
Transcriptional and Metabolic Response of a Strain of Escherichia coli PTS− to a Perturbation of the Energetic Level by Modification of [ATP]/[ADP] Ratio
 and
and
Abstract
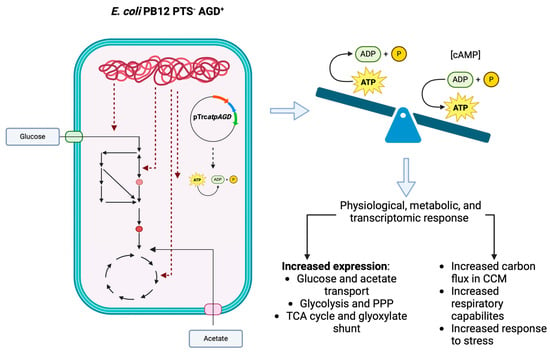
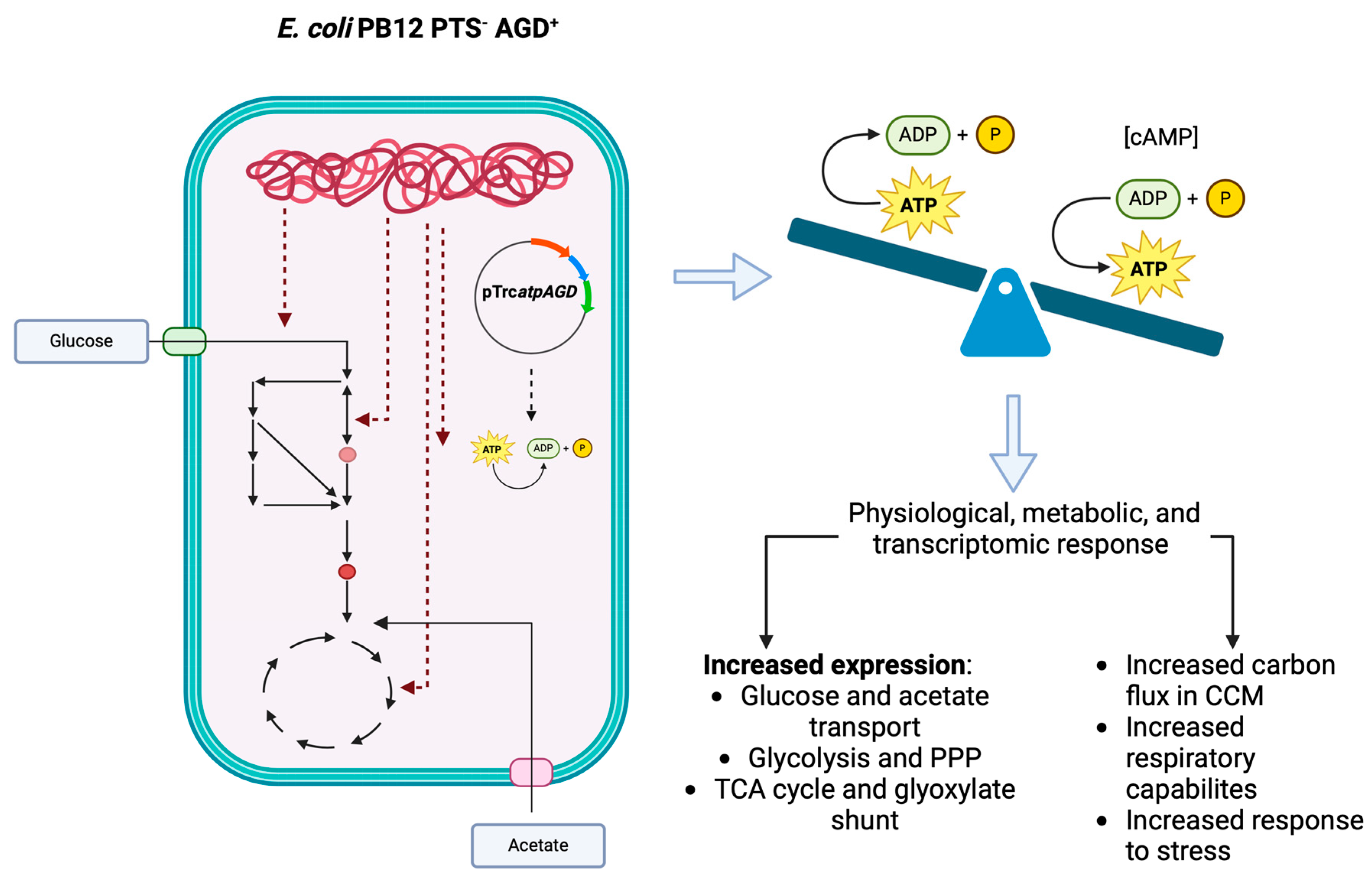
:
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plasmids, Culture Media, and Growing Conditions
2.2. Determination of Uncoupled ATPase Activity and Determination of [ATP]/[ADP] Ratio and cAMP Concentration
2.3. Measurement of Glucose and Acetate from Fermentation Supernatants
2.4. Total RNA Extraction and cDNA Synthesis
2.5. Transcriptomic Analysis Using RT–qPCR
3. Results
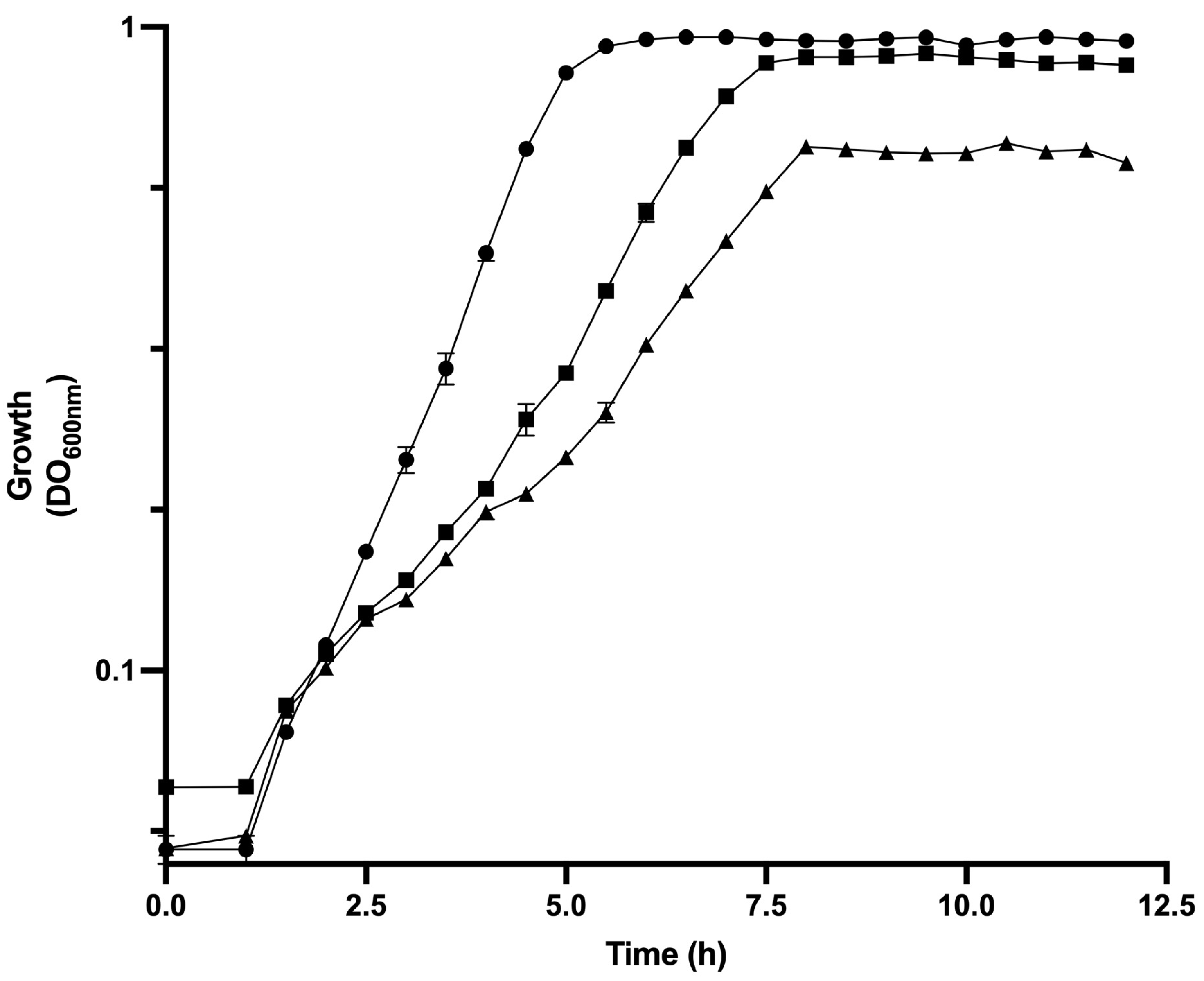
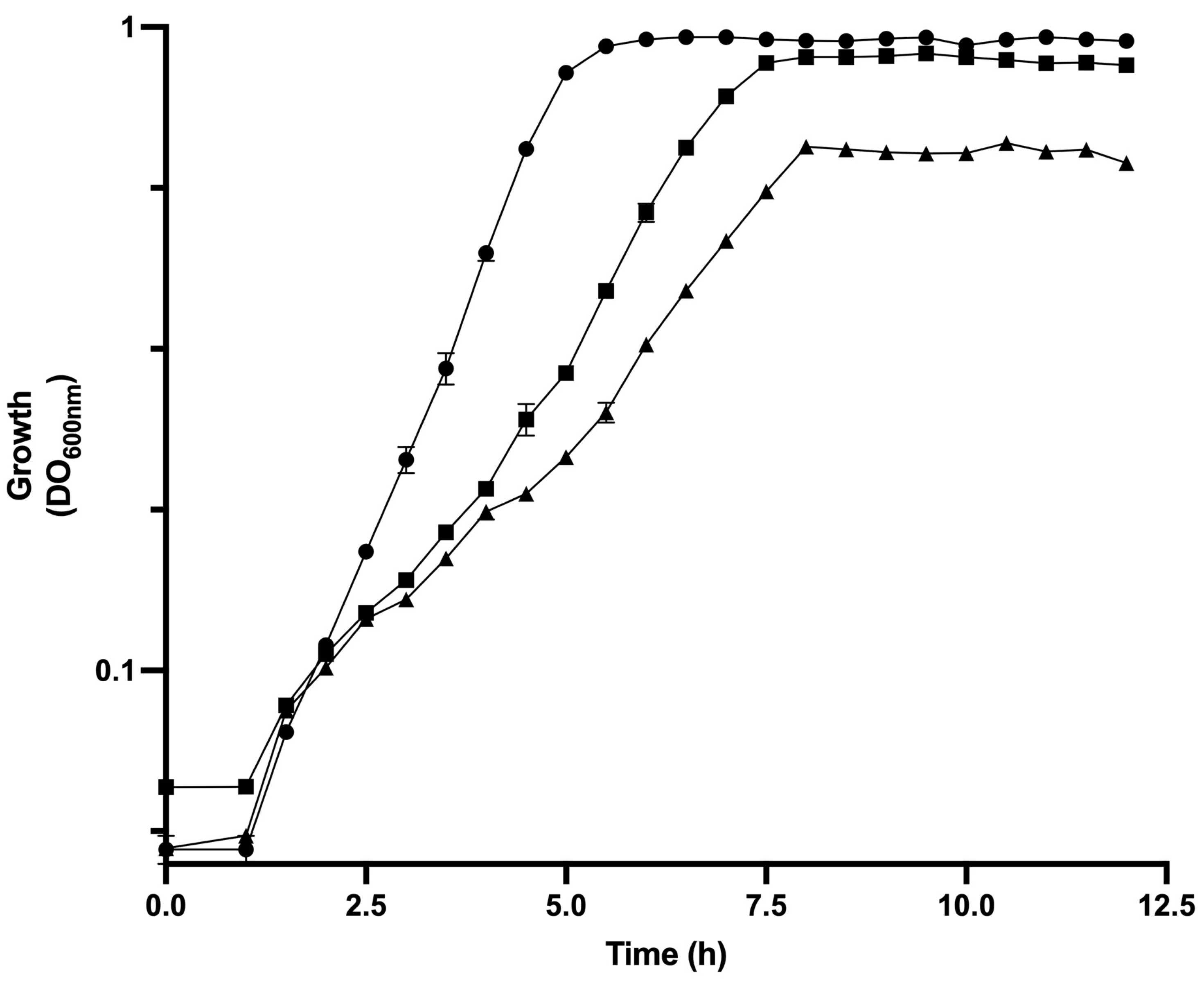
3.1. Physiological Parameters of Analyzed Strains of E. coli in Bioreactor Cultures
3.2. [ATP]/[ADP] Ratio, ATPase Activity, and cAMP Concentration Determinations
3.3. Differentially Expressed Genes in Response to the Disruption of the [ATP]/[ADP] Ratio in the Derivative Strain PB12AGD+
4. Discussion
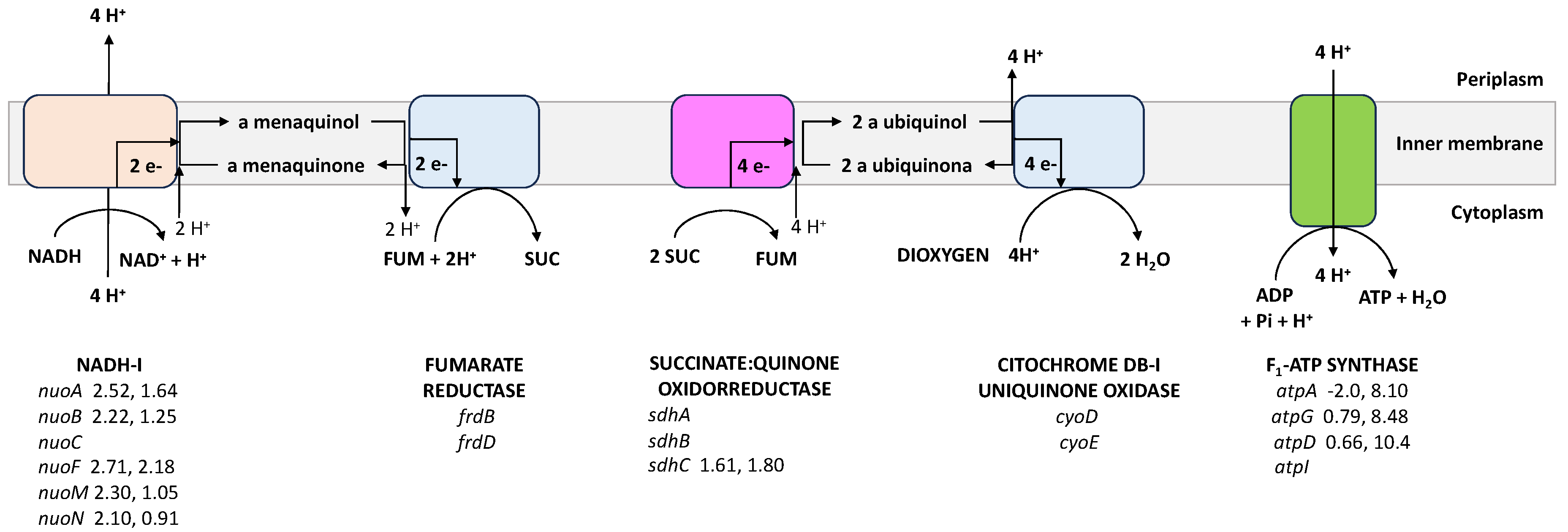
4.1. Expression of the atpAGD Operon, ATPase Activities, [ATP]/[ADP] Ratio, and Cyclic AMP (cAMP) Concentrations
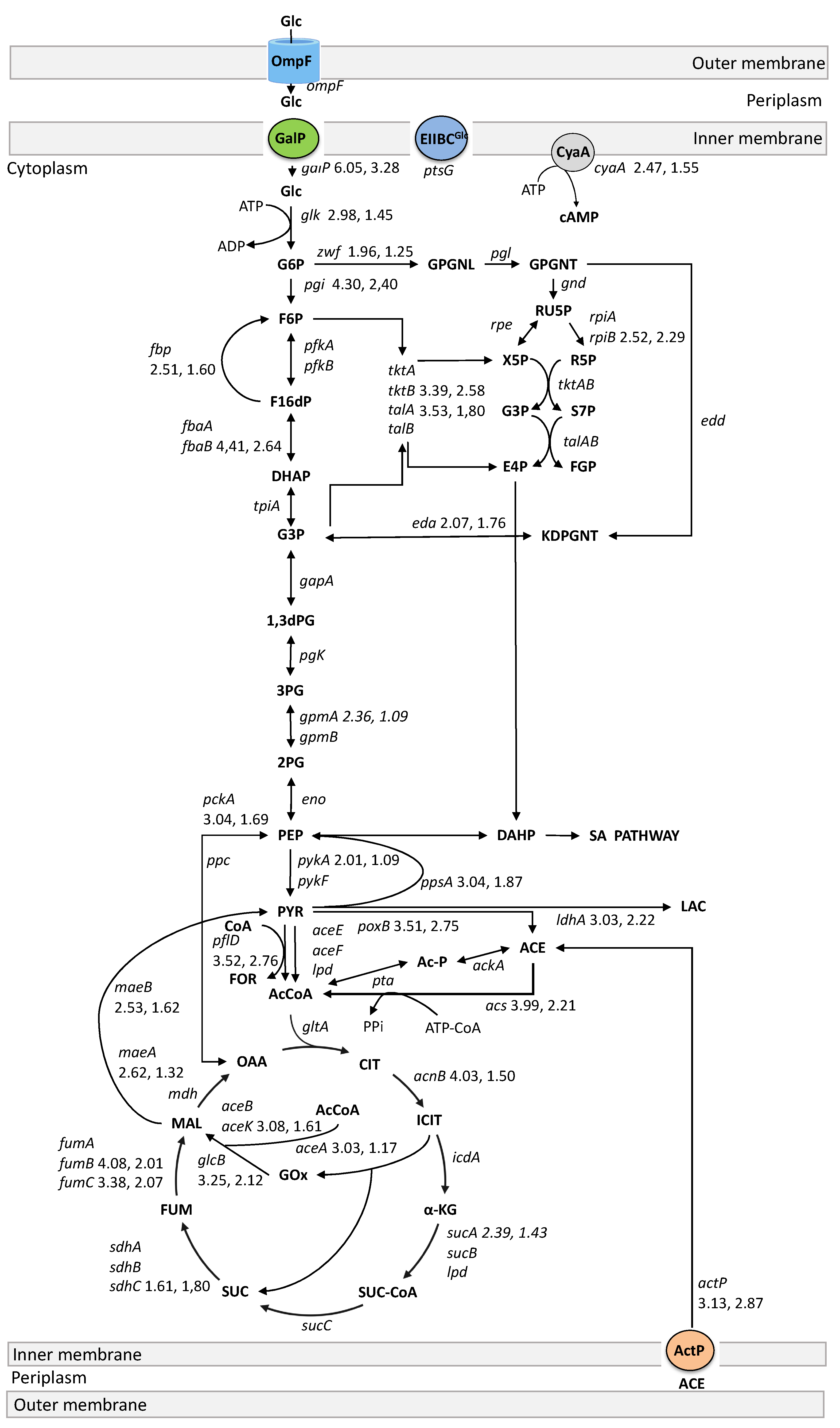
4.2. Glucose Transport and Phosphorylation
4.3. Glycolysis, Gluconeogenic, Pentose Phosphate, and Entner-Doudoroff Pathways
4.4. The TCA Cycle and the Glyoxylate Shunt
4.5. Fermentation, Acetate Production, and Utilization Genes
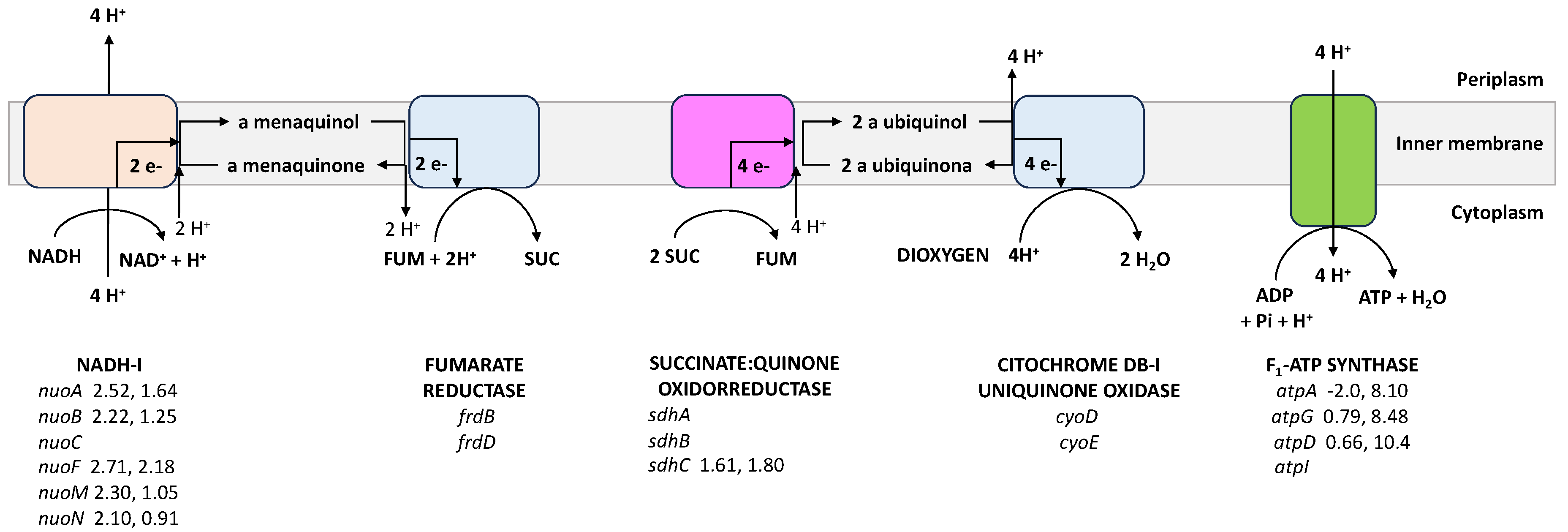
4.6. The Respiratory Chain
4.7. Regulatory Proteins and Sigma Factors
4.8. Expression of Stress Response Genes
4.9. Physiological Significance of [ATP]/[ADP] Ratio Disruption in E. coli PTS−
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Senior, A.E. ATP Synthesis by Oxidative Phosphorylation. Physiol. Rev. 1988, 68, 177–231. [Google Scholar] [CrossRef] [PubMed]
- Boogerd, F.C.; Boe, L.; Michelsen, O.; Jensen, P.R. Atp Mutants of Escherichia coli Fail To Grow on Succinate Due to a Transport Deficiency. J. Bacteriol. 1998, 180, 5855–5859. [Google Scholar] [CrossRef] [PubMed]
- Arechaga, I.; Miroux, B.; Runswick, M.J.; Walker, J.E. Over-expression of Escherichia coli F1 Fo–ATPase Subunit a Is Inhibited by Instability of the uncB Gene Transcript. FEBS Lett. 2003, 547, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Keseler, I.M.; Gama-Castro, S.; Mackie, A.; Billington, R.; Bonavides-Martínez, C.; Caspi, R.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Muñiz-Rascado, L.; et al. The EcoCyc Database in 2021. Front. Microbiol. 2021, 12, 711077. [Google Scholar] [CrossRef] [PubMed]
- Pérez, I.; Heitkamp, T.; Börsch, M. Mechanism of ADP-Inhibited ATP Hydrolysis in Single Proton-Pumping FoF1-ATP Synthase Trapped in Solution. Int. J. Mol. Sci. 2023, 24, 8442. [Google Scholar] [CrossRef] [PubMed]
- Soria, S.; De Anda, R.; Flores, N.; Romero-Garcia, S.; Gosset, G.; Bolívar, F.; Báez-Viveros, J.L. New Insights on Transcriptional Responses of Genes Involved in Carbon Central Metabolism, Respiration and Fermentation to Low ATP Levels in Escherichia coli. J. Basic Microbiol. 2013, 53, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Trchounian, A. Escherichia coli Proton-Translocating F0F1-ATP Synthase and Its Association with Solute Secondary Transporters and/or Enzymes of Anaerobic Oxidation–Reduction under Fermentation. Biochem. Biophys. Res. Commun. 2004, 315, 1051–1057. [Google Scholar] [CrossRef]
- Boecker, S.; Zahoor, A.; Schramm, T.; Link, H.; Klamt, S. Broadening the Scope of Enforced ATP Wasting as a Tool for Metabolic Engineering in Escherichia coli. Biotechnol. J. 2019, 14, 1800438. [Google Scholar] [CrossRef]
- Koebmann, B.J.; Westerhoff, H.V.; Snoep, J.L.; Nilsson, D.; Jensen, P.R. The Glycolytic Flux in Escherichia coli Is Controlled by the Demand for ATP. J. Bacteriol. 2002, 184, 3909–3916. [Google Scholar] [CrossRef]
- Aguilar, C.; Escalante, A.; Flores, N.; de Anda, R.; Riveros-McKay, F.; Gosset, G.; Morett, E.; Bolívar, F. Genetic Changes during a Laboratory Adaptive Evolution Process That Allowed Fast Growth in Glucose to an Escherichia coli Strain Lacking the Major Glucose Transport System. BMC Genom. 2012, 13, 385. [Google Scholar] [CrossRef]
- Flores, N.; Flores, S.; Escalante, A.; de Anda, R.; Leal, L.; Malpica, R.; Georgellis, D.; Gosset, G.; Bolívar, F. Adaptation for Fast Growth on Glucose by Differential Expression of Central Carbon Metabolism and Gal Regulon Genes in an Escherichia coli Strain Lacking the Phosphoenolpyruvate:Carbohydrate Phosphotransferase System. Metab. Eng. 2005, 7, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Amann, E.; Ochs, B.; Abel, K.-J. Tightly Regulated Tac Promoter Vectors Useful for the Expression of Unfused and Fused Proteins in Escherichia Coli. Gene 1988, 69, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Ausubel, F.R. Short Protocols in Molecular Biology, 4th ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1999; ISBN 0-471-32938-X. [Google Scholar]
- Messing, J. A Multipurpose Cloning System Based on the Single-Stranded DNA Bacteriophage M13. Recomb. DNA Tech. Bull. 1979, 2, 43–48. [Google Scholar]
- Escalante, A.; Astudillo, F.; Larios, V.; Flores, N.; Gosset, G.; Bolívar, F. The Complete Genome of Escherichia coli JM101 Assembled with a Combination of Nanopore and Illumina Platforms. Microbiol. Resour. Announc. 2024, 13, e00973-23. [Google Scholar] [CrossRef]
- Rodriguez, A.; Martínez, J.A.; Báez-Viveros, J.L.; Flores, N.; Hernández-Chávez, G.; Ramírez, O.T.; Gosset, G.; Bolivar, F. Constitutive Expression of Selected Genes from the Pentose Phosphate and Aromatic Pathways Increases the Shikimic Acid Yield in High-Glucose Batch Cultures of an Escherichia coli Strain Lacking PTS and pykF. Microb. Cell Fact 2013, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Carreón-Rodríguez, O.E.; Gosset, G.; Escalante, A.; Bolívar, F. Glucose Transport in Escherichia coli: From Basics to Transport Engineering. Microorganisms 2023, 11, 1588. [Google Scholar] [CrossRef]
- Martínez, K.; De Anda, R.; Hernández, G.; Escalante, A.; Gosset, G.; Ramírez, O.T.; Bolívar, F.G. Coutilization of Glucose and Glycerol Enhances the Production of Aromatic Compounds in an Escherichia coli Strain Lacking the Phosphoenolpyruvate: Carbohydrate Phosphotransferase System. Microb. Cell Fact 2008, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Akhova, A.V.; Tkachenko, A.G. ATP/ADP Alteration as a Sign of the Oxidative Stress Development in Escherichia coli Cells under Antibiotic Treatment. FEMS Microbiol. Lett. 2014, 353, 69–76. [Google Scholar] [CrossRef]
- Meyrat, A.; von Ballmoos, C. ATP Synthesis at Physiological Nucleotide Concentrations. Sci. Rep. 2019, 9, 3070. [Google Scholar] [CrossRef]
- Usui, Y.; Hirasawa, T.; Furusawa, C.; Shirai, T.; Yamamoto, N.; Mori, H.; Shimizu, H. Investigating the Effects of Perturbations to pgi and eno Gene Expression on Central Carbon Metabolism in Escherichia coli Using 13C Metabolic Flux Analysis. Microb. Cell Fact 2012, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Yamaki, M.; Sarada, M.; Nakayama, S.; Vibat, C.R.T.; Gennis, R.B.; Nakayashiki, T.; Inokuchi, H.; Kojima, S.; Kita, K. Two Hydrophobic Subunits Are Essential for the Heme b Ligation and Functional Assembly of Complex II (Succinate-Ubiquinone Oxidoreductase) from Escherichia coli(∗). J. Biol. Chem. 1996, 271, 521–527. [Google Scholar] [CrossRef]
- Tran, Q.M.; Rothery, R.A.; Maklashina, E.; Cecchini, G.; Weiner, J.H. The Quinone Binding Site in Escherichia coli Succinate Dehydrogenase Is Required for Electron Transfer to the Heme B*. J. Biol. Chem. 2006, 281, 32310–32317. [Google Scholar] [CrossRef]
- Simon, J.; van Spanning, R.J.M.; Richardson, D.J. The Organisation of Proton Motive and Non-Proton Motive Redox Loops in Prokaryotic Respiratory Systems. Biochim. Biophys. Acta 2008, 1777, 1480–1490. [Google Scholar] [CrossRef]
- Park, S.J.; Gunsalus, R.P. Oxygen, Iron, Carbon, and Superoxide Control of the Fumarase fumA and fumC Genes of Escherichia coli: Role of the arcA, fnr, and soxR Gene Products. J. Bacteriol. 1995, 177, 6255–6262. [Google Scholar] [CrossRef]
- Abdel-Hamid, A.M.; Attwood, M.M.; Guest, J.R. Pyruvate Oxidase Contributes to the Aerobic Growth Efficiency of Escherichia coli. Microbiology 2001, 147, 1483–1498. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, C.R.; Bennett, G.N.; San, K.-Y. Characterization of the Acetate-Producing Pathways in Escherichia coli. Biotechnol. Prog. 2005, 21, 1062–1067. [Google Scholar] [CrossRef]
- Hengge-Aronis, R.; Klein, W.; Lange, R.; Rimmele, M.; Boos, W. Trehalose Synthesis Genes Are Controlled by the Putative Sigma Factor Encoded by rpoS and Are Involved in Stationary-Phase Thermotolerance in Escherichia coli. J. Bacteriol. 1991, 173, 7918–7924. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.D.; Jones-Mortimer, M.C.; Kornberg, H.L. The Enzymic Interconversion of Acetate and Acetyl-Coenzyme A in Escherichia coli. J. Gen. Microbiol. 1977, 102, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Tishel, R.; Eisenbach, M.; Wolfe, A.J. Cloning, Characterization, and Functional Expression of acs, the Gene Which Encodes Acetyl Coenzyme A Synthetase in Escherichia coli. J. Bacteriol. 1995, 177, 2878–2886. [Google Scholar] [CrossRef]
- Valgepea, K.; Adamberg, K.; Nahku, R.; Lahtvee, P.-J.; Arike, L.; Vilu, R. Systems Biology Approach Reveals That Overflow Metabolism of Acetate in Escherichia coli Is Triggered by Carbon Catabolite Repression of Acetyl-CoA Synthetase. BMC Syst. Biol. 2010, 4, 166. [Google Scholar] [CrossRef] [PubMed]
- Unden, G.; Dünnwald, P. The Aerobic and Anaerobic Respiratory Chain of Escherichia coli and Salmonella Enterica: Enzymes and Energetics. EcoSal Plus 2008, 3, 1–37. [Google Scholar] [CrossRef]
- Friedrich, T.; Dekovic, D.K.; Burschel, S. Assembly of the Escherichia coli NADH:Ubiquinone Oxidoreductase (Respiratory Complex I). Biochim. Biophys. Acta 2016, 1857, 214–223. [Google Scholar] [CrossRef]
- Lee, P.T.; Hsu, A.Y.; Ha, H.T.; Clarke, C.F. A C-Methyltransferase Involved in Both Ubiquinone and Menaquinone Biosynthesis: Isolation and Identification of the Escherichia coli ubiE Gene. J. Bacteriol. 1997, 179, 1748–1754. [Google Scholar] [CrossRef]
- Potter, L.C.; Millington, P.; Griffiths, L.; Thomas, G.H.; Cole, J.A. Competition between Escherichia coli Strains Expressing Either a Periplasmic or a Membrane-Bound Nitrate Reductase: Does Nap Confer a Selective Advantage during Nitrate-Limited Growth? Biochem. J. 1999, 344, 77–84. [Google Scholar] [CrossRef] [PubMed]
- McNicholas, P.M.; Gunsalus, R.P. The Molybdate-Responsive Escherichia coli ModE Transcriptional Regulator Coordinates Periplasmic Nitrate Reductase (napFDAGHBC) Operon Expression with Nitrate and Molybdate Availability. J. Bacteriol. 2002, 184, 3253–3259. [Google Scholar] [CrossRef]
- Wang, H.; Tseng, C.-P.; Gunsalus, R.P. The napF and narG Nitrate Reductase Operons in Escherichia coli Are Differentially Expressed in Response to Submicromolar Concentrations of Nitrate but Not Nitrite. J. Bacteriol. 1999, 181, 5303–5308. [Google Scholar] [CrossRef]
- Pellicer, M.T.; Badía, J.; Aguilar, J.; Baldomà, L. clc Locus of Escherichia coli: Characterization of Genes Encoding the Subunits of Glycolate Oxidase and the Glc Regulator Protein. J. Bacteriol. 1996, 178, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Zhang, C. Integration Host Factor Is Required for the Induction of Acid Resistance in Escherichia coli. Curr. Microbiol. 2014, 69, 218–224. [Google Scholar] [CrossRef]
- Molina-Henares, A.J.; Krell, T.; Eugenia Guazzaroni, M.; Segura, A.; Ramos, J.L. Members of the IclR Family of Bacterial Transcriptional Regulators Function as Activators and/or Repressors. FEMS Microbiol. Rev. 2006, 30, 157–186. [Google Scholar] [CrossRef]
- Cronan, J.E. The Escherichia Coli FadR Transcription Factor: Too Much of a Good Thing? Mol. Microbiol. 2021, 115, 1080–1085. [Google Scholar] [CrossRef]
- Maloy, S.R.; Nunn, W.D. Role of Gene fadR in Escherichia coli Acetate Metabolism. J. Bacteriol. 1981, 148, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Gui, L.; Sunnarborg, A.; LaPorte, D.C. Regulated Expression of a Repressor Protein: FadR Activates iclR. J. Bacteriol. 1996, 178, 4704–4709. [Google Scholar] [CrossRef] [PubMed]
- Ireland, W.T.; Beeler, S.M.; Flores-Bautista, E.; McCarty, N.S.; Röschinger, T.; Belliveau, N.M.; Sweredoski, M.J.; Moradian, A.; Kinney, J.B.; Phillips, R. Deciphering the Regulatory Genome of Escherichia coli, One Hundred Promoters at a Time. eLife 2020, 9, e55308. [Google Scholar] [CrossRef] [PubMed]
- Sutton, V.R.; Mettert, E.L.; Beinert, H.; Kiley, P.J. Kinetic Analysis of the Oxidative Conversion of the [4Fe-4S]2+ Cluster of FNR to a [2Fe-2S]2+ Cluster. J. Bacteriol. 2004, 186, 8018–8025. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, T. Hungry Bacteria—Definition and Properties of a Nutritional State. Environ. Microbiol. 2001, 3, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Franchini, A.G.; Ihssen, J.; Egli, T. Effect of Global Regulators RpoS and Cyclic-AMP/CRP on the Catabolome and Transcriptome of Escherichia coli K12 during Carbon- and Energy-Limited Growth. PLoS ONE 2015, 10, e0133793. [Google Scholar] [CrossRef] [PubMed]
- Tierrafría, V.H.; Rioualen, C.; Salgado, H.; Lara, P.; Gama-Castro, S.; Lally, P.; Gómez-Romero, L.; Peña-Loredo, P.; López-Almazo, A.G.; Alarcón-Carranza, G.; et al. RegulonDB 11.0: Comprehensive High-Throughput Datasets on Transcriptional Regulation in Escherichia Coli K-12. Microb. Genom. 2022, 8, mgen000833. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Krishna, S.; Seshasayee, A.S.N. Regulation of Global Transcription in Escherichia coli by Rsd and 6S RNA. G3 2018, 8, 2079–2089. [Google Scholar] [CrossRef]
- Park, Y.-H.; Lee, C.-R.; Choe, M.; Seok, Y.-J. HPr Antagonizes the Anti-σ70 Activity of Rsd in Escherichia coli. Proc. Natl. Acad. Sci. USA 2013, 110, 21142–21147. [Google Scholar] [CrossRef]
- Noda, S.; Takezawa, Y.; Mizutani, T.; Asakura, T.; Nishiumi, E.; Onoe, K.; Wada, M.; Tomita, F.; Matsushita, K.; Yokota, A. Alterations of Cellular Physiology in Escherichia coli in Response to Oxidative Phosphorylation Impaired by Defective F1-ATPase. J. Bacteriol. 2006, 188, 6869–6876. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Weiss, B. Two-Stage Induction of the soxRS (Superoxide Response) Regulon of Escherichia coli. J. Bacteriol. 1992, 174, 3915–3920. [Google Scholar] [CrossRef] [PubMed]
- Touati, D. Sensing and Protecting against Superoxide Stress in Escherichia coli—How Many Ways Are There to Trigger soxRS Response? Redox Rep. 2000, 5, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of Reactive Oxygen Species Generation in Cell Signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Hansel, C.M.; Diaz, J.M. Production of Extracellular Reactive Oxygen Species by Marine Biota. Annu. Rev. Mar. Sci. 2021, 13, 177–200. [Google Scholar] [CrossRef] [PubMed]
- Patacq, C.; Chaudet, N.; Létisse, F. Crucial Role of ppGpp in the Resilience of Escherichia coli to Growth Disruption. mSphere 2020, 5, e01132-20. [Google Scholar] [CrossRef] [PubMed]
- Ono, S.; Suzuki, S.; Ito, D.; Tagawa, S.; Shiina, T.; Masuda, S. Plastidial (p)ppGpp Synthesis by the Ca2+-Dependent RelA–SpoT Homolog Regulates the Adaptation of Chloroplast Gene Expression to Darkness in Arabidopsis. Plant Cell Physiol. 2020, 61, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Spira, B.; Ospino, K. Diversity in E. coli (p)ppGpp Levels and Its Consequences. Front. Microbiol. 2020, 11, 1759. [Google Scholar] [CrossRef]
- Boecker, S.; Slaviero, G.; Schramm, T.; Szymanski, W.; Steuer, R.; Link, H.; Klamt, S. Deciphering the Physiological Response of Escherichia coli under High ATP Demand. Mol. Syst. Biol. 2021, 17, e10504. [Google Scholar] [CrossRef]
- Boecker, S.; Harder, B.-J.; Kutscha, R.; Pflügl, S.; Klamt, S. Increasing ATP Turnover Boosts Productivity of 2,3-Butanediol Synthesis in Escherichia coli. Microb. Cell Fact 2021, 20, 63. [Google Scholar] [CrossRef]
- Yatabe, F.; Seike, T.; Okahashi, N.; Ishii, J.; Matsuda, F. Improvement of Ethanol and 2,3-Butanediol Production in Saccharomyces cerevisiae by ATP Wasting. Microb. Cell Fact 2023, 22, 204. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.A.; Bolívar, F.; Escalante, A. Shikimic Acid Production in Escherichia coli: From Classical Metabolic Engineering Strategies to Omics Applied to Improve Its Production. Front. Biotechnol. Bioeng. 2015, 3, 145. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmids | Relevant Features | References |
|---|---|---|
| pTrc99A | Cloning vector under the trc promoter. Carries bla and lacIq genes. Replication origin from pBR322. | [12] |
| pTrcatpAGD | atpAGD operon cloned under the control of trc promoter into the pTrc99A vector. | [6] |
| Bacterial strains | ||
| JM101 | F′ traD36 proA+ proB+ lacIq lacZΔ15/supE thi Δ(lac-proAB). | [14,15] |
| PB12 | JM101 ΔptsHptsIcrr::Km, PTS− Glc+. | [10,11] |
| PB12AGD+ | PB12 transformed with pTrcatpAGD. | This study |
| PB12pTrc99A | PB12 transformed with pTrc99A. | This study |
| Strain | µ (h−1) | qGlc (gGlc/gDCW h) | YX/Glc (g/g) | qAce (g/gDCW h) | qBase (mmol/gdcw h) | qO2 (mmol/gDcw h) | [ATP]/[ADP] Ratio | Total ATPase Activity (U/mgprotein) | Intracellular cAMP (pmol/mg Protein) |
|---|---|---|---|---|---|---|---|---|---|
| JM101 | 0.71 ± 0.04 | 1.74 ± 0.04 | 0.41 | 0.10 ± 0.01 | 8.41 ± 0.36 | 1.58 ± 0.06 | 7.32 ± 0.14 | 1.46 ± 0.05 | 24.94 ± 1.25 |
| PB12 | 0.43 ± 0.02 | 0.81 ± 0.08 | 0.53 | 0.02 ± 0.00 | 5.60 ± 0.02 | 0.5 ± 0.01 | 2.53 ± 0.03 | 1.47 ± 0.01 | 29.92 ± 1.98 |
| PB12AGD+ | 0.24 ± 0.04 | 1.42 ± 0.07 | 0.17 | 0.06 ± 0.00 | 7.34 ± 0.07 | 1.19 ± 0.03 | 1.20 ± 0.01 | 1.97 ± 0.01 | 21.44 ± 2.02 |
|
Pathway, Group of Genes
or Cellular Process | Encoded Protein 1 |
Relative Transcription
Levels as 2−ΔΔCt | |
|---|---|---|---|
| PB12 | PB12AGD+ | ||
| ATP synthase | |||
| atpI | Subunit I | 1.61 ± 0.18 | 0.69 ± 0.05 |
| atpA | Subunit α | −2.0 ± 0.03 | 8.10 ± 2.82 |
| atpG | Subunit γ | 0.79 ± 0.17 | 8.48 ± 1.60 |
| atpD | Subunit β | 0.66 ± 0.11 | 10.4 ± 1.89 |
| Glucose transport | |||
| galP * | Galactose:H+ symporter | 6.05 ± 0.63 | 3.28 ± 0.33 |
| ompF * | Outer membrane porin F | 0.54 ± 0.08 | 0.44 ± 0.09 |
| ptsH * | Phosphocarrier protein Hpr | 0 | 0 |
| ptsG * | Glucose-specific PTS enzyme IIBC component | 1.53 ± 0.22 | 0.89 ± 0.23 |
| Acetate transport | |||
| actP (yjcG) * | Acetate/glycolate:cation symporter | 3.13 ± 0.07 | 2.87 ± 0.35 |
| Glycolysis and gluconeogenesis | |||
| Eno | Enolase | 0.66 ± 0.07 | 0.33 ± 0.03 |
| fbaA * | Fructose-bisphosphate aldolase class II | 0.64 ± 0.13 | 0.21 ± 0.09 |
| fbaB | Fructose-bisphosphate aldolase class I | 4.41 ± 0.94 | 2.64 ± 0.59 |
| fbp | Fructose-1,6-bisphosphatase 1 | 2.51 ± 0.00 | 1.60 ± 0.02 |
| gapA * | Glyceraldehyde-3-phosphate dehydrogenase | 1.04 ± 0.16 | 0.47 ± 0.04 |
| glk | Glucokinase | 2.98 ± 0.31 | 1.45 ± 0.30 |
| gpmA | 2,3-bisphosphoglycerate-dependent phosphoglycerate mutase | 2.36 ± 0.67 | 1.09 ± 0.06 |
| gpmB | Putative phosphatase | 1.59 ± 0.05 | 1.42 ± 0.05 |
| pfkA | 6-phosphofructokinase 1 | 0.96 ± 0.03 | 0.48 ± 0.04 |
| pgi | Glucose-6-phosphate isomerase | 4.30 ± 0.35 | 2.40 ± 0.25 |
| pgk * | Phosphoglycerate kinase | 0.77 ± 0.04 | 0.55 ± 0.02 |
| ppc | Phosphoenolpyruvate carboxylase | 1.53 ± 0.02 | 1.63 ± 0.11 |
| pykA | Pyruvate kinase 2 | 2.01 ± 0.24 | 1.09 ± 0.06 |
| tpiA | Triose-phosphate isomerase | 1.45 ± 0.01 | 0.68 ± 0.02 |
| Pentose phosphate pathway | |||
| eda | KHG/KDPG aldolase | 2.07 ± 0.18 | 1.76 ± 0.09 |
| gnd | 6-Phosphogluconate dehydrogenase, decarboxylating | 1.29 ± 0.49 | 0.63 ± 0.10 |
| rpiA | Ribose-5-phosphate isomerase A | 1.63 ± 0.40 | 1.39 ± 0.06 |
| rpiB | Ribose-5-phosphate isomerase B | 2.52 ± 0.49 | 2.29 ± 0.55 |
| talA | Transaldolase A | 3.53 ± 0.37 | 1.80 ± 0.32 |
| talB | Transaldolase B | 1.49 ± 0.05 | 0.76 ± 0.25 |
| tktB | Transketolase 2 | 3.39 ± 0.06 | 2.58 ± 0.57 |
| zwf | NADP+-dependent glucose-6-phosphate dehydrogenase | 1.96 ± 0.24 | 1.25 ± 0.08 |
| TCA cycle and the glyoxylate shunt | |||
| aceB * | Malate synthase A | 1.56 ± 0.00 | 0.78 ± 0.11 |
| aceA * | Isocitrate lyase | 3.03 ± 0.32 | 1.17 ± 0.03 |
| aceK * | Isocitrate dehydrogenase kinase | 3.08 ± 0.20 | 1.61 ± 0.04 |
| aceE * | Pyruvate dehydrogenase | 1.36 ± 0.25 | 0.78 ± 0.11 |
| aceF * | Pyruvate dehydrogenase E2 subunit | 1.32 ± 0.25 | 0.93 ± 0.15 |
| acnB * | Aconitate hydratase B | 4.03 ± 0.59 | 1.50 ± 0.12 |
| fumA * | Fumarase A | 0.77 ± 0.03 | 1.44 ± 0.03 |
| fumC * | Fumarase C | 3.38 ± 1.01 | 2.07 ± 0.69 |
| fumB * | Fumarase B | 4.08 ± 0.70 | 2.01 ± 0.31 |
| glcB | Malate synthase G | 3.25 ± 0.18 | 2.12 ± 0.22 |
| icdA | Isocitrate dehydrogenase | 1.16 ± 0.31 | 0.39 ± 0.01 |
| lpd * | Lipoamide dehydrogenase | 1.56 ± 0.16 | 1.19 ± 0.08 |
| mdh * | Malate dehydrogenase | 1.48 ± 0.17 | 0.75 ± 0.15 |
| sdhC * | Succinate:quinone oxidoreductase, SdhC | 1.61 ± 0.15 | 1.80 ± 0.20 |
| sdhA * | Succinate:quinone oxidoreductase, FAD binding protein | 0.93 ± 0.01 | 1.37 ± 0.19 |
| sdhB * | Succinate:quinone oxidoreductase, iron-sulfur cluster binding protein | 1.37 ± 0.27 | 0.91 ± 0.06 |
| sucA * | 2-Oxoglutarate decarboxylase, thiamine-requiring | 2.39 ± 0.44 | 1.43 ± 0.48 |
| sucB * | 2-Oxoglutarate dehydrogenase E2 subunit | 1.17 ± 0.26 | 0.84 ± 0.31 |
| sucC * | Succinyl-CoA synthetase subunit β | 1.41 ± 0.01 | 1.14 ± 0.25 |
| Anaplerotic genes | |||
| maeA (sfcA) | Malate dehydrogenase (oxaloacetate-decarboxylating) | 2.62 ± 0.00 | 1.32 ± 0.02 |
| maeB | Malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+) | 2.53 ± 0.47 | 1.62 ± 0.43 |
| pckA * | Phosphoenolpyruvate carboxykinase (ATP) | 3.04 ± 0.69 | 1.69 ± 0.07 |
| ppsA | Phosphoenolpyruvate synthetase | 3.04 ± 0.29 | 1.87 ± 0.08 |
| Respiratory chain | |||
| cyoD * | Cytochrome bo3, subunit 4 | 1.02 ± 0.03 | 0.63 ± 0.03 |
| cyoE | Heme O synthase | 1.41 ± 0.31 | 0.68 ± 0.00 |
| frdB | Fumarate reductase iron-sulfur protein | 1.49 ± 0.09 | 0.79 ± 0.08 |
| frdD | Fumarate reductase membrane protein FrdD | 1.63 ± 0.24 | 1.16 ± 0.20 |
| napA | Periplasmic nitrate reductase subunit NapA | 3.83 ± 0.08 | 2.76 ± 0.12 |
| narG | Nitrate reductase A subunit α | 4.22 ± 0.58 | 3.51 ± 0.94 |
| ndh | NADH:quinone oxidoreductase II | 1.74 ± 0.03 | 1.10 ± 0.21 |
| nuoA | NADH:quinone oxidoreductase subunit A | 2.52 ± 0.09 | 1.64 ± 0.18 |
| nuoB | NADH:quinone oxidoreductase subunit B | 2.22 ± 0.72 | 1.25 ± 0.65 |
| nuoC | NADH:quinone oxidoreductase subunit CB | 1.60 ± 0.23 | 1.50 ± 0.20 |
| nuoF | NADH:quinone oxidoreductase subunit F | 2.71 ± 0.24 | 2.18 ± 0.35 |
| nuoM | NADH:quinone oxidoreductase subunit M | 2.30 ± 0.32 | 1.05 ± 0.16 |
| nuoN | NADH:quinone oxidoreductase subunit N | 2.10 ± 0.65 | 0.91 ± 0.01 |
| ubiE | Bifunctional 2-octaprenyl-6-methoxy-1,4-benzoquinol methylase and demethyl menaquinone methyltransferase | 1.79 ± 0.00 | 1.60 ± 0.33 |
| Acetate production and utilization | |||
| ackA | Acetate kinase | 1.59 ± 0.05 | 0.82 ± 0.08 |
| acs * | Acetyl-CoA synthetase (AMP-forming) | 3.99 ± 0.25 | 2.21 ± 0.35 |
| ldhA | D-lactate dehydrogenase | 3.03 ± 0.16 | 2.22 ± 0.15 |
| pflD | Putative formate acetyltransferase 2 | 3.52 ± 0.24 | 2.76 ± 0.39 |
| pflB * | Pyruvate formate-lyase (inactive) | 1.41 ± 0.08 | 1.42 ± 0.24 |
| poxB | Pyruvate oxidase | 3.51 ± 0.58 | 2.75 ± 0.22 |
| Sigma factors | |||
| rpoA | RNA polymerase subunit α | 0.91 ± 0.13 | 0.69 ± 0.09 |
| rpoC | RNA polymerase subunit β′ | 2.13 ± 0.04 | 1.01 ± 0.18 |
| rpoZ | RNA polymerase subunit ω | 2.70 ± 0.00 | 2.02 ± 0.46 |
| rpoD | RNA polymerase sigma factor RpoD (σ70) | 5.41 ± 0.40 | 9.87 ± 2.09 |
| rpoE * | RNA polymerase sigma factor RpoE (σ24) | 1.77 ± 0.23 | 1.27 ± 0.07 |
| rpoH * | RNA polymerase sigma factor RpoH (σ32) | 4.21 ± 0.49 | 2.42 ± 0.44 |
| rpoN | RNA polymerase sigma factor RpoN (σ54) | 4.84 ± 0.26 | 3.75 ± 0.92 |
| rpoS * | RNA polymerase sigma factor RpoS (σ38) | 5.74 ± 0.81 | 1.92 ± 0.01 |
| Regulators | |||
| arcA | DNA-binding transcriptional dual regulator ArcA | 1.96 ± 0.09 | 0.88 ± 0.04 |
| arcB | Sensor histidine kinase ArcB | 3.10 ± 0.03 | 2.00 ± 0.25 |
| Cra | DNA-binding transcriptional dual regulator Cra | 3.25 ± 0.05 | 1.82 ± 0.04 |
| cyaA * | Adenylate cyclase | 2.47 ± 0.03 | 1.55 ± 0.03 |
| glcC * | DNA-binding transcriptional dual regulator GlcC | 3.26 ± 0.26 | 2.30 ± 0.51 |
| iclR | DNA-binding transcriptional repressor IcIR | 2.50 ± 0.33 | 1.18 ± 0.29 |
| ihfA | Integration host factor subunit α | 1.33 ± 0.05 | 1.38 ± 0.04 |
| fadR | DNA-binding transcriptional dual regulator FadR | 3.95 ± 0.20 | 2.22 ± 0.04 |
| Fnr | DNA-binding transcriptional dual regulator FNR | 3.22 ± 0.18 | 2.81 ± 0.36 |
| narL | DNA-binding transcriptional dual regulator NarL | 1.10 ± 0.20 | 1.02 ± 0.29 |
| Regulators to stress response | |||
| Rsd | Regulator of sigma D | 8.42 ± 0.33 | 18.1 ± 2.19 |
| soxR | DNA-binding transcriptional dual regulator SoxR | 30.6 ± 4.20 | 107.3 ± 10.3 |
| soxS | DNA-binding transcriptional dual regulator SoxS | 20.1 ± 0.55 | 81.8 ± 0.97 |
| spot | Bifunctional (p)ppGpp synthase/hydrolase SpoT | 75.1 ± 2.74 | 268.6 ± 30.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soria, S.; Carreón-Rodríguez, O.E.; de Anda, R.; Flores, N.; Escalante, A.; Bolívar, F. Transcriptional and Metabolic Response of a Strain of Escherichia coli PTS− to a Perturbation of the Energetic Level by Modification of [ATP]/[ADP] Ratio. BioTech 2024, 13, 10. https://doi.org/10.3390/biotech13020010
Soria S, Carreón-Rodríguez OE, de Anda R, Flores N, Escalante A, Bolívar F. Transcriptional and Metabolic Response of a Strain of Escherichia coli PTS− to a Perturbation of the Energetic Level by Modification of [ATP]/[ADP] Ratio. BioTech. 2024; 13(2):10. https://doi.org/10.3390/biotech13020010
Chicago/Turabian StyleSoria, Sandra, Ofelia E. Carreón-Rodríguez, Ramón de Anda, Noemí Flores, Adelfo Escalante, and Francisco Bolívar. 2024. "Transcriptional and Metabolic Response of a Strain of Escherichia coli PTS− to a Perturbation of the Energetic Level by Modification of [ATP]/[ADP] Ratio" BioTech 13, no. 2: 10. https://doi.org/10.3390/biotech13020010
APA StyleSoria, S., Carreón-Rodríguez, O. E., de Anda, R., Flores, N., Escalante, A., & Bolívar, F. (2024). Transcriptional and Metabolic Response of a Strain of Escherichia coli PTS− to a Perturbation of the Energetic Level by Modification of [ATP]/[ADP] Ratio. BioTech, 13(2), 10. https://doi.org/10.3390/biotech13020010