
Anti-Inflammatory and Anti-Oxidative Effects of GLP1-RAs and SGLT2i: The Guiding Star Towards Cardiovascular Protection in Type 2 Diabetes


Abstract
1. Introduction
2. The Cardiovascular Damage Mediated by Inflammation and Oxidative Stress in T2DM
2.1. ROS Production
2.2. Autophagy
2.3. Endothelial Dysfunction
2.4. EAT
3. The Role of SGLT2i and GLP-1 RAs in Cardiovascular Protection
3.1. The Anti-Inflammatory and Anti-Oxidative Effects of SGLT2i
3.2. The Anti-Inflammatory and Anti-Oxidative Effects of GLP-1 RAs
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Matheus, A.S.d.M.; Tannus, L.R.M.; Cobas, R.A.; Palma, C.C.S.; Negrato, C.A.; Gomes, M.d.B. Impact of diabetes on cardiovascular disease: An update. Int. J. Hypertens. 2013, 2013, 653789. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Dewanjee, S.; Vallamkondu, J.; Kalra, R.S.; John, A.; Reddy, P.H.; Kandimalla, R. Autophagy in the diabetic heart: A potential pharmacotherapeutic target in diabetic cardiomyopathy. Ageing Res. Rev. 2021, 68, 101338. [Google Scholar] [CrossRef] [PubMed]
- Myasoedova, V.A.; Bozzi, M.; Valerio, V.; Moschetta, D.; Massaiu, I.; Rusconi, V.; Di Napoli, D.; Ciccarelli, M.; Parisi, V.; Agostoni, P.; et al. Anti-Inflammation and Anti-Oxidation: The Key to Unlocking the Cardiovascular Potential of SGLT2 Inhibitors and GLP1 Receptor Agonists. Antioxidants 2023, 13, 16. [Google Scholar] [CrossRef]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2020, 83, 503–528. [Google Scholar] [CrossRef]
- Brown, E.; Heerspink, H.J.L.; Cuthbertson, D.J.; Wilding, J.P.H. SGLT2 inhibitors and GLP-1 receptor agonists: Established and emerging indications. Lancet 2021, 398, 262–276. [Google Scholar] [CrossRef]
- Kobayashi, S.; Liang, Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 252–261. [Google Scholar] [CrossRef]
- Balogh, D.B.; Wagner, L.J.; Fekete, A. An Overview of the Cardioprotective Effects of Novel Antidiabetic Classes: Focus on Inflammation, Oxidative Stress, and Fibrosis. Int. J. Mol. Sci. 2023, 24, 7789. [Google Scholar] [CrossRef]
- Wang, D.; Jiang, L.; Feng, B.; He, N.; Zhang, Y.; Ye, H. Protective effects of glucagon-like peptide-1 on cardiac remodeling by inhibiting oxidative stress through mammalian target of rapamycin complex 1/p70 ribosomal protein S6 kinase pathway in diabetes mellitus. J. Diabetes Investig. 2020, 11, 39–51. [Google Scholar] [CrossRef]
- Li, L.; Xu, J.; He, L.; Peng, L.; Zhong, Q.; Chen, L.; Jiang, Z. The role of autophagy in cardiac hypertrophy. Acta Biochim. Biophys. Sin. 2016, 48, 491–500. [Google Scholar] [CrossRef]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2015, 12, 144–153. [Google Scholar] [CrossRef]
- Xie, Z.; Lau, K.; Eby, B.; Lozano, P.; He, C.; Pennington, B.; Li, H.; Rathi, S.; Dong, Y.; Tian, R.; et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 2011, 60, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Xie, Z. Regulation of interplay between autophagy and apoptosis in the diabetic heart: New role of AMPK. Autophagy 2013, 9, 624–625. [Google Scholar] [CrossRef]
- Malik, R.A.; Zippel, N.; Frömel, T.; Heidler, J.; Zukunft, S.; Walzog, B.; Ansari, N.; Pampaloni, F.; Wingert, S.; Rieger, M.A.; et al. AMP-Activated Protein Kinase α2 in Neutrophils Regulates Vascular Repair via Hypoxia-Inducible Factor-1α and a Network of Proteins Affecting Metabolism and Apoptosis. Circ. Res. 2017, 120, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Glazer, H.P.; Osipov, R.M.; Clements, R.T.; Sellke, F.W.; Bianchi, C. Hypercholesterolemia is associated with hyperactive cardiac mTORC1 and mTORC2 signaling. Cell Cycle 2009, 8, 1738–1746. [Google Scholar] [CrossRef]
- Vincent, M.A.; Clerk, L.H.; Lindner, J.R.; Klibanov, A.L.; Clark, M.G.; Rattigan, S.; Barrett, E.J. Microvascular recruitment is an early insulin effect that regulates skeletal muscle glucose uptake in vivo. Diabetes 2004, 53, 1418–1423. [Google Scholar] [CrossRef]
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial cell–derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 2010, 121, 2407–2418. [Google Scholar] [CrossRef]
- Muniyappa, R.; Sowers, J.R. Role of insulin resistance in endothelial dysfunction. Rev. Endocr. Metab. Disord. 2013, 14, 5–12. [Google Scholar] [CrossRef]
- Ferrannini, E.; Cushman, W.C. Diabetes and hypertension: The bad companions. Lancet 2012, 380, 601–610. [Google Scholar] [CrossRef]
- Iacobellis, G. Epicardial adipose tissue in contemporary cardiology. Nat. Rev. Cardiol. 2022, 19, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G. Local and systemic effects of the multifaceted epicardial adipose tissue depot. Nat. Rev. Endocrinol. 2015, 11, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, T.; Zhang, L.; Zalewski, A.; Mannion, J.D.; Diehl, J.T.; Arafat, H.; Sarov-Blat, L.; O’Brien, S.; Keiper, E.A.; Johnson, A.G.; et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation 2003, 108, 2460–2466. [Google Scholar] [CrossRef] [PubMed]
- Langheim, S.; Dreas, L.; Veschini, L.; Maisano, F.; Foglieni, C.; Ferrarello, S.; Sinagra, G.; Zingone, B.; Alfieri, O.; Ferrero, E.; et al. Increased expression and secretion of resistin in epicardial adipose tissue of patients with acute coronary syndrome. Am. J. Physiol. Circ. Physiol. 2010, 298, H746–H753. [Google Scholar] [CrossRef]
- Gao, X.; Mi, S.; Zhang, F.; Gong, F.; Lai, Y.; Gao, F.; Zhang, X.; Wang, L.; Tao, H. Association of chemerin mRNA expression in human epicardial adipose tissue with coronary atherosclerosis. Cardiovasc. Diabetol. 2011, 10, 87. [Google Scholar] [CrossRef]
- Imoto-Tsubakimoto, H.; Takahashi, T.; Ueyama, T.; Ogata, T.; Adachi, A.; Nakanishi, N.; Mizushima, K.; Naito, Y.; Matsubara, H. Serglycin is a novel adipocytokine highly expressed in epicardial adipose tissue. Biochem. Biophys. Res. Commun. 2013, 432, 105–110. [Google Scholar] [CrossRef]
- Baker, A.R.; Harte, A.L.; Howell, N.; Pritlove, D.C.; Ranasinghe, A.M.; Da Silva, N.F.; Youssef, E.M.; Khunti, K.; Davies, M.J.; Bonser, R.S.; et al. Epicardial adipose tissue as a source of nuclear factor-kappaB and c-Jun N-terminal kinase mediated inflammation in patients with coronary artery disease. J. Clin. Endocrinol. Metab. 2009, 94, 261–267. [Google Scholar] [CrossRef]
- Du, Y.; Ji, Q.; Cai, L.; Huang, F.; Lai, Y.; Liu, Y.; Yu, J.; Han, B.; Zhu, E.; Zhang, J.; et al. Association between omentin-1 expression in human epicardial adipose tissue and coronary atherosclerosis. Cardiovasc. Diabetol. 2016, 15, 90. [Google Scholar] [CrossRef]
- Iacobellis, G.; Pistilli, D.; Gucciardo, M.; Leonetti, F.; Miraldi, F.; Brancaccio, G.; Gallo, P.; Digioia, C. Adiponectin expression in human epicardial adipose tissue in vivo is lower in patients with coronary artery disease. Cytokine 2005, 29, 251–255. [Google Scholar] [CrossRef]
- Karastergiou, K.; Evans, I.; Ogston, N.; Miheisi, N.; Nair, D.; Kaski, J.-C.; Jahangiri, M.; Mohamed-Ali, V. Epicardial adipokines in obesity and coronary artery disease induce atherogenic changes in monocytes and endothelial cells. Arter. Thromb. Vasc. Biol. 2010, 30, 1340–1346. [Google Scholar] [CrossRef]
- Kremen, J.; Dolinkova, M.; Krajickova, J.; Blaha, J.; Anderlova, K.; Lacinova, Z.; Haluzikova, D.; Bosanska, L.; Vokurka, M.; Svacina, S.; et al. Increased subcutaneous and epicardial adipose tissue production of proinflammatory cytokines in cardiac surgery patients: Possible role in postoperative insulin resistance. J. Clin. Endocrinol. Metab. 2006, 91, 4620–4627. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.-H.; Chu, C.-S.; Lee, K.-T.; Lin, T.-H.; Hsieh, C.-C.; Chiu, C.-C.; Voon, W.-C.; Sheu, S.-H.; Lai, W.-T. Adipocytokines and proinflammatory mediators from abdominal and epicardial adipose tissue in patients with coronary artery disease. Int. J. Obes. 2007, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jia, Y.; Wang, L.; Li, D.; Wang, L.; Zhu, Y.; Liu, J.; Gong, J. Vasodilator-Stimulated Phosphoprotein: Regulators of Adipokines Resistin and Phenotype Conversion of Epicardial Adipocytes. Med. Sci. Monit. 2018, 24, 6010–6020. [Google Scholar] [CrossRef] [PubMed]
- Camarena, V.; Sant, D.; Mohseni, M.; Salerno, T.; Zaleski, M.; Wang, G.; Iacobellis, G. Novel atherogenic pathways from the differential transcriptome analysis of diabetic epicardial adipose tissue. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 739–750. [Google Scholar] [CrossRef]
- McAninch, E.A.; Fonseca, T.L.; Poggioli, R.; Panos, A.L.; Salerno, T.A.; Deng, Y.; Li, Y.; Bianco, A.C.; Iacobellis, G. Epicardial adipose tissue has a unique transcriptome modified in severe coronary artery disease. Obesity 2015, 23, 1267–1278. [Google Scholar] [CrossRef]
- Salgado-Somoza, A.; Teijeira-Fernández, E.; Rubio, J.; Couso, E.; González-Juanatey, J.R.; Eiras, S. Coronary artery disease is associated with higher epicardial retinol-binding protein 4 (RBP4) and lower glucose transporter (GLUT) 4 levels in epicardial and subcutaneous adipose tissue. Clin. Endocrinol. 2011, 76, 51–58. [Google Scholar] [CrossRef]
- Suthahar, N.; Meijers, W.C.; Silljé, H.H.W.; de Boer, R.A. From Inflammation to Fibrosis—Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr. Heart Fail. Rep. 2017, 14, 235–250. [Google Scholar] [CrossRef]
- Byrne, N.J.; Soni, S.; Takahara, S.; Ferdaoussi, M.; Al Batran, R.; Darwesh, A.M.; Levasseur, J.L.; Beker, D.; Vos, D.Y.; Schmidt, M.A.; et al. Chronically Elevating Circulating Ketones Can Reduce Cardiac Inflammation and Blunt the Development of Heart Failure. Circ. Heart Fail. 2020, 13, e006573. [Google Scholar] [CrossRef]
- Withaar, C.; Meems, L.M.G.; Markousis-Mavrogenis, G.; Boogerd, C.J.; Silljé, H.H.W.; Schouten, E.M.; Dokter, M.M.; A Voors, A.; Westenbrink, B.D.; Lam, C.S.P.; et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc. Res. 2020, 117, 2108–2124. [Google Scholar] [CrossRef]
- Mazzieri, A.; Basta, G.; Calafiore, R.; Luca, G. GLP-1 RAs and SGLT2i: Two antidiabetic agents associated with immune and inflammation modulatory properties through the common AMPK pathway. Front. Immunol. 2023, 14, 1163288. [Google Scholar] [CrossRef]
- Wallenius, K.; Kroon, T.; Hagstedt, T.; Löfgren, L.; Sörhede-Winzell, M.; Boucher, J.; Lindén, D.; Oakes, N.D. The SGLT2 inhibitor dapagliflozin promotes systemic FFA mobilization, enhances hepatic β-oxidation, and induces ketosis. J. Lipid Res. 2022, 63, 100176. [Google Scholar] [CrossRef] [PubMed]
- Cowie, M.R.; Fisher, M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. SGLT2 inhibitors: Role in protective reprogramming of cardiac nutrient transport and metabolism. Nat. Rev. Cardiol. 2023, 20, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Ota, T. Emerging roles of SGLT2 inhibitors in obesity and insulin resistance: Focus on fat browning and macrophage polarization. Adipocyte 2018, 7, 121–128. [Google Scholar] [CrossRef]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 Inhibition by Empagliflozin Promotes Fat Utilization and Brownin and Attenuates Inflammation and Insulin Resistance by Polarizing M2 Macrophages in Diet-induced Obese Mice. EBioMedicine 2017, 20, 137–149. [Google Scholar] [CrossRef]
- Kitade, H.; Sawamoto, K.; Nagashimada, M.; Inoue, H.; Yamamoto, Y.; Sai, Y.; Takamura, T.; Yamamoto, H.; Miyamoto, K.I.; Ginsberg, H.N.; et al. CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes 2012, 61, 1680–1690. [Google Scholar] [CrossRef]
- Sato, T.; Aizawa, Y.; Yuasa, S.; Kishi, S.; Fuse, K.; Fujita, S.; Ikeda, Y.; Kitazawa, H.; Takahashi, M.; Sato, M.; et al. The effect of dapagliflozin treatment on epicardial adipose tissue volume. Cardiovasc. Diabetol. 2018, 17, 6. [Google Scholar] [CrossRef]
- Díaz-Rodríguez, E.; Agra, R.M.; Fernández, Á.L.; Adrio, B.; García-Caballero, T.; González-Juanatey, J.R.; Eiras, S. Effects of dapagliflozin on human epicardial adipose tissue: Modulation of insulin resistance, inflammatory chemokine production, and differentiation ability. Cardiovasc. Res. 2017, 114, 336–346. [Google Scholar] [CrossRef]
- Katsuumi, G.; Shimizu, I.; Suda, M.; Yoshida, Y.; Furihata, T.; Joki, Y.; Hsiao, C.-L.; Jiaqi, L.; Fujiki, S.; Abe, M.; et al. SGLT2 inhibition eliminates senescent cells and alleviates pathological aging. Nat. Aging 2024, 4, 926–938. [Google Scholar] [CrossRef]
- Dai, X.; Bu, X.; Gao, Y.; Guo, J.; Hu, J.; Jiang, C.; Zhang, Z.; Xu, K.; Duan, J.; He, S.; et al. Energy status dictates PD-L1 protein abundance and anti-tumor immunity to enable checkpoint blockade. Mol. Cell 2021, 81, 2317–2331.e6. [Google Scholar] [CrossRef]
- Wang, T.-W.; Johmura, Y.; Suzuki, N.; Omori, S.; Migita, T.; Yamaguchi, K.; Hatakeyama, S.; Yamazaki, S.; Shimizu, E.; Imoto, S.; et al. Blocking PD-L1–PD-1 improves senescence surveillance and ageing phenotypes. Nature 2022, 611, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.H.; Poffenberger, M.C.; Wong, A.H.-T.; Jones, R.G. The role of AMPK in T cell metabolism and function. Curr. Opin. Immunol. 2017, 46, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, B.J.; Blagih, J.; Ponce-Garcia, F.M.; Canavan, M.; Gudgeon, N.; Eastham, S.; Hill, D.; Hanlon, M.M.; Ma, E.H.; Bishop, E.L.; et al. Canagliflozin impairs T cell effector function via metabolic suppression in autoimmunity. Cell Metab. 2023, 35, 1132–1146.e9. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, X.; Ilyas, I.; Zheng, X.; Luo, S.; Little, P.J.; Kamato, D.; Sahebkar, A.; Wu, W.; Weng, J.; et al. Impact of sodium glucose cotransporter 2 (SGLT2) inhibitors on atherosclerosis: From pharmacology to pre-clinical and clinical therapeutics. Theranostics 2021, 11, 4502–4515. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, S.G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Kokubo, H.; Bumdelger, B.; Yoshizumi, M.; Yamamotoya, T.; Matsunaga, Y.; Ueda, K.; Inoue, Y.; Inoue, M.-K.; Fujishiro, M.; et al. The SGLT2 Inhibitor Luseogliflozin Rapidly Normalizes Aortic mRNA Levels of Inflammation-Related but Not Lipid-Metabolism-Related Genes and Suppresses Atherosclerosis in Diabetic ApoE KO Mice. Int. J. Mol. Sci. 2017, 18, 1704. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.K.; Nasiri-Ansari, N.; Agorogiannis, G.; Perrea, D.; Kostakis, I.D.; Papavassiliou, A.G.; Kaltsas, G.; Kassi, E.; Randeva, H.S. Canagliflozin attenuates the progression of atherosclerosis and inflammation process in APOE knockout mice. Endocr. Abstr. 2018, 17, 106. [Google Scholar] [CrossRef]
- Spigoni, V.; Fantuzzi, F.; Carubbi, C.; Pozzi, G.; Masselli, E.; Gobbi, G.; Solini, A.; Bonadonna, R.C.; Cas, A.D. Sodium-glucose cotransporter 2 inhibitors antagonize lipotoxicity in human myeloid angiogenic cells and ADP-dependent activation in human platelets: Potential relevance to prevention of cardiovascular events. Cardiovasc. Diabetol. 2020, 19, 46. [Google Scholar] [CrossRef]
- Adingupu, D.D.; Göpel, S.O.; Grönros, J.; Behrendt, M.; Sotak, M.; Miliotis, T.; Dahlqvist, U.; Gan, L.M.; Jönsson-Rylander, A.C. SGLT2 inhibition with empagliflozin improves coronary microvascular function and cardiac contractility in prediabetic ob/ob-/- mice. Cardiovasc. Diabetol. 2019, 18, 16. [Google Scholar] [CrossRef]
- Cappetta, D.; De Angelis, A.; Ciuffreda, L.P.; Coppini, R.; Cozzolino, A.; Miccichè, A.; Dell’Aversana, C.; D’amario, D.; Cianflone, E.; Scavone, C.; et al. Amelioration of diastolic dysfunction by dapagliflozin in a non-diabetic model involves coronary endothelium. Pharmacol. Res. 2020, 157, 104781. [Google Scholar] [CrossRef]
- Cessario, J.; Pierre-Louis, V.; Wahl, J.; Li, Z. Empagliflozin, alone or in combination with liraglutide, limits cell death in vitro: Role of oxidative stress and nitric oxide. Pharmacol. Rep. 2021, 73, 858–867. [Google Scholar] [CrossRef]
- Uthman, L.; Homayr, A.; Juni, R.P.; Spin, E.L.; Kerindongo, R.; Boomsma, M.; Hollmann, M.W.; Preckel, B.; Koolwijk, P.; Van Hinsbergh, V.W.M.; et al. Empagliflozin and Dapagliflozin Reduce ROS Generation and Restore NO Bioavailability in Tumor Necrosis Factor α-Stimulated Human Coronary Arterial Endothelial Cells. Cell. Physiol. Biochem. 2019, 53, 865–886. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Li, X.; Baartscheer, A.; Schumacher, C.A.; Baumgart, P.; Hermanides, J.; Preckel, B.; Hollmann, M.W.; Coronel, R.; Zuurbier, C.J.; et al. Empagliflozin reduces oxidative stress through inhibition of the novel inflammation/NHE/[Na+]c/ROS-pathway in human endothelial cells. Biomed. Pharmacother. 2022, 146, 112515. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Baratta, F.; Buzzetti, R.; D’amico, A.; Castellani, V.; Bartimoccia, S.; Siena, A.; D’onofrio, L.; Maddaloni, E.; Pingitore, A.; et al. The Sodium–Glucose Co-Transporter-2 (SGLT2) Inhibitors Reduce Platelet Activation and Thrombus Formation by Lowering NOX2-Related Oxidative Stress: A Pilot Study. Antioxidants 2022, 11, 1878. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Verma, S.; Hassanabad, A.F.; Teng, G.; Belke, D.D.; Dundas, J.A.; Guzzardi, D.G.; Svystonyuk, D.A.; Pattar, S.S.; Park, D.S.; et al. Direct Effects of Empagliflozin on Extracellular Matrix Remodelling in Human Cardiac Myofibroblasts: Novel Translational Clues to Explain EMPA-REG OUTCOME Results. Can. J. Cardiol. 2020, 36, 543–553. [Google Scholar] [CrossRef]
- Al Thani, N.A.; Hasan, M.; Yalcin, H.C. Use of Animal Models for Investigating Cardioprotective Roles of SGLT2 Inhibitors. J. Cardiovasc. Transl. Res. 2023, 16, 975–986. [Google Scholar] [CrossRef]
- Byrne, N.J.; Parajuli, N.; Levasseur, J.L.; Boisvenue, J.; Beker, D.L.; Masson, G.; Fedak, P.W.; Verma, S.; Dyck, J.R. Empagliflozin Prevents Worsening of Cardiac Function in an Experimental Model of Pressure Overload-Induced Heart Failure. JACC Basic. Transl. Sci. 2017, 2, 347–354. [Google Scholar] [CrossRef]
- Connelly, K.A.; Zhang, Y.; Visram, A.; Advani, A.; Batchu, S.N.; Desjardins, J.-F.; Thai, K.; Gilbert, R.E. Empagliflozin Improves Diastolic Function in a Nondiabetic Rodent Model of Heart Failure with Preserved Ejection Fraction. JACC Basic. Transl. Sci. 2019, 4, 27–37. [Google Scholar] [CrossRef]
- Marx, N.; Husain, M.; Lehrke, M.; Verma, S.; Sattar, N. GLP-1 Receptor Agonists for the Reduction of Atherosclerotic Cardiovascular Risk in Patients with Type 2 Diabetes. Circulation 2022, 146, 1882–1894. [Google Scholar] [CrossRef]
- Iacobellis, G. Epicardial fat: A new cardiovascular therapeutic target. Curr. Opin. Pharmacol. 2016, 27, 13–18. [Google Scholar] [CrossRef]
- Chen, J.; Mei, A.; Wei, Y.; Li, C.; Qian, H.; Min, X.; Yang, H.; Dong, L.; Rao, X.; Zhong, J. GLP-1 receptor agonist as a modulator of innate immunity. Front. Immunol. 2022, 13, 997578. [Google Scholar] [CrossRef] [PubMed]
- Vinué, Á.; Navarro, J.; Herrero-Cervera, A.; García-Cubas, M.; Andrés-Blasco, I.; Martínez-Hervás, S.; Real, J.T.; Ascaso, J.F.; González-Navarro, H. The GLP-1 analogue lixisenatide decreases atherosclerosis in insulin-resistant mice by modulating macrophage phenotype. Diabetologia 2017, 60, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Hadjiyanni, I.; Siminovitch, K.A.; Danska, J.S.; Drucker, D.J. Glucagon-like peptide-1 receptor signalling selectively regulates murine lymphocyte proliferation and maintenance of peripheral regulatory T cells. Diabetologia 2010, 53, 730–740. [Google Scholar] [CrossRef]
- Liu, G.; Ma, H.; Qiu, L.; Li, L.; Cao, Y.; Ma, J.; Zhao, Y. Phenotypic and functional switch of macrophages induced by regulatory CD4+CD25+ T cells in mice. Immunol. Cell Biol. 2011, 89, 130–142. [Google Scholar] [CrossRef]
- Luna-Marco, C.; de Marañon, A.M.; Hermo-Argibay, A.; Rodriguez-Hernandez, Y.; Hermenejildo, J.; Fernandez-Reyes, M.; Apostolova, N.; Vila, J.; Sola, E.; Morillas, C.; et al. Effects of GLP-1 receptor agonists on mitochondrial function, inflammatory markers and leukocyte-endothelium interactions in type 2 diabetes. Redox Biol. 2023, 66, 102849. [Google Scholar] [CrossRef]
- Noyan-Ashraf, M.H.; Shikatani, E.A.; Schuiki, I.; Mukovozov, I.; Wu, J.; Li, R.-K.; Volchuk, A.; Robinson, L.A.; Billia, F.; Drucker, D.J.; et al. A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation 2013, 127, 74–85. [Google Scholar] [CrossRef]
- Inoue, T.; Inoguchi, T.; Sonoda, N.; Hendarto, H.; Makimura, H.; Sasaki, S.; Yokomizo, H.; Fujimura, Y.; Miura, D.; Takayanagi, R. GLP-1 analog liraglutide protects against cardiac steatosis, oxidative stress and apoptosis in streptozotocin-induced diabetic rats. Atherosclerosis 2015, 240, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Qi, W.; Yu, Y.; Du, S.; Wu, J.; Liu, J. Effect of exenatide on the cardiac expression of adiponectin receptor 1 and NADPH oxidase subunits and heart function in streptozotocin-induced diabetic rats. Diabetol. Metab. Syndr. 2014, 6, 29. [Google Scholar] [CrossRef]
- Cao, Y.-Y.; Chen, Z.-W.; Gao, Y.-H.; Wang, X.-X.; Ma, J.-Y.; Chang, S.-F.; Qian, J.-Y.; Ge, J.-B. Exenatide Reduces Tumor Necrosis Factor-α-induced Apoptosis in Cardiomyocytes by Alleviating Mitochondrial Dysfunction. Chin. Med. J. 2015, 128, 3211–3218. [Google Scholar] [CrossRef]
- Zhang, L.; Tian, J.; Diao, S.; Zhang, G.; Xiao, M.; Chang, D. GLP-1 receptor agonist liraglutide protects cardiomyocytes from IL-1β-induced metabolic disturbance and mitochondrial dysfunction. Chem. Interact. 2020, 332, 109252. [Google Scholar] [CrossRef]
- Biran, A.; Zada, L.; Karam, P.A.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Qian, P.; Tian, H.; Wang, Y.; Lu, W.; Li, Y.; Ma, T.; Gao, X.; Yao, W. A novel oral glucagon-like peptide 1 receptor agonist protects against diabetic cardiomyopathy via alleviating cardiac lipotoxicity induced mitochondria dysfunction. Biochem. Pharmacol. 2020, 182, 114209. [Google Scholar] [CrossRef] [PubMed]
- Monji, A.; Mitsui, T.; Bando, Y.K.; Aoyama, M.; Shigeta, T.; Murohara, T. Glucagon-like peptide-1 receptor activation reverses cardiac remodeling via normalizing cardiac steatosis and oxidative stress in type 2 diabetes. Am. J. Physiol. Circ. Physiol. 2013, 305, H295–H304. [Google Scholar] [CrossRef]
- Liu, Q.; Anderson, C.; Broyde, A.; Polizzi, C.; Fernandez, R.; Baron, A.; Parkes, D.G. Glucagon-like peptide-1 and the exenatide analogue AC3174 improve cardiac function, cardiac remodeling, and survival in rats with chronic heart failure. Cardiovasc. Diabetol. 2010, 9, 76. [Google Scholar] [CrossRef]
- McCormick, L.M.; Hoole, S.P.; White, P.A.; Read, P.A.; Axell, R.G.; Clarke, S.J.; O’sullivan, M.; West, N.E.; Dutka, D.P. Pre-treatment with glucagon-like peptide-1 protects against ischemic left ventricular dysfunction and stunning without a detected difference in myocardial substrate utilization. JACC Cardiovasc. Interv. 2015, 8, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Trombara, F.; Cosentino, N.; Bonomi, A.; Ludergnani, M.; Poggio, P.; Gionti, L.; Baviera, M.; Colacioppo, P.; Roncaglioni, M.C.; Leoni, O.; et al. Impact of chronic GLP-1 RA and SGLT-2I therapy on in-hospital outcome of diabetic patients with acute myocardial infarction. Cardiovasc. Diabetol. 2023, 22, 26. [Google Scholar] [CrossRef]
- Wilcox, T.; De Block, C.; Schwartzbard, A.Z.; Newman, J.D. Diabetic Agents, From Metformin to SGLT2 Inhibitors and GLP1 Receptor Agonists. J. Am. Coll. Cardiol. 2020, 75, 1956–1974. [Google Scholar] [CrossRef]
- Giugliano, D.; Maiorino, M.I.; Bellastella, G.; Chiodini, P.; Esposito, K. Glycemic Control, Preexisting Cardiovascular Disease, and Risk of Major Cardiovascular Events in Patients with Type 2 Diabetes Mellitus: Systematic Review with Meta-Analysis of Cardiovascular Outcome Trials and Intensive Glucose Control Trials. J. Am. Heart Assoc. 2019, 8, e012356. [Google Scholar] [CrossRef]
- Singh, G.; Krauthamer, M.; Bjalme-Evans, M. Wegovy (semaglutide): A new weight loss drug for chronic weight management. J. Investig. Med. 2022, 70, 5–13. [Google Scholar] [CrossRef]
- Ding, Q.-Y.; Tian, J.-X.; Li, M.; Lian, F.-M.; Zhao, L.-H.; Wei, X.-X.; Han, L.; Zheng, Y.-J.; Gao, Z.-Z.; Yang, H.-Y.; et al. Interactions Between Therapeutics for Metabolic Disease, Cardiovascular Risk Factors, and Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 530160. [Google Scholar] [CrossRef]
- van Raalte, D.H.; Bjornstad, P.; Cherney, D.Z.I.; de Boer, I.H.; Fioretto, P.; Gordin, D.; Persson, F.; Rosas, S.E.; Rossing, P.; Schaub, J.A.; et al. Combination therapy for kidney disease in people with diabetes mellitus. Nat. Rev. Nephrol. 2024, 20, 433–446. [Google Scholar] [CrossRef] [PubMed]
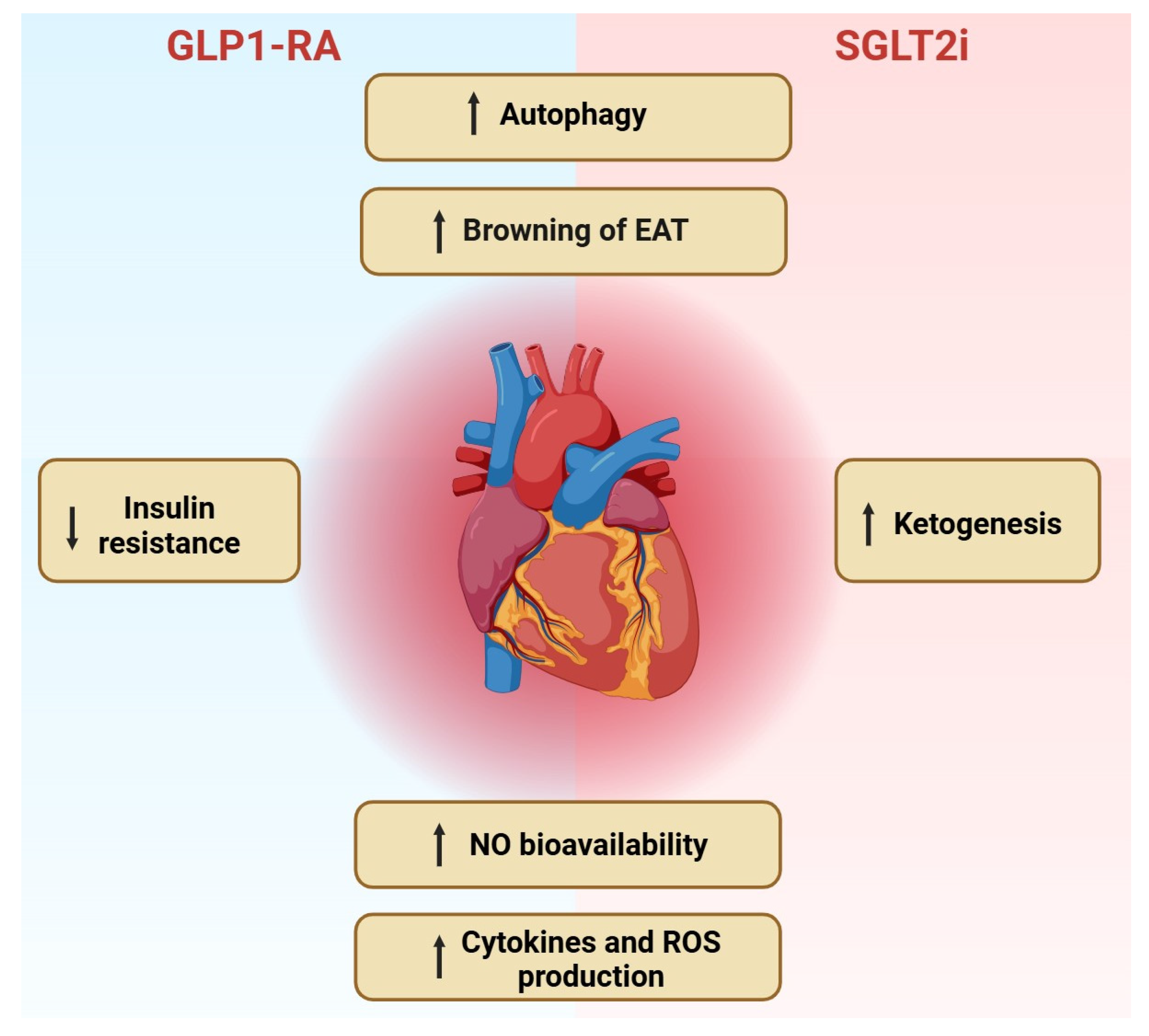

{kind=link}
| SGLT2i | |
|---|---|
| Metabolic Changes | - Promote a metabolic shift towards FFA utilization and ketogenesis, increasing cardiac efficiency (depending on the metabolic state of the patient) - Provide a negative caloric balance, promoting autophagy and weight loss - Promote fat-browning effect, providing polarization of M1 to M2 macrophages in adipose tissue |
| Oxidative Stress | - Decrease apoptosis - Increase NO production - Provide cytoprotective effects in endothelial cells - Reduce hydrogen peroxide and NADPH formation |
| Inflammation and Fibrosis | - Decrease the expression of profibrotic factors, decreasing extracellular matrix remodeling - Lower activation of NLRP3 inflammasomes - Reduce left ventricular mass and provide beneficial effects on diastolic function - Reduce cytokines, chemokines, and adhesion molecules - Promote plaque stabilization by antiproliferative effects and prevent endothelial dysfunction, reducing vascular stiffness |
| GLP1-RAs | |
|---|---|
| Metabolic Changes | - Promote fat-browning effect and brown adipose tissue thermogenesis - Reduce body weight, providing satiety signaling and increasing insulin sensitivity |
| Oxidative Stress | - Reduce intracellular and mitochondrial ROS production - Suppress NOX-4, increasing SOD-1 and glutathione peroxidase levels - Reduce myocardial triglyceride and diacylglycerol levels by the activation of AMPK pathway |
| Inflammation and Fibrosis | - Reduce collagen deposition, decreasing cardiac hypertrophy and myocardial fibrosis - Arrest adverse cardiac ischemic remodeling and improve the recovery of ventricular function after myocardial infarction - Provide antiatherogenic effects, reducing pro-inflammatory cytokines and delivering antiproliferative actions in vascular smooth muscle cells |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcon, L.M.R.; Mazzieri, A. Anti-Inflammatory and Anti-Oxidative Effects of GLP1-RAs and SGLT2i: The Guiding Star Towards Cardiovascular Protection in Type 2 Diabetes. Immuno 2025, 5, 11. https://doi.org/10.3390/immuno5010011
Marcon LMR, Mazzieri A. Anti-Inflammatory and Anti-Oxidative Effects of GLP1-RAs and SGLT2i: The Guiding Star Towards Cardiovascular Protection in Type 2 Diabetes. Immuno. 2025; 5(1):11. https://doi.org/10.3390/immuno5010011
Chicago/Turabian StyleMarcon, Livia M. R., and Alessio Mazzieri. 2025. "Anti-Inflammatory and Anti-Oxidative Effects of GLP1-RAs and SGLT2i: The Guiding Star Towards Cardiovascular Protection in Type 2 Diabetes" Immuno 5, no. 1: 11. https://doi.org/10.3390/immuno5010011
APA StyleMarcon, L. M. R., & Mazzieri, A. (2025). Anti-Inflammatory and Anti-Oxidative Effects of GLP1-RAs and SGLT2i: The Guiding Star Towards Cardiovascular Protection in Type 2 Diabetes. Immuno, 5(1), 11. https://doi.org/10.3390/immuno5010011