
Closing the Door with CRISPR: Genome Editing of CCR5 and CXCR4 as a Potential Curative Solution for HIV


Abstract
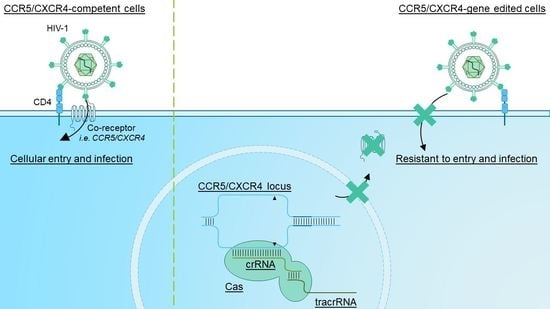
:
1. Introduction
2. The HIV Infection Cycle—Targets for Therapy?
3. Gene Editing Basics—Mode of Action and Different Tools
4. Targeting CCR5 via CRISPR/Cas9-Mediated Genome Editing
5. Targeting CXCR4 via CRISPR/Cas9-Mediated Genome Editing
6. Simultaneous Deletion of CCR5 and CXCR4 via CRISPR/Cas9-Mediated Genome Editing
7. Investigating Other Targets That Could Contribute to a Functional HIV Cure
8. Targeting the HIV Viral Reservoir
9. Considerations for Monitoring Cellular and Viral Compartments after CRISPR/Cas9-Mediated Genome Editing
10. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baxter, A.E.; O’Doherty, U.; Kaufmann, D.E. Beyond the Replication-Competent HIV Reservoir: Transcription and Translation-Competent Reservoirs. Retrovirology 2018, 15, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulick, R.M.; Flexner, C. Long-Acting HIV Drugs for Treatment and Prevention. Annu. Rev. Med. 2019, 70, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Parekh, B.S.; Ou, C.Y.; Fonjungo, P.N.; Kalou, M.B.; Rottinghaus, E.; Puren, A.; Alexander, H.; Cox, M.H.; Nkengasong, J.N. Diagnosis of Human Immunodeficiency Virus Infection. Clin. Microbiol. Rev. 2019, 32, e00064-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freen-van Heeren, J.J. Addressing HIV-1 Latency with Flow-FISH: Finding, Characterizing and Targeting HIV-1 Infected Cells. Cytom. Part A 2021, 99, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Wang, C.; Patton, C.; Murray, M. A Review of Long-Term Toxicity of Antiretroviral Treatment Regimens and Implications for an Aging Population. Infect. Dis. Ther. 2018, 7, 183–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boasso, A.; Shearer, G.M.; Chougnet, C. Immune Dysregulation in Human Immunodeficiency Virus Infection: Know It, Fix It, Prevent It? J. Intern. Med. 2009, 256, 78–96. [Google Scholar] [CrossRef]
- Bunjun, R.; Riou, C.; Soares, A.P.; Thawer, N.; Müller, T.L.; Kiravu, A.; Ginbot, Z.; Oni, T.; Goliath, R.; Kalsdorf, B.; et al. Effect of HIV on the Frequency and Number of Mycobacterium Tuberculosis-Specific CD4+ T Cells in Blood and Airways during Latent M. Tuberculosis Infection. J. Infect. Dis. 2017, 216, 1550–1560. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, K.V.; Karthigeyan, K.P.; Tripathi, S.P.; Hanna, L.E. Pathophysiology of CD4+ T-Cell Depletion in HIV-1 and HIV-2 Infections. Front. Immunol. 2017, 8, 580. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.P.; Kitchen, S.G.; Pugach, P.; Zack, J.A. The CCR5 and CXCR4 Coreceptors—Central to Understanding the Transmission and Pathogenesis of Human Immunodeficiency Virus Type 1 Infection. AIDS Res. Hum. Retrovir. 2004, 20, 111–126. [Google Scholar] [CrossRef]
- Barton, K.; Winckelmann, A.; Palmer, S. HIV-1 Reservoirs During Suppressive Therapy Kirston. Trends Microbiol. 2016, 24, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Ganepola, S.; Müßig, A.; Allers, K.; Ph, D.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; Blau, I.W.; Hofmann, W.K.; et al. Long-Term Control of HIV by CCR5 Delta32/Delta32 Stem-Cell Transplantation. N. Engl. J. Med. 2009, 360, 692–697. [Google Scholar]
- Duarte, R.F.; Salgado, M.; Sánchez-Ortega, I.; Arnan, M.; Canals, C.; Domingo-Domenech, E.; Fernández-de-Sevilla, A.; González-Barca, E.; Morón-López, S.; Nogues, N.; et al. Ccr5Δ32 Homozygous Cord Blood Allogeneic Transplantation in a Patient with Hiv: A Case Report. Lancet HIV 2015, 2, e236–e242. [Google Scholar] [CrossRef]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. HIV-1 Remission Following CCR5Δ32/Δ32 Haematopoietic Stem-Cell Transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 Infection in Caucasian Individuals Bearing Mutant Alleles of the CCR-5 Chemokine Receptor Gene. Nature 1996, 382, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Allers, K.; Hütter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the Cure of HIV Infection by CCR5δ32/Δ32 Stem Cell Transplantation. Blood 2011, 117, 2791–2799. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Peppa, D.; Hill, A.L.; Gálvez, C.; Salgado, M.; Pace, M.; McCoy, L.E.; Griffith, S.A.; Thornhill, J.; Alrubayyi, A.; et al. Evidence for HIV-1 Cure after CCR5Δ32/Δ32 Allogeneic Haemopoietic Stem-Cell Transplantation 30 Months Post Analytical Treatment Interruption: A Case Report. Lancet HIV 2020, 7, e340–e347. [Google Scholar] [CrossRef] [Green Version]
- Martinson, J.; Chapman, N.; Rees, D.; Liu, Y.; Clegg, J. Global Distribution of the CCR5 Gene 32-Basepair Deletion. Nat. Genet. 1997, 16, 100–103. [Google Scholar] [CrossRef]
- Hütter, G.; Thiel, E. Allogeneic Transplantation of CCR5-Deficient Progenitor Cells in a Patient with HIV Infection: An Update after 3 Years and the Search for Patient No. 2. Aids 2011, 25, 273–274. [Google Scholar] [CrossRef]
- Claireaux, M.; Robinot, R.; Kervevan, J.; Patgaonkar, M.; Staropoli, I.; Brelot, A.; Nouël, A.; Gellenoncourt, S.; Tang, X.; Héry, M.; et al. Low CCR5 Expression Protects HIV-Specific CD4+ T Cells of Elite Controllers from Viral Entry. Nat. Commun. 2022, 13, 521. [Google Scholar] [CrossRef]
- Didigu, C.A.; Wilen, C.B.; Wang, J.; Duong, J.; Secreto, A.J.; Danet-Desnoyers, G.A.; Riley, J.L.; Gregory, P.D.; June, C.H.; Holmes, M.C.; et al. Simultaneous Zinc-Finger Nuclease Editing of the HIV Coreceptors Ccr5 and Cxcr4 Protects CD4+ T Cells from HIV-1 Infection. Blood 2014, 123, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, R.; Bergis, B.; Solis-Leal, A.; Igbinedion, O.; Strong, C.; Schiller, M. TALEN Gene Editing Takes Aim on HIV. Hum. Genet. 2016, 135, 1059–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted Genome Engineering in Human Cells with the Cas9 RNA-Guided Endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ye, C.; Liu, J.; Zhang, D.; Kimata, J.T. CCR5 Gene Disruption via Lentiviral Vectors Expressing Cas9 and Single Guided RNA Renders Cells Resistant to HIV-1 Infection. PLoS ONE 2014, 12, e115987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Liang, J.; Chen, S.; Wang, K.; Liu, X.; Liu, B.; Xia, Y.; Guo, M.; Zhang, X.; Sun, G.; et al. Genome Editing of CCR5 by AsCpf1 Renders CD4+T Cells Resistance to HIV-1 Infection. Cell Biosci. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Hou, P.; Chen, S.; Wang, S.; Yu, X.; Chen, Y.; Jiang, M.; Zhuang, K.; Ho, W.; Hou, W.; Huang, J.; et al. Genome Editing of CXCR4 by CRISPR/Cas9 Confers Cells Resistant to HIV-1 Infection. Sci. Rep. 2015, 5, 15577. [Google Scholar] [CrossRef]
- Tebas, P.; Jadlowsky, J.K.; Shaw, P.A.; Tian, L.; Esparza, E.; Brennan, A.; Kim, S.; SY, N.; Richardson, M.; Vogel, A.; et al. CCR5-Edited CD4 T Cells Augment HIV-Specific Immunity to Enable Post Rebound Control of HIV Replication. J. Clin. Investig. 2021, 131, 7. [Google Scholar] [CrossRef]
- Knipping, F.; Newby, G.A.; Eide, C.R.; McElroy, A.N.; Nielsen, S.C.; Smith, K.; Fang, Y.; Cornu, T.I.; Costa, C.; Gutierrez-Guerrero, A.; et al. Disruption of HIV-1 Co-Receptors CCR5 and CXCR4 in Primary Human T Cells and Hematopoietic Stem and Progenitor Cells Using Base Editing. Mol. Ther. 2022, 30, 130–144. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-Target Effects in CRISPR/Cas9-Mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Gupta, R.M.; Musunuru, K. Expanding the Genetic Editing Tool Kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Investig. 2014, 124, 4154–4161. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and Feasibility of CRISPR-Edited T Cells in Patients with Refractory Non-Small-Cell Lung Cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Berger, E.A.; Murphy, P.M.; Farber, J.M. Chemokine Receptors as HIV-1 Coreceptors: Roles in Viral Entry, Tropism, and Disease. Annu. Rev. Immunol. 1999, 17, 657–700. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O. HIV-1 Assembly, Release and Maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [Google Scholar] [CrossRef]
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef]
- Feng, Y.; Broder, C.C.; Kennedy, P.A.; Berger, E.A. HIV-1 Entry Cofactor: Functional CDNA Cloning of a Seven-Transmembrane, G Protein-Coupled Receptor. Science 1996, 272, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Aiken, C.; Rousso, I. The HIV-1 Capsid and Reverse Transcription. Retrovirology 2021, 18, e00289-17. [Google Scholar] [CrossRef]
- Hu, W.S.; Hughes, S.H. HIV-1 Reverse Transcription. Cold Spring Harb. Perspect. Med. 2012, 2, a006882. [Google Scholar] [CrossRef] [Green Version]
- Santa-Marta, M.; de Brito, P.M.; Godinho-Santos, A.; Goncalves, J. Host Factors and HIV-1 Replication: Clinical Evidence and Potential Therapeutic Approaches. Front. Immunol. 2013, 4, 343. [Google Scholar] [CrossRef] [Green Version]
- Rebensburg, S.V.; Wei, G.; Larue, R.C.; Lindenberger, J.; Francis, A.C.; Annamalai, A.S.; Morrison, J.; Shkriabai, N.; Huang, S.W.; KewalRamani, V.; et al. Sec24C Is an HIV-1 Host Dependency Factor Crucial for Virus Replication. Nat. Microbiol. 2021, 6, 435–444. [Google Scholar] [CrossRef]
- Hiatt, J.; Hultquist, J.F.; McGregor, M.J.; Bouhaddou, M.; Leenay, R.T.; Simons, L.M.; Young, J.M.; Haas, P.; Roth, T.L.; Tobin, V.; et al. A Functional Map of HIV-Host Interactions in Primary Human T Cells. Nat. Commun. 2022, 13, 1752. [Google Scholar] [CrossRef]
- Cleret-Buhot, A.; Zhang, Y.; Planas, D.; Goulet, J.P.; Monteiro, P.; Gosselin, A.; Wacleche, V.S.; Tremblay, C.L.; Jenabian, M.A.; Routy, J.P.; et al. Identification of Novel HIV-1 Dependency Factors in Primary CCR4+CCR6+Th17 Cells via a Genome-Wide Transcriptional Approach. Retrovirology 2015, 12, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glatzová, D.; Cebecauer, M. Dual Role of CD4 in Peripheral T Lymphocytes. Front. Immunol. 2019, 10, 618. [Google Scholar] [CrossRef] [PubMed]
- De Silva, E.; Stumpf, M.P.H. HIV and the CCR5-Δ32 Resistance Allele. FEMS Microbiol. Lett. 2004, 241, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Wang, J.; Crain, K.; Fearns, C.; Kim, K.A.; Hua, K.L.; Gregory, P.D.; Holmes, M.C.; Torbett, B.E. Zinc-Finger Nuclease Editing of Human Cxcr4 Promotes HIV-1 CD4 T Cell Resistance and Enrichment. Mol. Ther. 2012, 20, 849–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarze, L.I.; Głów, D.; Sonntag, T.; Uhde, A.; Fehse, B. Optimisation of a TALE Nuclease Targeting the HIV Co-Receptor CCR5 for Clinical Application. Gene Ther. 2021, 28, 588–601. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Adli, M. The CRISPR Tool Kit for Genome Editing and Beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef]
- Cebrian-Serrano, A.; Davies, B. CRISPR-Cas Orthologues and Variants: Optimizing the Repertoire, Specificity and Delivery of Genome Engineering Tools. Mamm. Genome 2017, 28, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, P.; Jakimo, N.; Jacobson, J.M. Minimal PAM Specificity of a Highly Similar SpCas9 Ortholog. Sci. Adv. 2018, 4, eaau0766. [Google Scholar] [CrossRef] [Green Version]
- Vicencio, J.; Sánchez-Bolaños, C.; Moreno-Sánchez, I.; Brena, D.; Vejnar, C.E.; Kukhtar, D.; Ruiz-López, M.; Cots-Ponjoan, M.; Rubio, A.; Melero, N.R.; et al. Genome Editing in Animals with Minimal PAM CRISPR-Cas9 Enzymes. Nat. Commun. 2022, 13, 2601. [Google Scholar] [CrossRef]
- Qi, C.; Li, D.; Jiang, X.; Jia, X.; Lu, L.; Wang, Y.; Sun, J.; Shao, Y.; Wei, M. Inducing CCR5Δ32/Δ32 Homozygotes in the Human Jurkat CD4+ Cell Line and Primary CD4+ Cells by CRISPR-Cas9 Genome-Editing Technology. Mol. Ther. Nucleic Acids 2018, 12, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Guan, X.; Du, T.; Jin, W.; Wu, B.; Liu, Y.; Wang, P.; Hu, B.; Griffin, G.E.; Shattock, R.J.; et al. Inhibition of HIV-1 Infection of Primary CD4+ T-Cells by Gene Editing of CCR5 Using CRISPR/Cas9. J. Gen. Virol. 2015, 96, 2381–2393. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Chen, S.; Wang, Q.; Liu, Z.; Liu, S.; Deng, H.; Hou, W.; Wu, D.; Xiong, Y.; Li, J.; et al. CCR5 Editing by Staphylococcus Aureus Cas9 in Human Primary CD4+ T Cells and Hematopoietic Stem/Progenitor Cells Promotes HIV-1 Resistance and CD4+ T Cell Enrichment in Humanized Mice. Retrovirology 2019, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, S.; Jin, X.; Wang, Q.; Yang, K.; Li, C.; Xiao, Q.; Hou, P.; Liu, S.; Wu, S.; et al. Genome Editing of the HIV Co-Receptors CCR5 and CXCR4 by CRISPR-Cas9 Protects CD4+ T Cells from HIV-1 Infection. Cell Biosci. 2017, 7, 47. [Google Scholar] [CrossRef]
- Herskovitz, J.; Hasan, M.; Patel, M.; Blomberg, W.R.; Cohen, J.D.; Machhi, J.; Shahjin, F.; Mosley, R.L.; McMillan, J.E.; Kevadiya, B.D.; et al. CRISPR-Cas9 Mediated Exonic Disruption for HIV-1 Elimination. EBioMedicine 2021, 73, 103678. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Kwaku Dad, A.B.; Beloor, J.; Gopalappa, R.; Lee, S.K.; Kim, H. Gene Disruption by Cell-Penetrating Peptide-Mediated Delivery of Cas9 Protein and Guide RNA. Genome Res. 2014, 24, 1020–1027. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Yang, H.; Gao, Y.; Chen, Z.; Xie, L.; Liu, Y.; Liu, Y.; Wang, X.; Li, H.; Lai, W.; et al. CRISPR/Cas9-Mediated CCR5 Ablation in Human Hematopoietic Stem/Progenitor Cells Confers HIV-1 Resistance In Vivo. Mol. Ther. 2017, 25, 1782–1789. [Google Scholar] [CrossRef]
- Seki, A.; Rutz, S. Optimized RNP Transfection for Highly Efficient CRISPR/Cas9-Mediated Gene Knockout in Primary T Cells. J. Exp. Med. 2018, 215, 985–997. [Google Scholar] [CrossRef]
- Hamilton, J.R.; Tsuchida, C.A.; Nguyen, D.N.; Shy, B.R.; McGarrigle, E.R.; Sandoval Espinoza, C.R.; Carr, D.; Blaeschke, F.; Marson, A.; Doudna, J.A. Targeted Delivery of CRISPR-Cas9 and Transgenes Enables Complex Immune Cell Engineering. Cell Rep. 2021, 35, 109207. [Google Scholar] [CrossRef]
- Freen-van Heeren, J.J. Exploiting HIV-1 Tropism to Target CD4+ T Cells for CRISPR. Immunol. Cell Biol. 2021, 99, 677–679. [Google Scholar] [CrossRef]
- Gurusamy, D.; Henning, A.N.; Yamamoto, T.N.; Yu, Z.; Zacharakis, N.; Krishna, S.; Kishton, R.J.; Vodnala, S.K.; Eidizadeh, A.; Jia, L.; et al. Multi-Phenotype CRISPR-Cas9 Screen Identifies P38 Kinase as a Target for Adoptive Immunotherapies. Cancer Cell 2020, 37, 818–833.e9. [Google Scholar] [CrossRef] [PubMed]
- Freen-van Heeren, J.J. Using CRISPR to Enhance T Cell Effector Function for Therapeutic Applications. Cytokine X 2021, 3, 100049. [Google Scholar] [CrossRef] [PubMed]
- Freen-van Heeren, J.J.; Popović, B.; Guislain, A.; Wolkers, M.C. Human T Cells Employ Conserved AU-Rich Elements to Fine-Tune IFN-γ Production. Eur. J. Immunol. 2020, 50, 949–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Zhou, P.; Wei, J.; Long, L.; Shi, H.; Dhungana, Y.; Palacios, G.; Wang, Y.; Qian, C.; Yu, J.; et al. In Vivo CRISPR Screening Reveals Nutrient Signaling Processes Underpinning CD8+ T Cell Fate Decisions. Cell 2021, 184, 1245–1261. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Anti-Tumor Efficacy of Human Chimeric Antigen Receptor T Cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Wang, J.; Liu, Y.; Xie, L.; Su, B.; Mou, D.; Wang, L.; Liu, T.; Wang, X.; Zhang, B.; et al. CRISPR-Edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 1240–1247. [Google Scholar] [CrossRef]
- Rouet, R.; Thuma, B.A.; Roy, M.D.; Lintner, N.G.; Rubitski, D.M.; Finley, J.E.; Wisniewska, H.M.; Mendonsa, R.; Hirsh, A.; De Oñate, L.; et al. Receptor-Mediated Delivery of CRISPR-Cas9 Endonuclease for Cell-Type-Specific Gene Editing. J. Am. Chem. Soc. 2018, 140, 6596–6603. [Google Scholar] [CrossRef]
- Tian, S.; Liu, Y.; Appleton, E.; Wang, H.; Church, G.M.; Dong, M. Targeted Intracellular Delivery of Cas13 and Cas9 Nucleases Using Bacterial Toxin-Based Platforms. Cell Rep. 2022, 38, 110476. [Google Scholar] [CrossRef]
- Choi, J.G.; Dang, Y.; Abraham, S.; Ma, H.; Zhang, J.; Guo, H.; Cai, Y.; Mikkelsen, J.G.; Wu, H.; Shankar, P.; et al. Lentivirus Pre-Packed with Cas9 Protein for Safer Gene Editing. Gene Ther. 2016, 23, 627–633. [Google Scholar] [CrossRef]
- Choe, H.; Farzan, M.; Sun, Y.; Sullivan, N.; Rollins, B.; Ponath, P.D.; Wu, L.; Mackay, C.R.; LaRosa, G.; Newman, W.; et al. The β-Chemokine Receptors CCR3 and CCR5 Facilitate Infection by Primary HIV-1 Isolates. Cell 1996, 85, 1135–1148. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.K.; Strelchenko, N.; Park, M.A.; Kim, Y.H.; Mean, K.D.; Schotzko, M.L.; Kang, H.J.; Golos, T.G.; Slukvin, I.I. Genome Editing of CCR5 by CRISPR-Cas9 in Mauritian Cynomolgus Macaque Embryos. Sci. Rep. 2020, 10, 18457. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.S.; Kumar, A.; Weinfurter, J.; Park, M.A.; Maufort, J.; Tao, L.; Kang, H.; Dettle, S.T.; Golos, T.; Thomson, J.A.; et al. Generation of SIV-Resistant T Cells and Macrophages from Nonhuman Primate Induced Pluripotent Stem Cells with Edited CCR5 Locus. Stem Cell Rep. 2022, 17, 953–963. [Google Scholar] [CrossRef]
- Cradick, T.J.; Fine, E.J.; Antico, C.J.; Bao, G. CRISPR/Cas9 Systems Targeting β-Globin and CCR5 Genes Have Substantial off-Target Activity. Nucleic Acids Res. 2013, 41, 9584–9592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Holguin, L.; Burnett, J.C. CRISPR-Cas9-Mediated Gene Disruption of HIV-1 Co-Receptors Confers Broad Resistance to Infection in Human T Cells and Humanized Mice. Mol. Ther. Methods Clin. Dev. 2022, 24, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Mock, U.; MacHowicz, R.; Hauber, I.; Horn, S.; Abramowski, P.; Berdien, B.; Hauber, J.; Fehse, B. MRNA Transfection of a Novel TAL Effector Nuclease (TALEN) Facilitates Efficient Knockout of HIV Co-Receptor CCR5. Nucleic Acids Res. 2015, 43, 5560–5571. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene Editing of CCR5 in Autologous CD4 T Cells of Persons Infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Sorokina, A.; Artyuhov, A.; Goltsova, A.; Dashinimaev, E. Detection of CCR5Δ32 Mutant Alleles in Heterogeneous Cell Mixtures Using Droplet Digital PCR. Front. Mol. Biosci. 2022, 9, 805931. [Google Scholar] [CrossRef]
- Scheller, S.H.; Rashad, Y.; Saleh, F.M.; Willingham, K.A.; Reilich, A.; Lin, D.; Izadpanah, R.; Alt, E.U.; Braun, S.E. Biallelic, Selectable, Knock-in Targeting of CCR5 via CRISPR-Cas9 Mediated Homology Directed Repair Inhibits HIV-1 Replication. Front. Immunol. 2022, 13, 821190. [Google Scholar] [CrossRef]
- Lin, D.; Scheller, S.H.; Robinson, M.M.; Izadpanah, R.; Alt, E.U.; Braun, S.E. Increased Efficiency for Biallelic Mutations of the CCR5 Gene by CRISPR-Cas9 Using Multiple Guide RNAs As a Novel Therapeutic Option for Human Immunodeficiency Virus. Cris. J. 2021, 4, 92–103. [Google Scholar] [CrossRef]
- Freisinger, E.; Cramer, C.; Xia, X.; Murthy, S.N.; Slakey, D.P.; Chiu, E.; Newsome, E.R.; Alt, E.U.; Izadpanah, R. Characterization of Hematopoietic Potential of Mesenchymal Stem Cells. J. Cell. Physiol. 2010, 225, 888–897. [Google Scholar] [CrossRef]
- Ning, H.; Lei, H.E.; Xu, Y.D.; Guan, R.L.; Venstrom, J.M.; Lin, G.; Lue, T.F.; Xin, Z.; Lin, C.S. Conversion of Adipose-Derived Stem Cells into Natural Killer-like Cells with Anti-Tumor Activities in Nude Mice. PLoS ONE 2014, 9, e106246. [Google Scholar] [CrossRef] [Green Version]
- Mandal, P.K.; Ferreira, L.M.R.; Collins, R.; Meissner, T.B.; Boutwell, C.L.; Friesen, M.; Vrbanac, V.; Garrison, B.S.; Stortchevoi, A.; Bryder, D.; et al. Efficient Ablation of Genes in Human Hematopoietic Stem and Effector Cells Using CRISPR/Cas9. Cell Stem Cell 2014, 15, 643–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Wang, J.; Beyer, A.I.; Teque, F.; Cradick, T.J.; Qi, Z.; Chang, J.C.; Bao, G.; Muench, M.O.; Yu, J.; et al. Seamless Modification of Wild-Type Induced Pluripotent Stem Cells to the Natural CCR5Δ32 Mutation Confers Resistance to HIV Infection. Proc. Natl. Acad. Sci. USA 2014, 111, 9591–9596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.J.; Minder, P.; Park, M.A.; Mesquitta, W.T.; Torbett, B.E.; Slukvin, I.I. CCR5 Disruption in Induced Pluripotent Stem Cells Using CRISPR/Cas9 Provides Selective Resistance of Immune Cells to CCR5-Tropic HIV-1 Virus. Mol. Ther. Nucleic Acids 2015, 4, e268. [Google Scholar] [CrossRef] [PubMed]
- Teque, F.; Ye, L.; Xie, F.; Wang, J.; Morvan, M.G.; Kan, Y.W.; Levy, J.A. Genetically-Edited Induced Pluripotent Stem Cells Derived from HIV-1-Infected Patients on Therapy Can Give Rise to Immune Cells Resistant to HIV-1 Infection. Aids 2020, 34, 1141–1149. [Google Scholar] [CrossRef]
- Park, R.J.; Wang, T.; Koundakjian, D.; Hultquist, J.F.; Lamothe-Molina, P.; Monel, B.; Schumann, K.; Yu, H.; Krupzcak, K.M.; Garcia-Beltran, W.; et al. A Genome-Wide CRISPR Screen Identifies a Restricted Set of HIV Host Dependency Factors. Nat. Genet. 2017, 49, 193–203. [Google Scholar] [CrossRef]
- OhAinle, M.; Helms, L.; Vermeire, J.; Roesch, F.; Humes, D.; Basom, R.; Delrow, J.J.; Overbaugh, J.; Emerman, M. A Virus-Packageable CRISPR Screen Identifies Host Factors Mediating Interferon Inhibition of HIV. eLife 2018, 7, e39823. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Malik, N.; Rao, M.S. A Review of the Methods for Human IPSC Derivation. Methods Mol. Biol. 2013, 997, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.; Varga, E.; Aarts, C.; Wust, T.; Kuijpers, T.; von Lindern, M.; van den Akker, E. Efficient Production of Erythroid, Megakaryocytic and Myeloid Cells, Using Single Cell-Derived IPSC Colony Differentiation. Stem Cell Res. 2018, 29, 232–244. [Google Scholar] [CrossRef]
- Kim, J.Y.; Nam, Y.; Rim, Y.A.; Ju, J.H. Review of the Current Trends in Clinical Trials Involving Induced Pluripotent Stem Cells. Stem Cell Rev. Rep. 2022, 18, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Sugai, K.; Sumida, M.; Shofuda, T.; Yamaguchi, R.; Tamura, T.; Kohzuki, T.; Abe, T.; Shibata, R.; Kamata, Y.; Ito, S.; et al. First-in-Human Clinical Trial of Transplantation of IPSC-Derived NS/PCs in Subacute Complete Spinal Cord Injury: Study Protocol. Regen. Ther. 2021, 18, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Ellwanger, J.; Kulmann-Leal, B.; de Lima Kaminski, V.; Gonçalves Rodrigues, A.; Alves de Souza Bragatte, M.; Chies, J.A.B. Beyond HIV Infection: Neglected and Varied Impacts of CCR5 and CCR5Δ32 on Viral Diseases. Virus Res. 2020, 286, 198040. [Google Scholar] [CrossRef] [PubMed]
- Schumann, K.; Lin, S.; Boyer, E.; Simeonov, D.R.; Subramaniam, M.; Gate, R.E.; Haliburton, G.E.; Ye, C.J.; Bluestone, J.A.; Doudna, J.A.; et al. Generation of Knock-in Primary Human T Cells Using Cas9 Ribonucleoproteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10437–10442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.A.; Seki, A.; Rutz, S. Ribonucleoprotein Transfection for CRISPR/Cas9-Mediated Gene Knockout in Primary T Cells. Curr. Protoc. Immunol. 2019, 124, e69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpova, D.; Bonig, H. Concise Review: CXCR4/CXCL12 Signaling in Immature Hematopoiesis—Lessons from Pharmacological and Genetic Models. Stem Cells 2015, 33, 2391–2399. [Google Scholar] [CrossRef]
- Karpova, D.; Ritchey, J.K.; Holt, M.S.; Abou-Ezzi, G.; Monlish, D.; Batoon, L.; Millard, S.; Spohn, G.; Wiercinska, E.; Chendamarai, E.; et al. Continuous Blockade of CXCR4 Results in Dramatic Mobilization and Expansion of Hematopoietic Stem and Progenitor Cells. Blood 2017, 129, 2939–2949. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Yao, Y.; Xiao, H.; Li, J.; Liu, Q.; Yang, Y.; Adah, D.; Lu, J.; Zhao, S.; Qin, L.; et al. Simultaneous Knockout of CXCR4 and CCR5 Genes in CD4+ T Cells via CRISPR/Cas9 Confers Resistance to Both X4- and R5-Tropic Human Immunodeficiency Virus Type 1 Infection. Hum. Gene Ther. 2018, 29, 51–67. [Google Scholar] [CrossRef]
- Cohn, L.B.; Chomont, N.; Deeks, S.G. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe 2020, 27, 519–530. [Google Scholar] [CrossRef]
- Grau-Expósito, J.; Serra-Peinado, C.; Miguel, L.; Navarro, J.; Curran, A.; Burgos, J.; Ocaña, I.; Ribera, E.; Torrella, A.; Planas, B.; et al. A Novel Single-Cell FISH-Flow Assay Identifies Effector Memory CD4+ T Cells as a Major Niche for HIV-1 Transcription in HIV-Infected Patients. MBio 2017, 8, e00876-17. [Google Scholar] [CrossRef] [Green Version]
- Batra, H.; Zhu, J.; Jain, S.; Ananthaswamy, N.; Mahalingam, M.; Tao, P.; Lange, C.; Zhong, C.; Kearney, M.F.; Hu, H.; et al. Engineered Bacteriophage T4 Nanoparticle as a Potential Targeted Activator of HIV-1 Latency in CD4+ Human T-Cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Freen-van Heeren, J.J.; Nicolet, B.P.; Wolkers, M.C. Measuring T Cell Responses by Flow Cytometry–Based Fluorescence In Situ Hybridization. Crit. Rev. Immunol. 2018, 38, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Baxter, A.E.; Niessl, J.; Fromentin, R.; Richard, J.; Porichis, F.; Massanella, M.; Brassard, N.; Alsahafi, N.; Routy, J.-P.; Finzi, A.; et al. Multiparametric Characterization of Rare HIV-Infected Cells Using an RNA-Flow FISH Technique. Nat. Protoc. 2017, 12, 2029–2049. [Google Scholar] [CrossRef] [PubMed]
- Baxter, A.E.; Niessl, J.; Fromentin, R.; Richard, J.; Porichis, F.; Charlebois, R.; Massanella, M.; Brassard, N.; Alsahafi, N.; Delgado, G.G.; et al. Single-Cell Characterization of Viral Translation-Competent Reservoirs in HIV-Infected Individuals. Cell Host Microbe 2016, 20, 368–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freen-van Heeren, J.J. Flow-FISH as a Tool for Studying Bacteria, Fungi and Viruses. BioTech 2021, 10, 21. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Method | Advantage(s) | Disadvantage(s) | Used in HIV Research? | Reference(s) |
|---|---|---|---|---|
| Cell-penetrating peptides | Little cell manipulation required In-house production | Batch-to-batch differences | Yes | [56] |
| Chemical transfection | Little cell manipulation required | Not every method is suitable for every cell type | Yes | [55] |
| Electroporation | Extensive protocols available Adaptable to cell type of interest | Can be cytotoxic [63] Costly | Yes | [27,55,57,58] |
| Lenti/retroviral vectors | Inclusion of (fluorescent) selection marker | Low knock-out efficacy Genomic integration | Yes | [51,52,53,54] |
| (Lipid) nanoparticles | Highly adaptable to specific (researcher) needs | Complex to engineer | Yes | [55] |
| Ligand fusion tags | Cell-type specific | Cells need to express receptor | No | [67,68] |
| Virus-like particles | Targetable to cell type of interest Potential for in vivo use | Dependent on viral mechanisms for specific cellular targeting | Yes | [55,69] |
| Target | Remarks | Reference(s) |
|---|---|---|
| ASCs | CRISPR-mediated KO feasible | [78] |
| Enhanced CCR5 KO when employing two crRNAs | [79] | |
| HSCs | Knockout confers in vitro resistance to HIV infection in differentiated macrophages | [74] |
| Multi-lineage differentiation in vitro | [53] | |
| Minimal off-target modifications detected Multi-lineage engraftment potential in animal model | [82] | |
| Multi-lineage engraftment potential in animal model In vivo resistance to HIV infection | [57] | |
| Multi-lineage engraftment potential after autologous HSC transplantation Persistence of low frequencies of CCR5 knockout CD4+ T cells | [66] | |
| iPSCs | No off-target modifications detected iPSC-derived monocytes/macrophages resistant to HIV infection | [83,84,85] |
| Primary CD4+ T cells | Low transduction efficiency with lentiviral vectors | [52] |
| Knockout confers in vitro resistance to HIV infection | [24,52,53,74] | |
| Introduction of Δ32/Δ32 mutation No off-target modifications detected | [51] | |
| Macrophage or T cell cell-lines | CRISPR-mediated KO feasible | [78,86,87] |
| Introduction of Δ32/Δ32 mutation High fidelity screening method | [77] |
| Target | Remarks | Reference(s) |
|---|---|---|
| Primary CD4+ T cells | Knockout is feasible | [27,58,94,95] |
| Knockout confers in vitro resistance to HIV infection | [25,40,74] | |
| Macrophage or T cell cell-lines | Knockout is feasible | [86,87] |
| Knockout confers minimal in vitro resistance to HIV infection | [74] |
| Target | Remarks | Reference(s) |
|---|---|---|
| Primary CD4+ T cells | Dual CXCR4 and CCR5 knockout feasible | [54,98] |
| No impact on survival and proliferation upon double knockout | ||
| Knockout confers in vitro resistance to HIV infection | [27,54,98] | |
| T cells retain in vitro cytokine production potential | [27] | |
| Knockout confers in vivo resistance to HIV infection in murine model Poor engraftment in murine model | [74] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freen-van Heeren, J.J. Closing the Door with CRISPR: Genome Editing of CCR5 and CXCR4 as a Potential Curative Solution for HIV. BioTech 2022, 11, 25. https://doi.org/10.3390/biotech11030025
Freen-van Heeren JJ. Closing the Door with CRISPR: Genome Editing of CCR5 and CXCR4 as a Potential Curative Solution for HIV. BioTech. 2022; 11(3):25. https://doi.org/10.3390/biotech11030025
Chicago/Turabian StyleFreen-van Heeren, Julian J. 2022. "Closing the Door with CRISPR: Genome Editing of CCR5 and CXCR4 as a Potential Curative Solution for HIV" BioTech 11, no. 3: 25. https://doi.org/10.3390/biotech11030025
APA StyleFreen-van Heeren, J. J. (2022). Closing the Door with CRISPR: Genome Editing of CCR5 and CXCR4 as a Potential Curative Solution for HIV. BioTech, 11(3), 25. https://doi.org/10.3390/biotech11030025