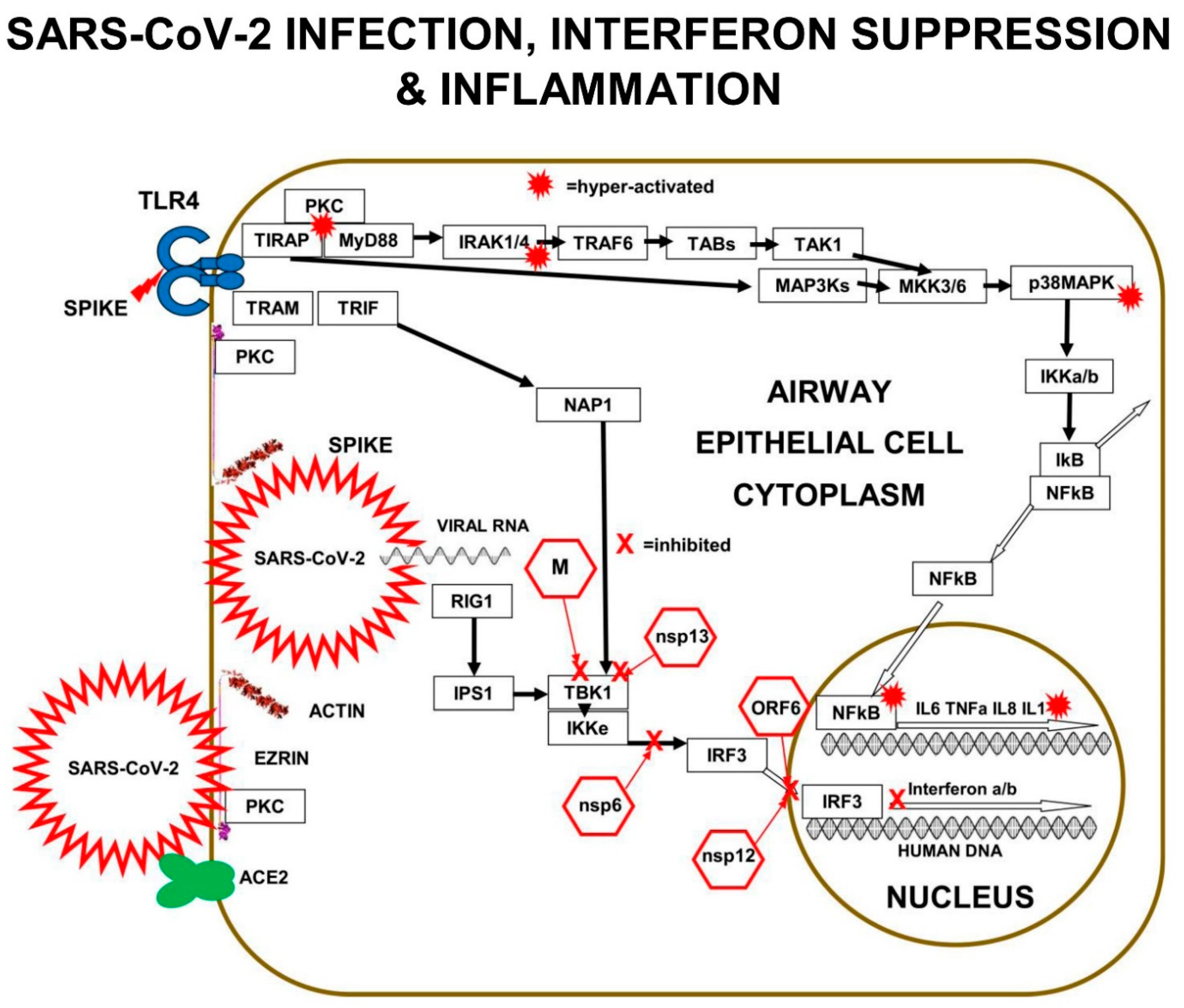
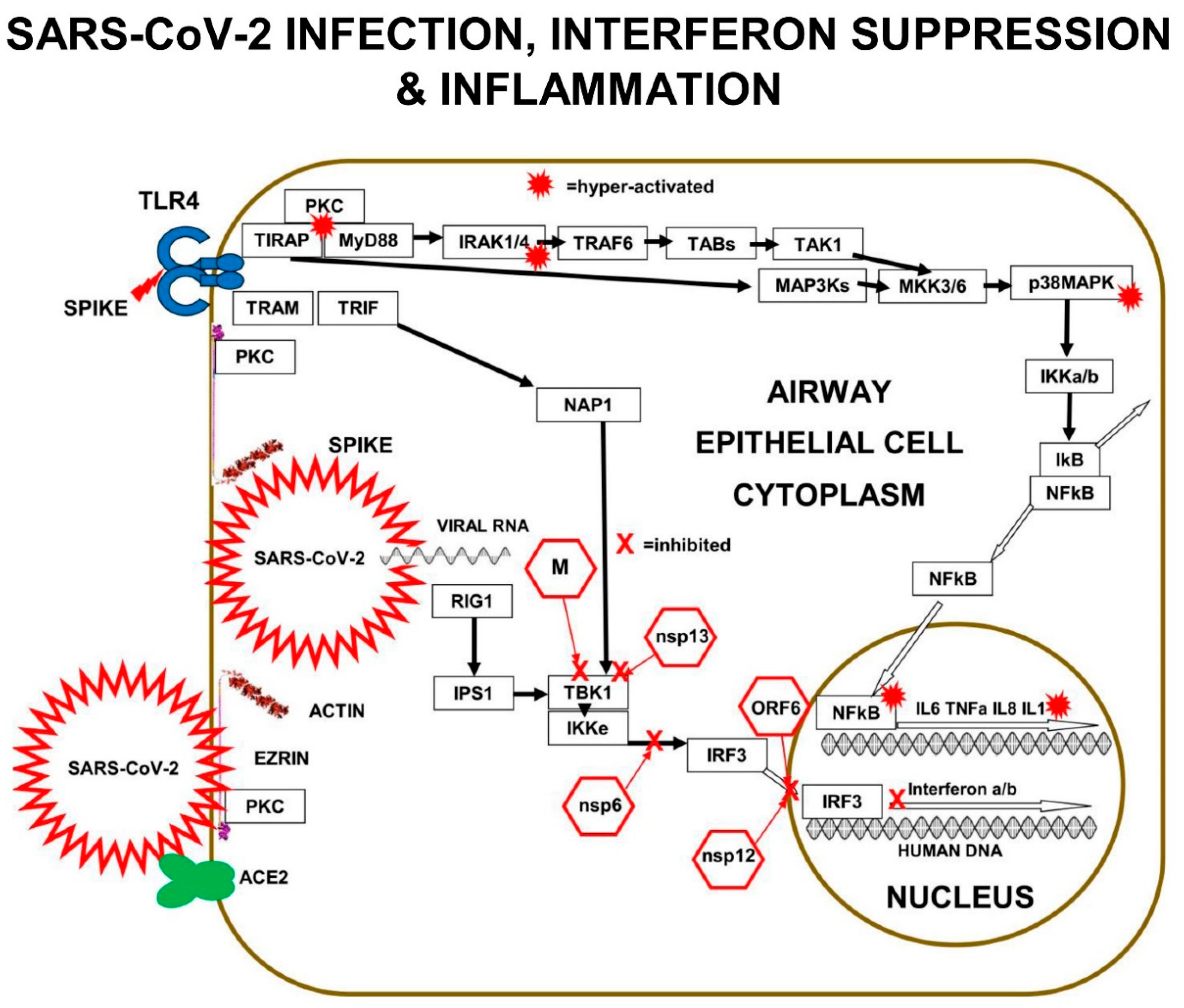
The main downstream pathway from m-RAGE resembles TLR signalling. Ligand triggering of m-RAGE causes phosphorylation and activation of p38MAPK and IKKa/b, followed by de-repression of IκB-inhibition, leading to activation of transcription-factor NFκB. m-RAGE does not induce interferon expression, because there is no binding site on the cytoplasmic domain of m-RAGE for TRAM.
Cell-surface m-RAGE is activated by serum AGEs, HMGB1, S100-proteins, and DAMPs. Ligand-binding to m-RAGE induces a conformational change in the receptor, which causes the binding and phosphorylation of sub-membrane TIRAP, a 221-amino-acid two-domain protein. TIRAP is a central mediator of innate-immune-responses and inflammatory signalling from both activated m-RAGE and from activated Toll Like Receptors (TLRs) [
26].
The charged cytoplasmic-domains of m-RAGE and TLRs bind to TIRAP, and the TIR domain of TIRAP binds to MyD88, forming an m-RAGE or TLR+TIRAP+MyD88 multi-protein-complex. This TIRAP-bridge to MyD88 is common to both m-RAGE signalling and TLR signalling. This is the first step in MyD88-dependent signalling downstream to intracellular signalling mediators and effector proteins, which cause inflammation.
Multi-protein-complex formation by both Toll Like Receptors and m-RAGE is dependent on PKC-z phosphorylation and activation. The PKC activation requires phosphorylation by the GTPase-p21Ras, the small signalling-switch which is complexed with the FERM-domain of ezrin. When extra-cellular ligands like S100 proteins or AGEs bind to cell-surface domain of m-RAGE, a conformation change induces the activation and binding of phosphorylated PKC-z or -f, to the cytoplasmic domain of m-RAGE, leading to phosphorylation of its Ser391.
Protein Kinase C-d also constitutively interacts with the TIR domain of TIRAP and phosphorylates its tyrosine-Y106. Phosphorylation of Ser391 on m-RAGE enhances RAGE-cytoplasmic-domain binding to the Y106-phosphorylated TIR domain of TIRAP, creating a multi-protein complex of TIRAP+m-RAGE+PKC+Ras+Ezrin. Protein Kinase C binds to both the activated ezrin-Alpha-domain and TIR domain of TIRAP [
27]. The TIRAP+PKC-d complex is required for both TLR2-induced and TLR4-induced activation of the p38MAPK>>NFκB>IL-6 and TNFa signalling pathway [
28].
3.6. Signals Further Downstream of TIRAP
TIRAP is the origin of many downstream pro-inflammatory signalling pathways [
29]. The best studied is LPS triggering of TLR4, which results in PKC-d phosphorylation of TIRAP at Y106, activation of MyD88, which then activates both IRAK4 and TRAF6, which branches signals down three different tracks. The first track is a kinase phosphorylation cascade from IRAK1/4 to TAB3 to TAK1 to MKK3/6 to p38MAPK to IKKa/b, leading to activation of transcription factor-NFκB. Direct inhibition of either PKC-z or TIRAP or MyD88 results in decreased production of pro-inflammatory cytokines IL-6, IL-8, TNFα, and Il-1β.
The second track is a kinase phosphorylation cascade from TRAF6 to TAB1/2 to TAK1 to MKK3/6 to p38MAPK to IKKa/b, again leading to activation of transcription factor-NFκB. The third track is another kinase phosphorylation cascade from TRAF6, to TAB1/2, to TAK1, to MEK1/2, to ERK1/2, to transcription-factor AP-1, a “Leucine Zipper” heterodimer of two of four alpha-helical proteins: c-Fos, c-Jun, ATF, and JDP.
In a similar manner to TLRs, m-RAGE also forms a multi-protein-complex with PKC (d or z) and TIRAP, which is followed by phosphorylation and activation of MyD88, then the activation of the same three signalling tracks to activate transcription-factors NFκB and AP-1.
Other signalling pathways also originate from the multi-protein-complexes of TLRs or m-RAGE+PKC+TIRAP+MyD88. Normally, RAGE and TLRs are cross-regulated and coordinate together to regulate the inflammatory signal through TIRAP and MyD88.
There are two other mitogen-activated-protein-kinase pathways involving ERK1/2 and JNK activation. In addition, there is activation of Ezrin+PI3K>PKB/Akt signalling; and pathways originating from the activation of small GTPases, such as ezrin-associated p21-Ras, Rac-1, or cdc42.
These various kinase-cascades cause the differential-activation of various transcription factors, such as NFκB, AP-1, CREB, and STAT-3. Activated transcription-factors migrate from the cytoplasm into the nucleus, where they interact with the DNA-promoters of genes. NFκB and AP-1 increase expression of pro-inflammatory cytokines such as IL-6, TNFα, IL-8, and IL-1β. In addition, m-RAGE gene-expression is controlled by NFκB, so triggering of m-RAGE results in a positive-feedback-loop that expresses more m-RAGE.
3.7. Signal-Transduction from AGE>RAGE to p38MAPK
Aging and age-related-diseases are associated with increases in AGEs in the blood plasma. Chemically modified Human-Serum-Albumin (CML-HSA) is a major AGE product, which progressively accumulates during aging, diabetes, and renal failure [
30].
In human monocyte THP-1 cells, CML-HSA triggers m-RAGE to activate the p38MAPK pathway, which not only activates transcription factor NFκB, but also induces
NFκB gene expression. CML-HSA Activation of NFκB increases secretion of pro-inflammatory cytokines such as IL-6, TNFα, IL-1b, MCP-1, M-CSF, and RAGE itself [
31].
Small p21 GTPase Ras, and ERK1/2, also have a role in regulating and amplifying the p38MAPK inflammatory signal. For example, HMGB1-induced RAGE-signalling in C6 glioma cells, activates both p38MAPK and ERK1/2. The signal-transduction from m-RAGE to NFκB involves a parallel transient-activation of tyrosine phosphorylation leading to protein-kinase ERK1/2 activation, but not protein-kinase JNK activation, even though environmental stress and cytokines normally activate JNK via an p38MAPK mediated pathway.
m-RAGE-mediated NFκB activation is suppressed by the selective p38-inhibitor SB203580, or by co-expression of a p38MAPK kinase-dead dominant-negative mutant: both methods block expression of IL-6, TNFa, and IL-1. p38MAPK appears to be the dominant downstream effector of m-RAGE, because there is an absolute need for activated p38MAPK in the stimulation of NFκB mediated transcription and secretion of pro-inflammatory cytokines.
AGE-induced cellular dysfunction related to m-RAGE>p38MAPK>NFκB signalling is part of the inflammatory disease mechanism in arthritis, septic shock, myocardial dysfunction after ischemia, atherosclerosis, diabetes, and Alzheimer’s. Elements of these diseases also appear in the symptoms of severe-COVID-19 induced by SARS-CoV-2.
3.8. SARS-CoV-2 Dysregulation of PKC>MAP3K>MAP2K>p38MAPK>NFκB Signals
In normal innate immune activation, PKC activates MAP3Ks to activate MKK3 and MKK6, which go on to activate p38MAPK, which together with MKK6 activates “Inhibitor-of-kappaB-Kinase” (IKK) in the cytoplasm. IKK then phosphorylates “Inhibitor-of-kappaB” (IkB), which dissociates from the two-subunits of NFκB, p65 (RelA) and p50, allowing the transcription factor subunits to translocate into the nucleus and bind to the DNA of gene-expression-promoters for inflammatory-cytokines [
22].
Multiple components of the PKC>MAP3K>MAP2K>p38MAPK pathway are involved in SARS-CoV-2 replication in human lung epithelial cells. PKC is activated within minutes of SARS-CoV-2 infection. In addition, PKC is also activated by RAGE>TIRAP signalling. In vitro experiments performed in A549 human lung epithelial cells expressing the ACE2 receptor (A549-ACE2 Cells) infected with SARS-CoV-2 revealed the signalling components corrupted by SARS-CoV-2.
p38MAPK has four functionally different p38-kinase isoforms: p38α/MAPK14, p38β/MAPK11, p38γ/MAPK12, and p38δ/MAPK13. When p38β/MAPK11 is knocked down by small-interfering-RNA (siRNA) inhibition in SARS-CoV-2 infected A549 cells, there is a 1000× reduction in SARS-CoV-2 titre, demonstrating that the p38β/MAPK11 is the isoform responsible for the SARS-CoV-2 corrupted signalling.
SARS-CoV-2 interferes with the p38MAPK kinase pathway, by activating p38β/MAPK11 to promote coronavirus “nucleo-capsid N-protein” (N) activation and viral protein translation. Virally induced p38β/MAPK11 activation also activates NFκB-dependent inflammation pathways, which result in chronically high levels of expression of IL-6 and TNFa.
Inhibition of p38β/MAPK11 activity in virally infected human lung epithelial cells reduces both viral replication and cytokine production. SARS-CoV-2 Nucleo-capsid protein (N) is the only viral protein identified with a significantly altered phosphorylation pattern, as a result of activation of p38β/MAPK11-dependent phosphorylation. N-protein performs many crucial functions in the virus life cycle, including oligomerising viral RNA for protection, enhancing viral polymerase activity, modulating template switching, and evading innate immunity [
11].
Small-interfering-RNA (siRNA) inhibition of MAP3Ks upstream of p38MAPKβ, show the SARS-CoV-2 viral replication depends on: MAP3K8/TPL2/COT, MAP3K9/MLK1, and MAP3K11/MLK3. These virally induced MAP3Ks go on to activate MAP2Ks, such as MKK3 that triggers p38MAPK. SARS-CoV-2 infection results in chronic NFκB hyper-activation and suppression of CREB. In viral-infected cells, IL-10 does not suppress pro-inflammatory cytokine production efficiently, because both CREB transcription and expression of IL-10 is inhibited.
3.9. Downstream Effectors of p38MAPK: MSK1/2, MNK1 and DUSP
Downstream of p38βMAPK11 in SARS-CoV-2-infected cells, siRNA knockdown of MSK2 reduces viral load by approximately 65% but does not lead to recovery of Type-1-interferon production. MSK2 is required for phosphorylation of transcription factor ATF-1 and is probably required for viral protein transcription. MSK2 can normally activate CREB, but in SARS-CoV-2 infected cells CREB activation is blocked.
In dendritic cells, the MST1>p38MAPK>MK2/MSK1>CREB pathway controls pro-inflammatory IL-6 production. However, it also self-regulates because CREB and transcription-factor AP-1 also bind to the IL-10-promoter and produce anti-inflammatory IL-10. In addition, this pathway is associated with T-cell proliferation and up-regulation of co-stimulatory receptor expression.
Mouse coronavirus MHV, human SARS-CoV-1, and human SARS-CoV-2 have similar effects on p38MAPK signalling. MHV-proteins induce p38MAPK activation followed by NFκB triggered IL-6 expression. It was discovered that activated mouse p38MAPK also activates MNK1, which in turn activates eIF4E, which is needed to translate viral proteins [
12]. Similarly, both SARS-CoV-1 and its spike protein also activate and phosphorylate the cascade of kinases of the p38MAPK pathway, resulting in NFκB activation and IL-6 and TNFa expression, as well as the MNK1 activation of human eukaryotic translation Initiation Factor 4G (eIF4G), which is needed by the SARS1 virus to translate its proteins. In SARS-CoV-2 infection, the critical function for viral replication gained by activating p38β/MAPK11 also includes the upregulation of MNK1, which amplifies cap-dependent viral protein translation by phosphorylating and activating eIF4G.
In addition, SARS-CoV-2 infection substantially reduces the expression of DUal-Specificity Phosphatases DUSP1 and DUSP5, which are negative-feed-back-regulators that de-phosphorylate p38MAPK and inhibit p38MAPK>>NFκB>IL-6 signalling [
32]. Activated-CREB is required for transcription and expression of DUSP1 and DUSP5, but transcription-factor CREB activation is suppressed by SARS-CoV-2 proteins hyper-activating NFκB [
33].
4. Ezrin Inflammation Control
4.1. Introduction to Ezrin
The structures and functions of multi-protein-complexes built by ezrin at the cell membrane are generally over-simplified in the literature, but the data suggests co-ordination of multiple conformations of proteins and multiple phosphorylations of specific residues.
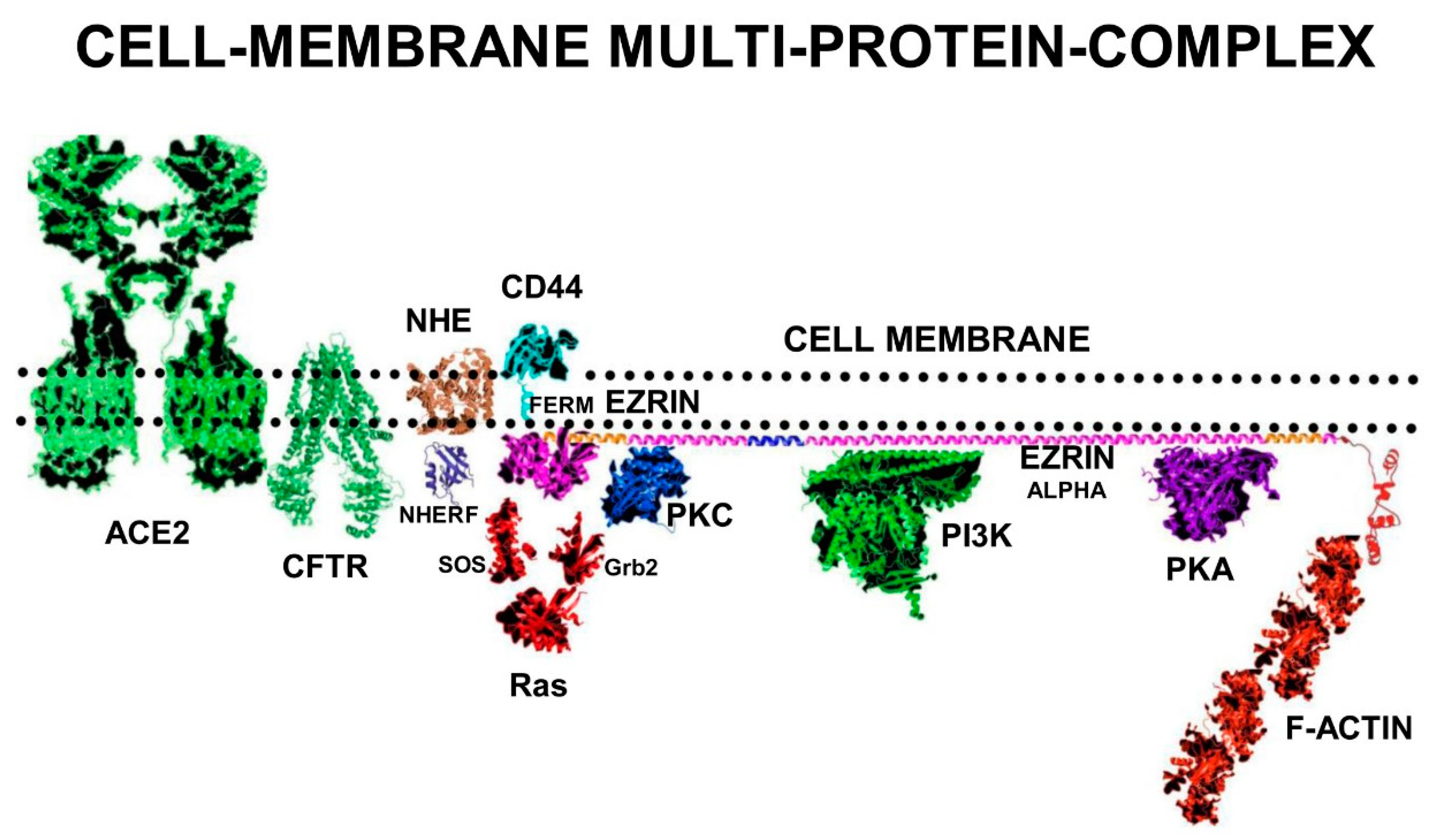
The evolution-conserved Ezrin, Radixin, and Moesin (ERM) adaptor-proteins are membrane-associated multi-protein-complex-formers, receptor and signalling proteins, which also regulate and anchor filamentous F-actin to the cell membrane. Ezrin has a key role in the regulation of alveolar structure and lung homeostasis.
Ezrin is a sub-membrane adaptor protein for multi-protein cell signalling complexes and depending on its conformation can also function as a cell-surface-receptor. Ezrin consists of three domains: The N-terminal FERM-domain formed into a clover-leaf-like structure with three lobes (F1 lobe; amino-acids 1–93: F2 lobe; amino-acids 94–200: F3 Lobe amino-acids 201–296). The FERM-domain is connected to a folded α-helical Alpha domain (ALPHA; amino-acids 297–468), a transmembrane proline-rich domain (TRANS amino-acids 469–497), and a C-terminal domain (C-terminal amino-acids 498–585) [
34].
Activation of ezrin results in a shift of ezrin from the cytoplasm to membrane microvilli, ruffles, pits, and adhesion sites. In the dormant soluble cytoplasmic form of ezrin, the C-terminal domain is bound to the FERM domain between the F2 and F3 lobes. The first stage of ezrin activation occurs at the sub-surface of the cell membrane, where the F1 and F3 lobes of the FERM domain binds to sub-membrane surface PIP2-lipid-rafts and partially unfolds.
Ezrin probably inserts the folded Alpha domain through the membrane at this step, creating the “Receptor” conformation of ezrin. Ezrin adopts its fully elongated conformation after kinase phosphorylation of Thr567 on the C-terminal domain. Depending on the signalling environment, this phosphorylation is performed by either ROCK, or PKCα, or Akt2 or GRK2 (
Figure 4).
In its fully unfolded “Active” conformation, ezrin stretches out under the sub-membrane surface and exposes multiple binding sites on its FERM-domain. Elongated ezrin can form multi-protein complexes with a range of receptors, adhesion proteins, ion channel proteins, and cell signalling kinases. Ezrin has specific binding sites on its FERM-domain for sub-membrane-PIP2 lipid-rafts, and the cytoplasmic tails of adhesion molecules and growth-factor receptors such as CD44, CD43, ICAM1&2, and EGFR.
In addition, there are also specific binding sites on the ezrin-FERM domain for cell-signalling proteins such as SOS and Ras. The FERM-domain also binds to NHERF, which binds the ion channel proteins NHE and CFTR. On the ezrin-Alpha domain there are binding sites for the cell signalling proteins PKC, PI3K, and PKA. On the c-terminal domain, there are binding sites for F-actin [
35].
Ezrin is inactive in the cytoplasm, but upon phosphorylation on serine and threonine residues by PKC or RhoA kinase, the protein translocates to the cell surface where it builds multi-protein-complexes that connect to the actin cytoskeleton.
Figure 4.
Ezrin has three main conformations: a soluble globular inactive form in the cytoplasm; a membrane associated “Receptor” form at the membrane where the Alpha domain is inserted through the membrane; and an elongated activated form which is attached to PIP2-lipid rafts on the submembrane and stabilised by T567 phosphorylation. The “Receptor” transition conformation of ezrin with part of the Alpha domain exposed on the cell surface can be detected by anti-ezrin alpha-domain monoclonal antibody [6F1A9] (ab205381) [
36]. RepG3, a peptide derived from the ezrin-Alpha domain, triggers a change in conformation from the “Receptor” form to the sub-membrane active form of ezrin. The main phosphorylation sites on ezrin are: tyrosine Y146, tyrosine Y354, Serine S366, tyrosine Y478, Serine S535, and Threonine T567 (phosphorylated by ROCK2 and PKC/PRKCI) [
37]. “*” means N-to-C amino acid sequence number.
Figure 4.
Ezrin has three main conformations: a soluble globular inactive form in the cytoplasm; a membrane associated “Receptor” form at the membrane where the Alpha domain is inserted through the membrane; and an elongated activated form which is attached to PIP2-lipid rafts on the submembrane and stabilised by T567 phosphorylation. The “Receptor” transition conformation of ezrin with part of the Alpha domain exposed on the cell surface can be detected by anti-ezrin alpha-domain monoclonal antibody [6F1A9] (ab205381) [
36]. RepG3, a peptide derived from the ezrin-Alpha domain, triggers a change in conformation from the “Receptor” form to the sub-membrane active form of ezrin. The main phosphorylation sites on ezrin are: tyrosine Y146, tyrosine Y354, Serine S366, tyrosine Y478, Serine S535, and Threonine T567 (phosphorylated by ROCK2 and PKC/PRKCI) [
37]. “*” means N-to-C amino acid sequence number.
4.2. Ezrin Regulates m-RAGE+TIRAP Mediated Inflammation
m-RAGE, CD44, and ezrin are important regulators that maintain normal alveolar epithelial structure and function in the lung. In resting alveolar-epithelial A549 cells, m-RAGE and ezrin are abundantly expressed together in the cell-surface microvilli and also co-localise as protein–protein complexes diffusely distributed in the cytoplasm. In these m-RAGE+ezrin complexes, the FERM domain and the C-terminal domain of ezrin both bind to the cytoplasmic tail of m-RAGE (ezrin is in its transition “Receptor” conformation). Pro-inflammatory cytokine treatment of A549 cells (for example a mixture of TGF-1, IL-6, IL-1, and TNFa) causes more ezrin to relocate from the cytoplasm to the sub-surface of the cell membrane, and triggers ROCK to phosphorylate C-terminal T567 of ezrin [
38]. This threonine-phosphorylation switches the conformation of ezrin from its “Receptor” conformation to stretch-out into its sub-membrane “Active” conformation, which disrupts ezrin+m-RAGE complexes clustered on the sub-membrane. The m-RAGE+ezrin complexes also disappear from the cytoplasm.
In addition, TLR4 mediated LPS-induced p38MAPK activation and nuclear NFκB activation is ezrin dependent. Ezrin associates with MyD88/IRAK-1 upon LPS challenge. Blockade of RhoA/ROCK inhibits LPS-induced ezrin phosphorylation and its translocation from the cytoplasm to the cell membrane. Moreover, the suppression of ezrin by siRNA or the blockade of ROCK activation with Y-27632 reduces the production of TNF-α, IL-1β, and HMGB1 in response to LPS [
39].
High concentrations of negatively charged PIP2 on the sub-surface of the plasma-membrane are required for multi-protein-complex formation. The local sub-membrane concentrations of PIP2 controls the activation state of both TLR4+TIRAP+MyD88 and m-RAGE+TIRAP+MyD88 multi-protein-complexes. Ezrin also occupies PIP2-lipid-rafts to anchor its own multi-protein-complexes.
Lipopolysaccharide (LPS) triggering of TLR4 leads to the formation of the TLR4+TIRAP+MyD88 complex. The association of TIRAP and MyD88 into a multi-protein-complex depends on the availability of lipid-rafts containing high concentrations of PIP2. A critical step is the electrostatic attraction of negatively charged PIP2 in the sub-membrane, for positively charged TIRAP. The N-terminal region of TIRAP contains a PIP2-binding domain (PBD) that has a strong binding affinity for PIP2, which induces conformational changes in TIRAP that results in its activation.
PIP2 is synthesized by membrane-associated PIP5Kα, which is an essential component of TIRAP-dependent TLR4-activation. LPS triggering of TLR4 increases PIP5Kα mediated synthesis of PIP2. Higher PIP2 concentrations in the lipid-rafts induces more TIRAP translocation to the plasma membrane. PIP5Kα knock-down results in lower PIP2 concentrations in the sub-membrane and significantly attenuates TLR4-induced phosphorylation of p38MAPK, JNK, and NFκB, which inhibits the expression of pro-inflammatory cytokines IL-6 and TNF-α. In addition, Phospholipase-γ2 (PLCγ2) decreases the lipid-raft concentration of PIP2. Low concentrations of PIP2 results in TLR4+TIRAP+MyD88 complexes being internalized by endocytosis and their dissociation into separate MyD88, TIRAP, and TLR4 proteins [
40].
TIRAP specifically localizes to the zones of the cell membrane where activated-ezrin binds PIP2. High concentrations of PIP2 in lipid rafts are also necessary for ezrin multi-protein-complex formation that are involved in actin-cytoskeleton re-arrangement and the anchoring of filamentous-actin at “membrane ruffles”.
The FERM-domain of activated-ezrin is a competitive-inhibitor of TIRAP and activated ezrin regulates the activity of the multi-protein-complexes of m-RAGE and TLR4+TIRAP+MyD88 by competing with TIRAP for space on the sub-membrane lipid-rafts of concentrated PIP2. This competition for PIP2-lipid-rafts between TIRAP and ezrin is a “protein-complex-switch” for turning inflammatory signalling on-and-off. Ezrin has also been demonstrated to regulate inflammatory cytokine expression in B-cells. Conditional deletion or chemical inhibition of active ezrin in B-cells increases LPS-triggered TLR4-induction of pro-inflammatory cytokine production [
41].
4.3. Ezrin+S100 Complexes Regulate S100+RAGE Inflammation
When S100 proteins bind to m-RAGE, they activate inflammation. When S100 proteins bind to ezrin, inflammatory signalling is suppressed [
42]. However, ezrin binds to S100-proteins involved in m-RAGE activation in an unexpected way. In HEK-293T cells, S100A4 co-localizes below the cell membrane in regions rich in ezrin. However, NMR spectroscopy in vitro of S100A4+ezrin interactions revealed that S100 proteins efficiently bind to both the F2-lobe of the isolated N-terminal FERM domains of ezrin and also to isolated small C-terminal actin-binding domains of ezrin, with similar high-affinity. However, S100A4 only very weakly binds to sub-membrane associated full-length ezrin, nor does it bind to the fully folded inactive soluble cytoplasmic conformation of ezrin either.
Fast kinetic measurements reveal that S100A4 binding induces structural changes to the whole of the ezrin protein. The physiological complex of S100+ezrin probably involves the ezrin “Receptor” conformation, where the FERM domain and C-terminal domain are loosely associated below the cell membrane and the ezrin-Alpha domain is exposed on the cell surface.
In the cytoplasm of many cell types, there are inactive complexes of ezrin+S100-Calgranulin-C (S100A12), where ezrin is in the “Receptor” transition-state between conformations. When intracellular calcium concentration increases, S100A12+ezrin binds Ca2+ in the cytoplasm and then translocates to the cell-membrane. Calcium-bound S100A12 forms dimeric or even hexameric structures on the sub-membrane surface. In the presence of ezrin, m-RAGE is also internalized into the cell and then recycled back to the cell membrane, after binding to S100+ezrin complexes.
4.4. The Ezrin-Protein-Complex and the Ras>Raf>MEK>ERK>RSK Pathway
Activated phospho-ezrin+actin catalyses the formation of a multi-protein-complex consisting of Receptor Tyrosine Kinases (RTKs) with Grb2, SOS, and Ras. The small G-protein p21-Ras binds to the ezrin FERM domain and other signalling proteins, where it functions as a molecular-switch, relaying extracellular stimuli to diverse intracellular effector pathways, which are responsible for controlling proliferation, motility, and differentiation [
43].
SOS catalyses the nucleotide-exchange on Ras but the activation of Ras also requires the participation of ezrin and co-receptors such as CD44 and actin. Disrupting the interaction between ezrin and co-receptors, for example by down-regulation of different ERM proteins by siRNA, or by expression of dominant-negative mutants, or by disruption of F-actin, abolishes growth-factor-induced Ras-activation. In addition, over-expression of ezrin-R579A mutant, that is unable to bind F-actin, also reduces RTK-Ras dependent signalling, and IL-6-dependent ERK phosphorylation. In contrast, ezrin-R579A does not interfere with Ezrin-PKC-induced ERK phosphorylation.
Triggering of Grd2+SOS+ezrin+CD44+actin+p21Ras>Raf>MEK>ERK>p90RSK signalling results in the activation of transcription-factor CREB, inhibition of NFκB, and inhibition of pro-inflammatory cytokine expression.
5. VASO-Active Intestinal Peptide and COVID-19
5.1. Natural Plasma-VIP Levels in Patients Correlate to Recovery from Severe COVID-19
Vasoactive Intestinal Peptide (VIP) is a 28-amino acid peptide released by intrinsic neurons in the human airways. The plasma concentration of natural-VIP is elevated in the plasma of individuals with manifestations of severe COVID-19, and the amount of VIP-elevation correlates to survival and recovery from severe COVID-19 [
44].
In a clinical study in April and May 2020, with 24 critically ill COVID-19 patients of median age 53 years, the plasma levels of VIP were measured and compared with patients with only mild COVID-19 symptoms, and with non-infected healthy individuals. As expected, comparing five pro-inflammatory markers (TNF-α IL-8, IL-12p40, IL-17A, CXCL10/IP-10) in mild COVID-19 and severe COVID-19 cases, there was a significant positive correlation between pro-inflammatory cytokine concentration and severity of disease symptoms. Severe COVID-19 was characterized by very elevated serum levels of pro-inflammatory cytokines, particularly IL-6, TNFa, IL-8, and IL-1β.
Plasma levels of VIP were significantly elevated in patients with severe COVID-19. However, in the group of severe COVID-19 patients, higher levels of VIP correlated with lower levels of pro-inflammatory cytokines. The critical discovery was that high VIP plasma levels significantly correlated with survival of patients from severe COVID-19, compared to patients who died of severe COVID-19.
5.2. VIP In Vitro Inhibits SARS-CoV-2-Induced NFκB Activation and IL-6 Expression
In vitro in infected monocytes, SARS-CoV-2 activates NFκB to increase the amounts of the pro-inflammatory cytokines above controls; IL-6 up 15x, IL-8 up 4x, and TNFa up 12x. Phosphorylated NFκBp65 subunit, a marker for activated NFκB, was also significantly up-regulated in SARS-CoV-2 infected cells. In vitro treatment with VIP reduced the NFκB p65 phosphorylation.
Calu-3 cells, a lung epithelial cell line which expresses VIP-receptor VPAC1, are highly susceptible to SARS-CoV-2 infection. In Calu-3 lung epithelial cells, VIP inhibited activation of NFκB and reduced pro-inflammatory cytokine expression by about fifty per cent, which correlated with reduced viral production and protection from cell death. A specific inhibitor of NFκB was found to mimic the anti-inflammatory activity of VIP. Treatment of SARS-CoV-2-infected monocytes with Bay11-7082, a pharmacological inhibitor of NFκB, resulted in decreased viral RNA synthesis, reduced cell death, and suppression of production of IL-6 and TNF-α.
5.3. VIP In Vitro Decreases SARS-CoV-2 Replication
In vitro, VIP decreases SARS-CoV-2 replication and viral mRNA expression in human monocytes and Calu-3 lung epithelial cells. Treatment of SARS-CoV-2 infected Calu-3 cells with 10 nM VIP results in a fifty per cent reduction in SARS-CoV-2 RNA synthesis. However, it was intriguing that when comparing mild COVID-19 and severe COVID-19 in patients, there was only a weak correlation between plasma VIP levels and SARS-CoV-2 viral load.
5.4. VIP Up-Regulates CREB to Inhibit NFκB Mediated Inflammation
Cyclic-AMP Response Element Binding-protein (CREB) is a phosphorylation-activated transcription-factor, part of the basic-leucine-zipper (bZIP) superfamily, comprising of CREB, ATF1, and CREM. CREB has an alpha-helical structure, which contains a Kinase Inducible Domain (KID) in its central region, two glutamate Q1 and Q2 domains and a bZIP domain. The phosphorylation site located at Serine-133 (Ser133) on CREB is critical for CREB transcriptional activity. CREB can be phosphorylated at Ser133 by the serine-threonine kinases PKA, PKC, and RSK. Activated CREB can form homodimers or heterodimers in a transcription-complex with CREB-Binding-Protein (CBP) and p300 (also called EP300 or E1A binding protein p300), which bind to DNA at Cyclic-AMP Response Element CRE-promotors, initiating transcription of CREB responsive genes.
Effective NFκB transcription of pro-inflammatory cytokine genes requires interaction of the NFκB subunit p65 (RelA) with either CBP or p300. However, phosphorylation of CREB-serine133 in its kinase-inducible-domain (KID) promotes the binding of the co-activator proteins CBP and p300 to activated-CREB to commence transcription of CREB-responsive genes. Activated-CREB inhibits NFκB mediated transcription by sterically hindering the binding of CBP to the NFκB complex. Activated-CREB also starves NFκB of CBP and p300, preventing it functioning as a transcription-factor.
In SARS-CoV-2-infected monocytes, CREB activation and its transcription-factor mediated gene-expression are suppressed by the viral proteins in control of cell-signalling, which chronically amplify NFκB activity. In contrast, treatment of SARS-CoV-2-infected monocytes with VIP triggers the activation of transcription-factor CREB. The activated-CREB in these virally infected cells inhibits the activation of transcription-factor NFκB that is responsible for the expression of pro-inflammatory cytokines. CREB activation is also connected to an anti-apoptotic response in monocytes and macrophages exposed to inflammatory stimuli. In addition, CREB may also directly suppress SARS-CoV-2 viral replication by inhibition of cell signalling pathways that lead to NFκB activation. CREB also promotes anti-inflammatory immune responses, through the induction of anti-inflammatory IL-10.
5.5. Intravenous VIP-Therapy Rescues Patients from Severe-COVID-19
Aviptadil, a synthetic form of Vasoactive Intestinal Peptide (VIP) has been granted Fast Track Designation for the treatment of Critical COVID-19 with Respiratory Failure.
In a controlled clinical trial at Houston Methodist Hospital, intravenous treatment of patients suffering severe COVID-19 with Vasoactive Intestinal Peptide (synthetic-VIP, brand-name Aviptadil) was performed in a prospective, open-label clinical study. The 45 patients enrolled in the study were admitted to the intensive care unit (ICU) in June and July 2020 with severe/critical COVID-19 and co-morbidities [
45,
46,
47].
The 21 patients in the VIP treatment group were unconscious when treated with Aviptadil, so the chance of placebo bias was discounted because they could not know which medication they were receiving through multiple intravenous lines. Patients in the VIP treatment group received three successive 12-h intravenous infusions of VIP-Aviptadil at 50 pmol/kg/h, 100 pmol/kg/h, and 150 pmol/kg/h. The main clinical outcomes measured included survival and recovery from respiratory failure.
However, Aviptadil has side-effects; it is known to cause serious hypotension and diarrhoea, so intravenous VIP should only be administered by critical-care physicians who have sufficient expertise to manage these side effects. In this study, about twenty per cent of patients exhibited VIP-related hypotension and/or diarrhoea, but no other serious VIP-related adverse events were recorded.
Patients treated with intravenous Aviptadil-VIP were followed for at least 60 days after ICU admission. They were compared with 24 individuals with comparable COVID-19 and co-morbidities, who received only standard-of-care treatment. Of the 21 patients treated with Aviptadil, 19 survived to day 28, 17 survived to day 60 compared to only five survivors of the 24 individuals in the standard-therapy group (81% vs. 21%; p < 0.0001). Improved radiographic appearance was seen in both lungs of 17 VIP treated patients but only one lung of two VIP treated patients who subsequently died. Four out of five patients treated with Aviptadil on Extracorporeal Membrane Oxygenation were successfully de-cannulated and survived. In comparison, only three out of 13 standard-therapy patients survived.
Initial release of the data revealed increased survival rates that correlated to a significant reduction in plasma IL-6 and improved lung-surfactant production.
In all patients, the reduction of the inflammatory markers C-reactive protein (−76% ± 3%) and IL-6 (−75% ± 3%) was very significant.
6. VIP>PKC>Ezrin>PKA>CREB; Suppresses NFκB Activity & COVID-19
6.1. VIP Triggering of VPAC1, Activation of PKC-e and Ezrin
VIP>VPAC1 signalling results in suppression of pro-inflammatory cytokines such as IL-6, IL-8, TNFα, and IL-1β, and the induction of the anti-inflammatory cytokine IL-10. The VIP-receptor (VPAC1) is constitutively expressed in the lung epithelia and macrophages and is the main receptor for VIP in the Alveolar Type II cells. In human bronchial cell line Calu-3, VIP triggered VPAC1 activates calcium-independent Protein Kinase C-e (PKC-e), which increases the concentration of activated T567-phosphorylated ezrin on the subsurface of the cell membrane. An inhibitor of PKC-e (Peptide-EAVSLKPT) blocks the effect [
48].
6.2. Multi-Protein-Complex: CFTR+NHERF+NHE+Ezrin+PKC+PKA in the Lungs
Cystic Fibrosis Trans-membrane Conductance Regulator (CFTR) and Na+/H+ Exchanger-1 (NHE) in tandem manage sodium and chloride ion secretion that results in the hydration of the mucus membranes of the lungs. T567-phosphorylated ezrin induces NHERF1, NHE, and CFTR to form the multi-protein complex, CFTR+NHERF+NHE+ezrin, at the N-terminal FERM domain of ezrin.
The activated complex also attracts PKC and PKA to bind the Alpha domain of ezrin, creating a larger complex of CFTR+NHERF+NHE+ezrin+PKC+PKA and actin. This multi-protein complex is vital for the management of fluid secretions in the lungs, airway maintenance, and gas-exchange. This larger multi-protein complex enhances stability, function, and density of CFTR in the cell membrane of airway epithelial cells. In addition, phosphorylation and activation of ezrin activates PKC bound to its Alpha-domain. This multi-protein complex is also sensitive to the intra-cellular cyclic-AMP concentration [cAMP] that activates PKA.
6.3. VIP>VPAC1>cAMP>Ezrin+PKA and Activation of CREB
VIP binding to VPAC activates Adenylyl-Cyclase (AC) at the membrane, resulting in increasing intra-cellular concentrations of cAMP and activation of PKA anchored on the Alpha domain of ezrin. PKA is commonly known as a cAMP-dependent-protein-kinase, because its activity depends on intra-cellular cyclic-AMP concentration [cAMP]. PKA Type II comprises of RIIa and RIIb regulatory sub-units, with two catalytic subunits stacked on top to form a tetramer. After activation of PKA by cAMP, a conformational change is induced so the catalytic domains detach and migrate to the nucleus, where they activate “cAMP-Responsive Element Binding-protein” (CREB). VIP triggering of VPAC1 also activates three closely related pathways, commencing with ezrin+PKA activation of Rap1>B-Raf>MEK, or EPAC>Rap1>B-Raf>MEK, or Ras>C-Raf>MEK, which result in ERK1/2 >RSK>CREB signalling [
48]. Ezrin and EPAC1 a cAMP-sensor protein cooperate together to activate various cell functions, in response to elevations in intracellular cAMP [
49,
50].
6.4. Direct-Inhibition of Pro-Inflammatory Responses by PKA and CREB
CREB and NFκB are general effectors of two competing expression programs, controlled by macro-molecular protein-complexes, which change gene-expression to favour either innate-immune-responses or adaptive-immune-responses. Efficient CREB, CBP, and p300 transcription-complex formation starves transcription factors AP-1, IRF, and NFκB of the CBP and p300 needed to form transcription complexes for pro-inflammatory target genes [
51].
Activation of CREB also results in the gene-expression of anti-inflammatory interleukin-10. IL-10 reduces inflammation by inducing the expression and activation of GSK-3b, a serine/threonine kinase which phosphorylates CBP and p300, decreasing transcription complex formation with the p65RelA subunit of NFκB. In contrast, GSK-3b phosphorylation of CREB favours binding of CREB to CRE, stimulating more IL-10 expression. PKA can also directly phosphorylate GSK-3b, causing inhibition of IKK, resulting in inhibition of the IkB phosphorylation needed for NFκB sub-unit dissociation and translocation to the nucleus [
52].
In T-cells, CREB phosphorylation is induced by a multi-protein-complex of ezrin, Ras, PKA, and PKC. For example, CD28-triggering of T cells can result in a 100× increase in phosphorylated CREB, which is halved when the cells are treated with a PKC inhibitor, and completely blocked by a PKA inhibitor. Generally, CREB regulates T-cell Th1, Th2, Th17 responses. In contrast, IFN-γ inhibits ezrin+PI3K>AKT activation of GSK-3b and down-regulates CREB-induced IL-10 production.
In B-cells, CREB phosphorylation induced by ezrin multi-protein-complexes containing PKA, PKC, and Ras induces B-Cell Receptor (BCR) triggering that promotes B-cell activation and proliferation. This process is dependent on activated ezrin, PKC-d, and ERK1/2>pp90RSK signalling, which results in downstream CREB phosphorylation and activation.
In dendritic cells, knock-out of CREB significantly reduces spontaneous B-Cell activation and results in a two-fold reduction in the formation of B-Cell germinal centres, reduction in antigen-specific B-cell responses and reduced IgG titres. In contrast, CREB activation in CD11c+ dendritic cells increases HLA expression, enhances B-Cell germinal centre responses and increases IgG titres [
53].
6.5. The Human Ezrin Peptide & VIP Anti-COVID-19 Mechanism of Action
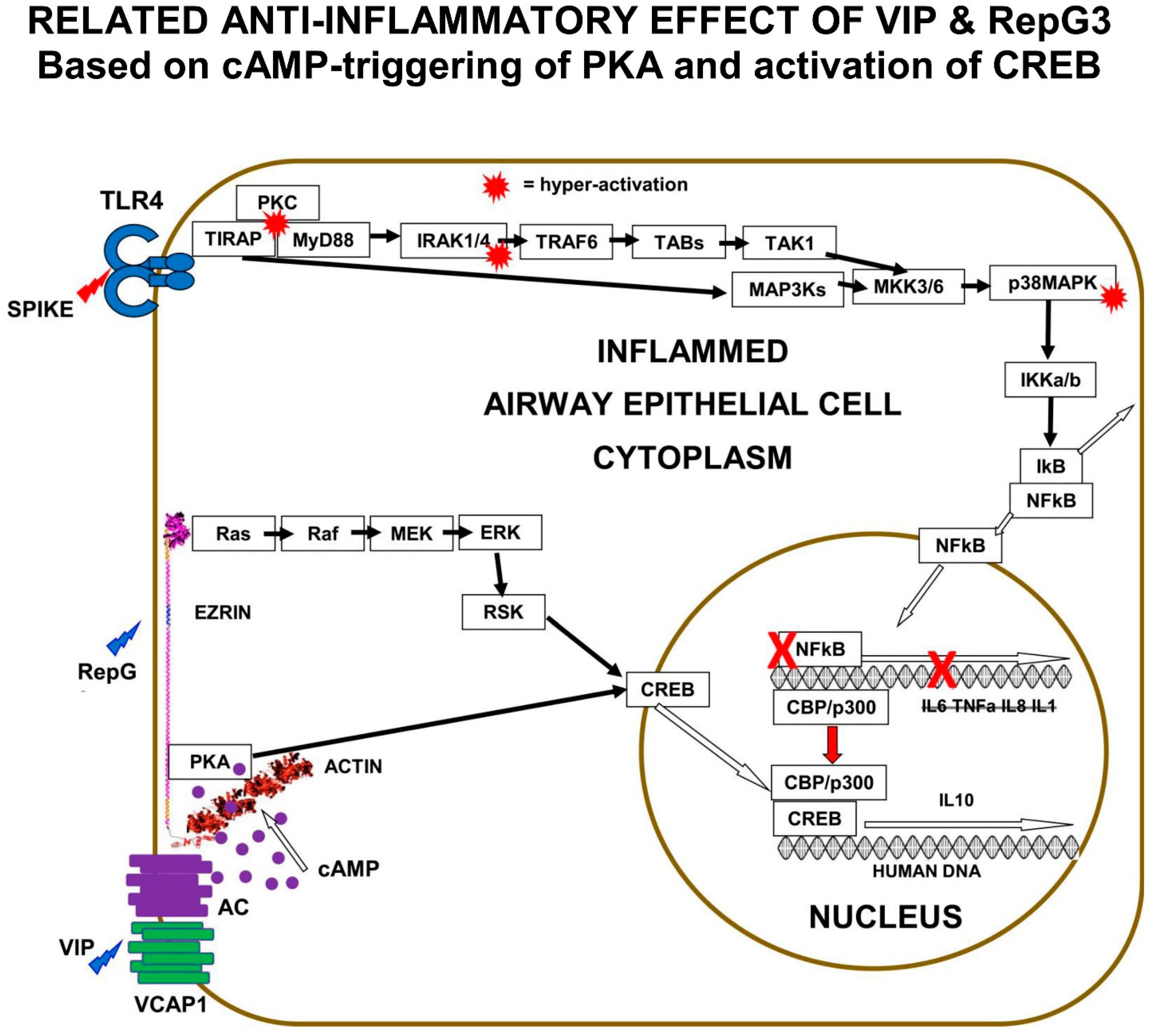
The clinical investigation of the role of natural VIP and the effect of VIP therapy in COVID-19 suggests that the VIP-activation of PKA results in the activation of CREB, suppression of NFκB activity, and the inhibition of the pro-inflammatory cytokine expression that causes COVID-19.
The “Receptor” conformation of ezrin is believed to be the binding target of human ezrin peptides: HEP-1 and RepG3. The hypothesis is that ezrin multi-protein-complexes can be induced by Human Ezrin Peptides, to activate PKA bound to ezrin, and also to signal down the Ras>Raf>MEK>ERK1/2>pp90 RSK pathway, resulting in CREB phosphorylation and activation. Both VIP and Human Ezrin Peptides seem to induce a similar mechanism to suppress COVID-19 (
Figure 5).
In 2020 and 2021, more than twenty individual human volunteers with mild-to-moderate COVID-19 self-administered a 1 mg per 1 mL solution of either Human Ezrin Peptide RepG3 or Human Ezrin Peptide HEP-1 by spray-inhalation (approximately 6 mg Human Ezrin Peptide per day). All volunteers recovered in approximately one week and became PCR negative. One volunteer who self-administered RepG3 in March 2020 for mild COVID-19 has been PCR negative for SARS-CoV-2 for almost two years without being vaccinated, despite travel exposure to SARS-CoV-2 variants.
In addition, doctors in Germany administered 0.2 mg per 0.2 mL intra-peritoneal injections of Human Ezrin Peptide HEP-1 to more than twenty volunteers with mild-to-moderate COVID-19 and achieved similar positive treatment results. Different human-ezrin-peptides are effective treatments for COVID-19 and seem to have the same mechanism of action [
54]. SARS-CoV-2 infection is characterized by elevated serum levels of pro-inflammatory cytokines particularly IL-6, TNFα, IL-8, IL-1β. Human Ezrin Peptides inhibit pro-inflammatory cytokine (IL-6, TNFα, IL-8, IL-1β) expression probably via the Ezrin>Ras>Raf>MEK>ERK>RSK>CREB and ezrin+PKA>CREB pathways. These pathways also directly amplify the induction of the anti-inflammatory cytokine IL-10, as well as T cell and B cell mediated adaptive immunity. Higher IgG antibody titres have also been observed with Human Ezrin Peptide treatment in combination with hepatitis B vaccination. Human Ezrin Peptides may bind the “Receptor” conformation of ezrin to trigger the formation and activation of the multi-protein-complex of CFTR+NHERF+NHE+ezrin+PKC+PKA [
54,
55,
56].
In addition, CREB activity is necessary for normal cognitive function and memory. SARS-CoV-2 interference of CREB signalling in the brain may be the origin of COVID-19-related “brain fog” [
57,
58].
7. Conclusions
Intravenous-VIP and spray-inhaled Human Ezrin Peptides appear to stop COVID-19 by related mechanisms of action. Human Ezrin Peptides (HEPs) such as HEP-1 and RepG3, and Vaso-active-Intestinal Peptide (VIP), appear to have a related anti-inflammatory and anti-COVID-19 activity by triggering PKA>CREB activation and inhibiting NFκB-mediated pro-inflammatory cytokine expression. However, this mechanism is still speculative and much more work needs to be done.
However, VIP therapy has the disadvantage that it can cause dangerous blood-pressure reduction (hypotension) and diarrhoea, and the larger peptide requires intravenous injection. In contrast, no side effects nor adverse events have been reported for Human Ezrin Peptides over the twenty years they have been in clinical use in Russia. RepG3 and Hep1 have been demonstrated to cure individual volunteers of COVID-19. RepG3 has double the anti-inflammatory activity of HEP-1 and seems to induce adaptive immune responses that appear to prevent re-infection by SARS-CoV-2 variants.
Generally, Human Ezrin Peptides are known to be safe and have broad spectrum efficacy against not only viral infections, but also bacterial infections, fungal infections, and chronic inflammatory diseases. RepG3 is suitable for self-administered spray inhalation therapy of mild-to-moderate COVID-19, and there is a great need for such a therapy to be available globally at affordable prices.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}