
From Sequence to Solution: Intelligent Learning Engine Optimization in Drug Discovery and Protein Analysis



Abstract
1. Introduction
2. Materials and Methods
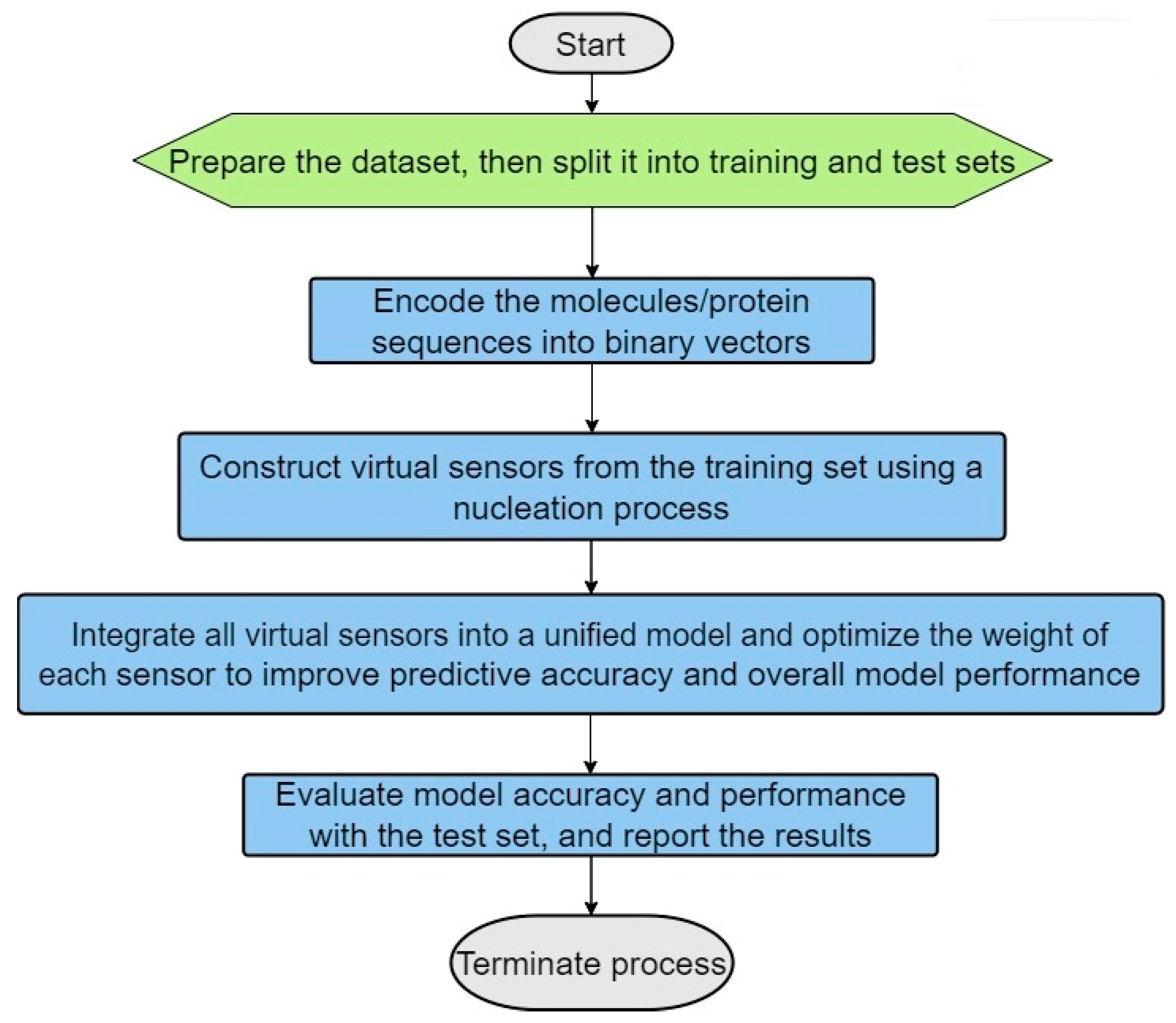
- (1)
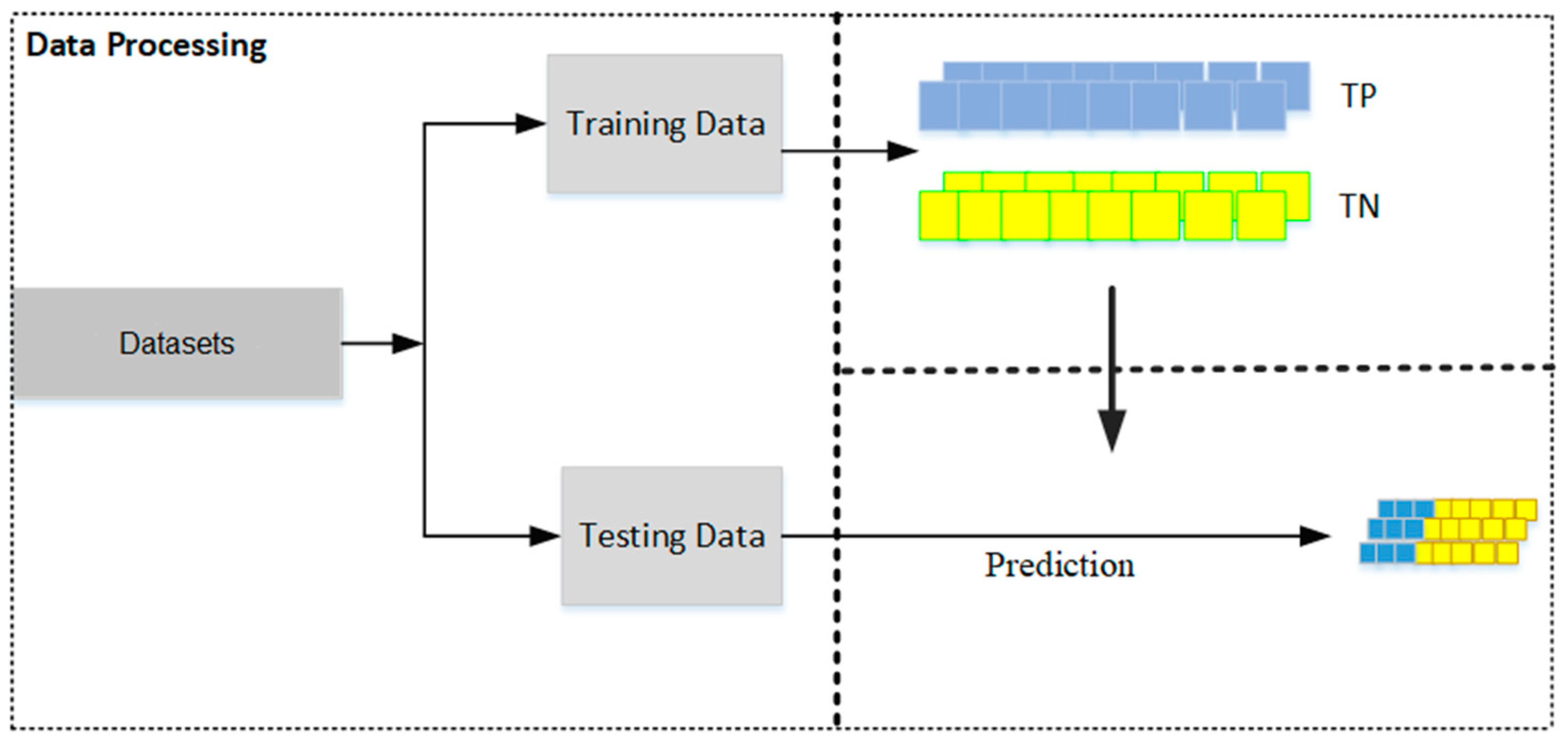
- Dataset Preparation: Two distinct datasets are prepared—one containing true positive (TP) matches and the other true negative (TN) matches. These datasets should then be split into training and testing sets, with a typical allocation of two-thirds for training and one-third for testing (see Figure 2). This division is crucial for facilitating effective model training and validation.
- (2)
- Encoding of the Molecules/Protein Sequences into Binary Vectors: The molecules’ protein sequences are encoded into binary vectors, such that each position along the vector is marked as 1 if it has the specific characteristic (for example if its molecular weight falls in a range of 155 to 220 daltons or if the amino acid type at a certain position is aspartic acid), and otherwise as 0.
- (3)
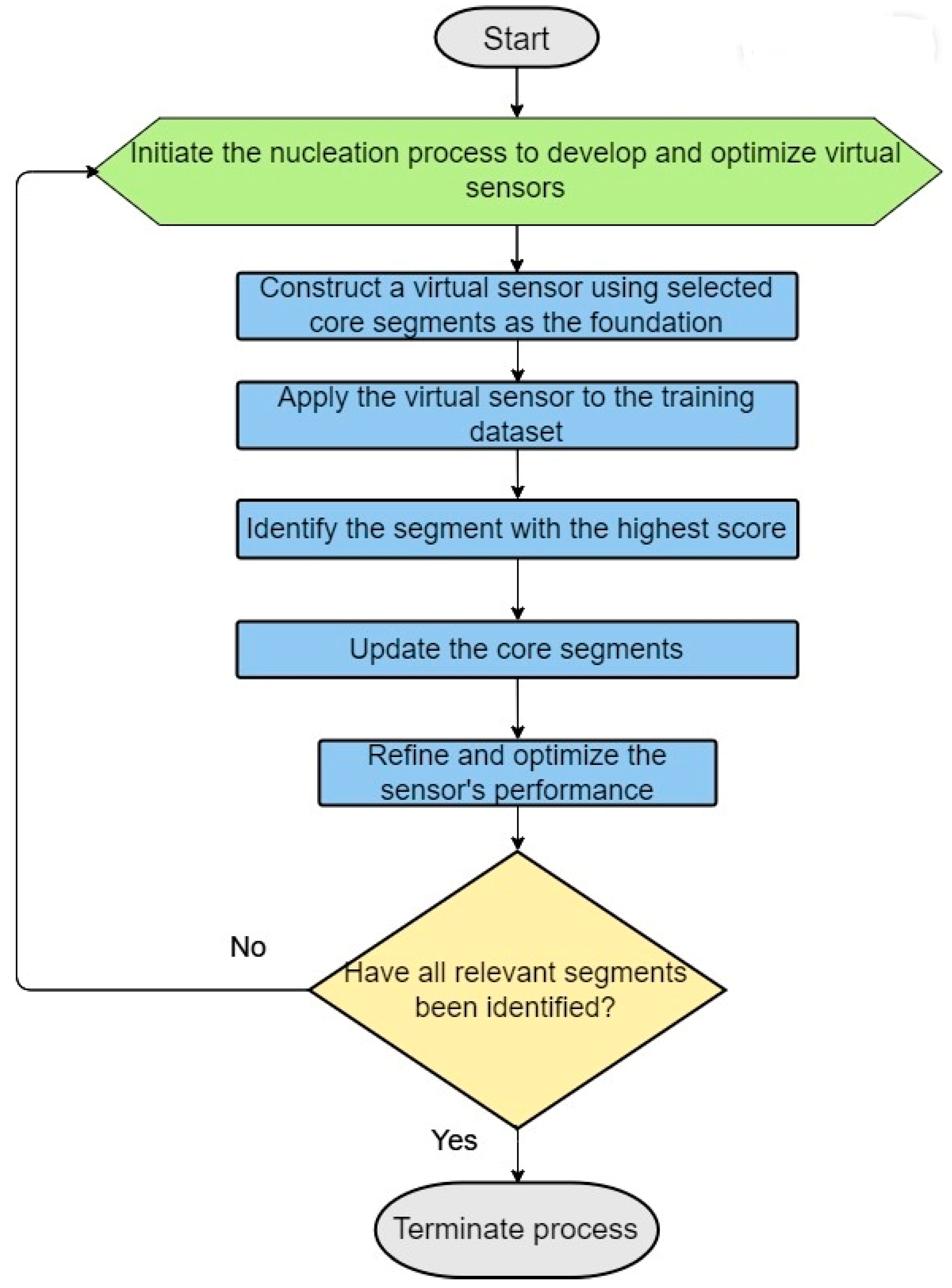
- Virtual Sensor Construction through Nucleation: Virtual sensors are defined by their sensor weight scores (SWSs), which are determined for specific segments of the binary vector. To calculate a sensor’s overall SWS, logical operations such as exclusive OR (XOR) and exclusive NOR (XNOR) are utilized to integrate the sensors with segments of the vector. This facilitates the dynamic generation of features capable of identifying distinct patterns in the binary vectors, especially patterns that mirror the intrinsic biological or chemical attributes of the molecules or proteins involved.
- (4)
- Sensor Optimization: The configuration of sensors is optimized using scoring functions such as specificity, sensitivity, and the Matthews correlation coefficient (MCC). The optimized sensors are then evaluated against the test set to verify their accuracy and reliability; this is aimed at minimizing false positives and false negatives.
- (5)
- Maximization of Virtual Sensor Efficiency: Factors are applied to the weights of the virtual sensors to boost their effectiveness and to augment the model’s capability to distinguish between true positive (TP) and true negative (TN) cases.
- (6)
- Usage of the Optimized Selected Sensors for Modeling Purposes: The refined model, which includes the selected optimized virtual sensors, is applied to specific tasks, such as indexing molecular activity, identifying and classifying proteins, modeling homology, and so forth.
3. Building Better Virtual Sensors: An Iterative Strategy of Nucleation and Updates
4. Results
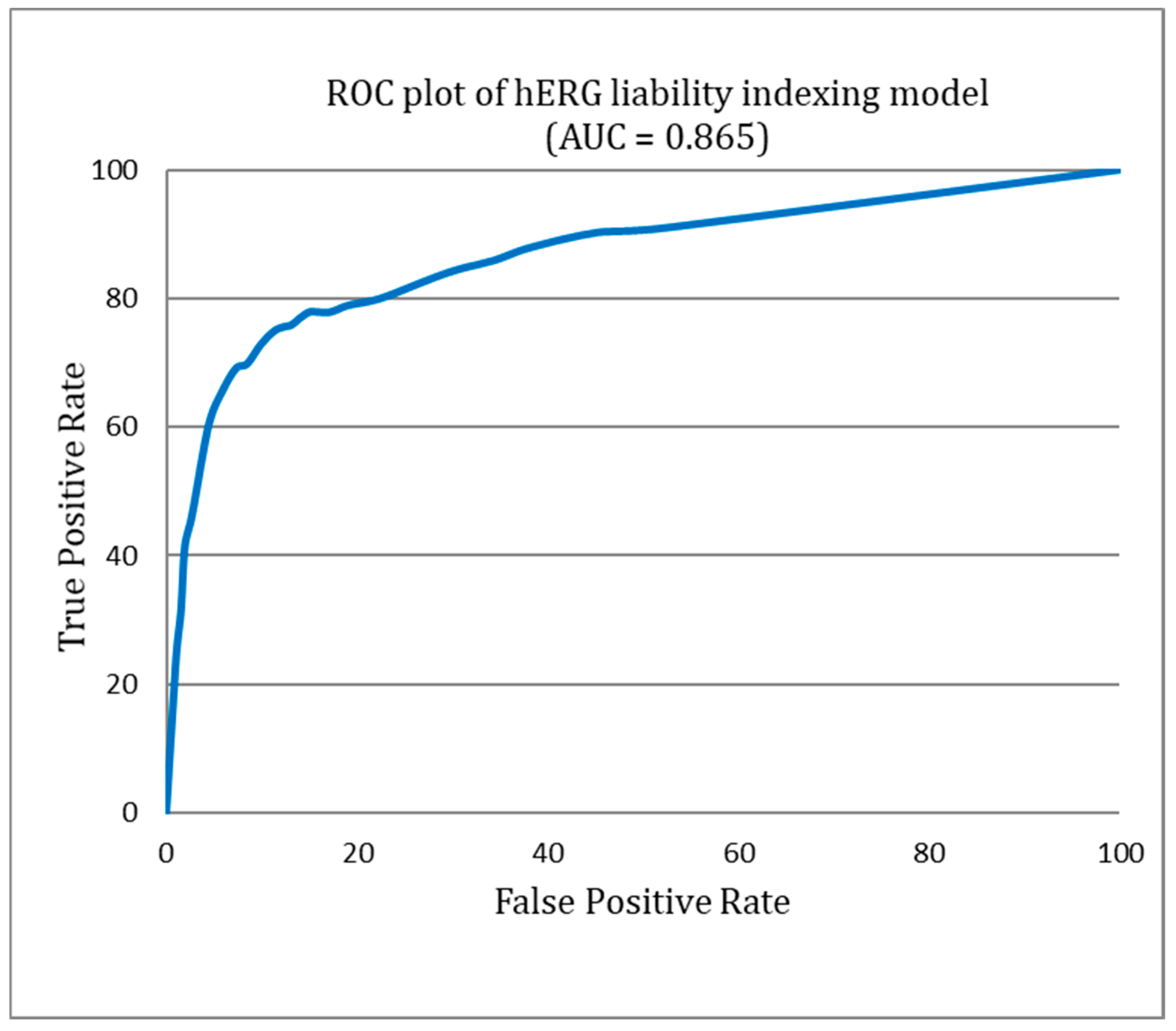
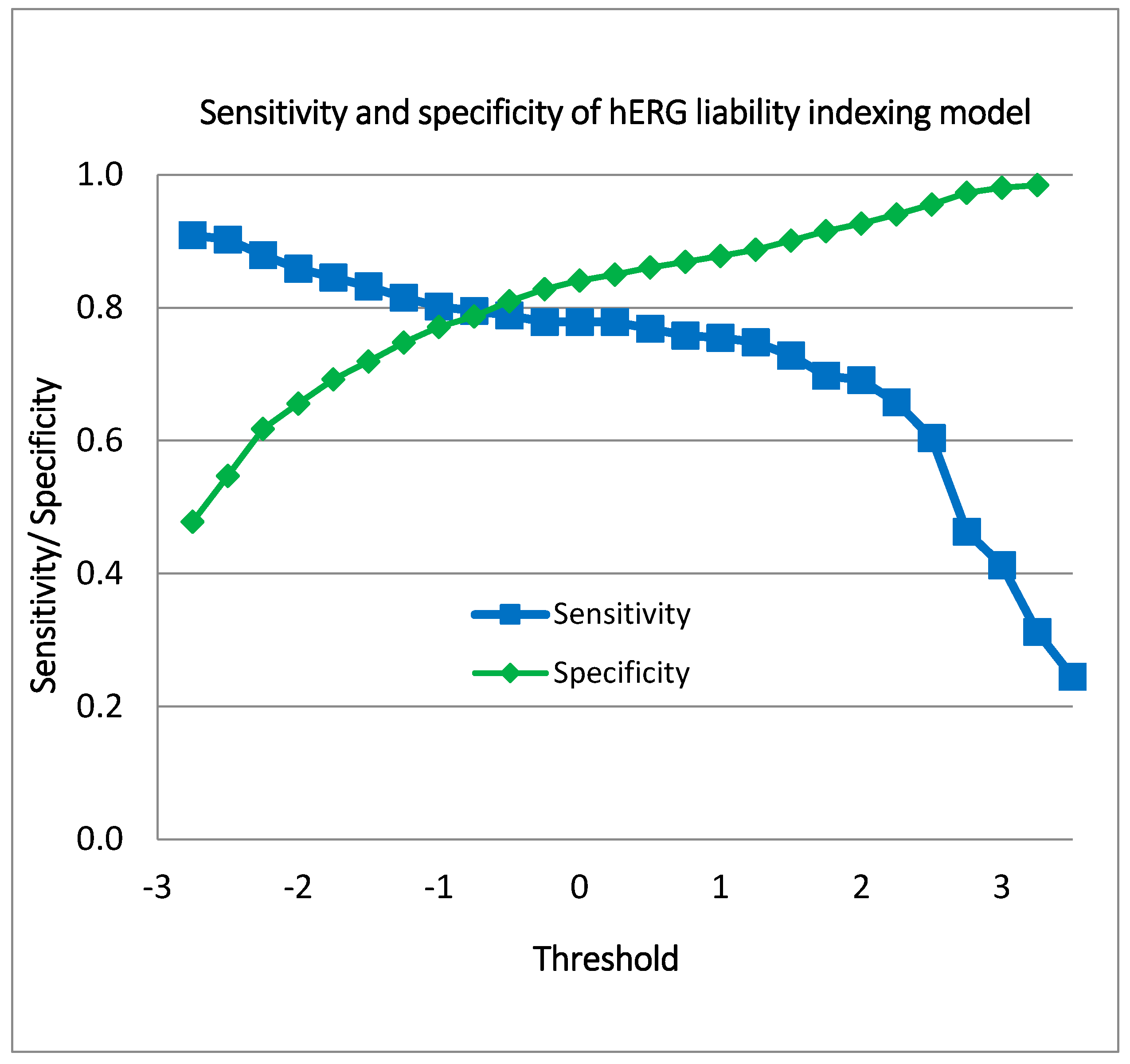
5. hERG Liability Indexing Model
Model Validation and Performance Assessment
6. Discussion
Model for hERG-Toxic Molecules
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Da’adoosh, B.; Marcus, D.; Rayan, A.; King, F.; Che, J.; Goldblum, A. Discovering highly selective and diverse PPAR-delta agonists by ligand based machine learning and structural modeling. Sci. Rep. 2019, 9, 1106. [Google Scholar] [CrossRef] [PubMed]
- Rayan, A.; Raiyn, J.; Falah, M. Nature is the best source of anticancer drugs: Indexing natural products for their anticancer bioactivity. PLoS ONE 2017, 12, e0187925. [Google Scholar] [CrossRef] [PubMed]
- Rayan, A. New vistas in GPCR 3D structure prediction. J. Mol. Model. 2010, 16, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Pappalardo, M.; Rayan, M.; Abu-Lafi, S.; Leonardi, M.E.; Milardi, D.; Guccione, S.; Rayan, A. Homology-based Modeling of Rhodopsin-like Family Members in the Inactive State: Structural Analysis and Deduction of Tips for Modeling and Optimization. Mol. Inform. 2017, 36, 1700014. [Google Scholar] [CrossRef] [PubMed]
- Aswad, M.; Rayan, M.; Abu-Lafi, S.; Falah, M.; Raiyn, J.; Abdallah, Z.; Rayan, A. Nature is the best source of anti-inflammatory drugs: Indexing natural products for their anti-inflammatory bioactivity. Inflamm. Res. 2018, 67, 67–75. [Google Scholar] [CrossRef]
- Awhangbo, L.; Schmitt, V.; Marcilhac, C.; Charnier, C.; Latrille, E.; Steyer, J.P. Determination of the optimal feed recipe of anaerobic digesters using a mathematical model and a genetic algorithm. Bioresour. Technol. 2023, 393, 130091. [Google Scholar] [CrossRef]
- Ghaleb, F.A.; Al-Rimy, B.A.S.; Boulila, W.; Saeed, F.; Kamat, M.; Foad Rohani, M.; Razak, S.A. Fairness-Oriented Semichaotic Genetic Algorithm-Based Channel Assignment Technique for Node Starvation Problem in Wireless Mesh Networks. Comput. Intell. Neurosci. 2021, 2021, 2977954. [Google Scholar] [CrossRef]
- Shi, K.; Huang, L.; Jiang, D.; Sun, Y.; Tong, X.; Xie, Y.; Fang, Z. Path Planning Optimization of Intelligent Vehicle Based on Improved Genetic and Ant Colony Hybrid Algorithm. Front. Bioeng. Biotechnol. 2022, 10, 905983. [Google Scholar] [CrossRef] [PubMed]
- Onizawa, N.; Katsuki, K.; Shin, D.; Gross, W.J.; Hanyu, T. Fast-Converging Simulated Annealing for Ising Models Based on Integral Stochastic Computing. IEEE Trans. Neural Netw. Learn. Syst. 2023, 34, 10999–11005. [Google Scholar] [CrossRef]
- Zhou, X.; Ling, M.; Lin, Q.; Tang, S.; Wu, J.; Hu, H. Effectiveness Analysis of Multiple Initial States Simulated Annealing Algorithm, A Case Study on the Molecular Docking Tool AutoDock Vina. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 3830–3841. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.B.; Jowett, T.; Yu, D.; Pahl, E.; Garden, A.L. Comparison of Taboo Search Methods for Atomic Cluster Global Optimization with a Basin-Hopping Algorithm. J. Chem. Inf. Model. 2023, 63, 5784–5793. [Google Scholar] [CrossRef]
- Rayan, A.; Falah, M.; Raiyn, J.; Da’adoosh, B.; Kadan, S.; Zaid, H.; Goldblum, A. Indexing molecules for their hERG liability. Eur. J. Med. Chem. 2013, 65, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Rayan, A.; Marcus, D.; Goldblum, A. Predicting oral druglikeness by iterative stochastic elimination. J. Chem. Inf. Model. 2010, 50, 437–445. [Google Scholar] [CrossRef]
- Rayan, M.; Abdallah, Z.; Abu-Lafi, S.; Masalha, M.; Rayan, A. Indexing Natural Products for their Antifungal Activity by Filters-based Approach: Disclosure of Discriminative Properties. Curr. Comput. Aided Drug Des. 2019, 15, 235–242. [Google Scholar] [CrossRef]
- Jensen, S.T.; Liu, J.S. BioOptimizer: A Bayesian scoring function approach to motif discovery. Bioinformatics 2004, 20, 1557–1564. [Google Scholar] [CrossRef]
- Carstens, S.; Nilges, M.; Habeck, M. Bayesian inference of chromatin structure ensembles from population-averaged contact data. Proc. Natl. Acad. Sci. USA 2020, 117, 7824–7830. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yao, H.; Lin, K. An overview of neural networks for drug discovery and the inputs used. Expert Opin. Drug Discov. 2018, 13, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ghose, U.; Buckley, N.J.; Engelborghs, S.; Sleegers, K.; Frisoni, G.B.; Wallin, A.; Lleó, A.; Popp, J.; Martinez-Lage, P.; et al. Predicting AT(N) pathologies in Alzheimer’s disease from blood-based proteomic data using neural networks. Front. Aging Neurosci. 2022, 14, 1040001. [Google Scholar] [CrossRef] [PubMed]
- Hasanzadeh, A.; Hamblin, M.R.; Kiani, J.; Noori, H.; Hardie, J.M.; Karimi, M.; Shafiee, H. Could artificial intelligence revolutionize the development of nanovectors for gene therapy and mRNA vaccines? Nano Today 2022, 47, 101665. [Google Scholar] [CrossRef]
- Barbero-Aparicio, J.A.; Olivares-Gil, A.; Díez-Pastor, J.F.; García-Osorio, C. Deep learning and support vector machines for transcription start site identification. PeerJ Comput. Sci. 2023, 9, e1340. [Google Scholar] [CrossRef]
- Machicao, J.; Craighero, F.; Maspero, D.; Angaroni, F.; Damiani, C.; Graudenzi, A.; Antoniotti, M.; Bruno, O.M. On the Use of Topological Features of Metabolic Networks for the Classification of Cancer Samples. Curr. Genom. 2021, 22, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Siddique, A.; Shirzaei, S.; Smith, A.E.; Valenta, J.; Garner, L.J.; Morey, A. Acceptability of Artificial Intelligence in Poultry Processing and Classification Efficiencies of Different Classification Models in the Categorisation of Breast Fillet Myopathies. Front. Physiol. 2021, 12, 712649. [Google Scholar] [CrossRef]
- Lepage, T.; Junier, I. Modeling Bacterial DNA: Simulation of Self-Avoiding Supercoiled Worm-Like Chains Including Structural Transitions of the Helix. Methods Mol. Biol. 2017, 1624, 323–337. [Google Scholar] [PubMed]
- Pan, X.; Chen, L.; Feng, K.Y.; Hu, X.H.; Zhang, Y.H.; Kong, X.Y.; Huang, T.; Cai, Y.D. Analysis of Expression Pattern of snoRNAs in Different Cancer Types with Machine Learning Algorithms. Int. J. Mol. Sci. 2019, 20, 2185. [Google Scholar] [CrossRef]
- Aryanti, C.; Uwuratuw, J.A.; Labeda, I.; Raharjo, W.; Lusikooy, R.E.; Rauf, M.A.; Mappincara, A.; Sampetoding, S.; Kusuma, M.I.; Syarifuddin, E. The Mutation Portraits of Oncogenes and Tumor Supressor Genes in Predicting the Overall Survival in Pancreatic Cancer: A Bayesian Network Meta-Analysis. Asian Pac. J. Cancer Prev. 2023, 24, 2895–2902. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Marchant, R.; Morris, R.W.; Baur, L.A.; Simpson, S.J.; Cripps, S. Bayesian network modelling to identify on-ramps to childhood obesity. BMC Med. 2023, 21, 105. [Google Scholar] [CrossRef] [PubMed]
- Emani, P.S.; Geradi, M.N.; Gürsoy, G.; Grasty, M.R.; Miranker, A.; Gerstein, M.B. Assessing and mitigating privacy risks of sparse, noisy genotypes by local alignment to haplotype databases. Genome Res. 2023, 33, 2156–2173. [Google Scholar] [CrossRef]
- Ringbauer, H.; Huang, Y.; Akbari, A.; Mallick, S.; Olalde, I.; Patterson, N.; Reich, D. Accurate detection of identity-by-descent segments in human ancient DNA. Nat. Genet. 2023, 56, 143–151. [Google Scholar] [CrossRef]
- Kwak, K.; Kostic, E.; Kim, D. Gait variability-based classification of the stages of the cognitive decline using partial least squares-discriminant analysis. Sci. Prog. 2023, 106, 368504231218604. [Google Scholar] [CrossRef]
- Teunissen, J.W.; Faber, I.R.; De Bock, J.; Slembrouck, M.; Verstockt, S.; Lenoir, M.; Pion, J. A machine learning approach for the classification of sports based on a coaches’ perspective of environmental, individual and task requirements: A sports profile analysis. J. Sports Sci. 2023, 1–10. [Google Scholar] [CrossRef]
- Zeidan, M.; Rayan, M.; Zeidan, N.; Falah, M.; Rayan, A. Indexing Natural Products for Their Potential Anti-Diabetic Activity: Filtering and Mapping Discriminative Physicochemical Properties. Molecules 2017, 22, 1563. [Google Scholar] [CrossRef]
- SoRelle, R. Warnings strengthened on tranquilizer inapsine (Droperidol). Circulation 2001, 104, E9061–E9062. [Google Scholar] [PubMed]
- Oyekan, P.J.; Gorton, H.C.; Copeland, C.S. Antihistamine-related deaths in England: Are the high safety profiles of antihistamines leading to their unsafe use? Br. J. Clin. Pharmacol. 2021, 87, 3978–3987. [Google Scholar] [CrossRef] [PubMed]
- Paterson, T.; Azizoglu, S.; Gokhale, M.; Chambers, M.; Suphioglu, C. Preserved Ophthalmic Anti-Allergy Medication in Cumulatively Increasing Risk Factors of Corneal Ectasia. Biology 2023, 12, 1036. [Google Scholar] [CrossRef] [PubMed]
- Madeira, L.; Queiroz, G.; Henriques, R. Prepandemic psychotropic drug status in Portugal: A nationwide pharmacoepidemiological profile. Sci. Rep. 2023, 13, 6912. [Google Scholar] [CrossRef]
- Abbas, S.; Ihle, P.; Adler, J.B.; Engel, S.; Günster, C.; Linder, R.; Lehmkuhl, G.; Schübert, I. Psychopharmacological Prescriptions in Children and Adolescents in Germany. Dtsch. Arztebl. Int. 2016, 113, 396–403. [Google Scholar] [CrossRef]
- Kobayashi, K.; Omuro, N.; Takahara, A. The conventional antihistamine drug cyproheptadine lacks QT-interval-prolonging action in halothane-anesthetized guinea pigs: Comparison with hydroxyzine. J. Pharmacol. Sci. 2014, 124, 92–98. [Google Scholar] [CrossRef]
- Binggeli, C.; Candinas, R.; Brunckhorst, C. Psychopharmaceuticals and arrhythmias. Ther. Umsch. 2004, 61, 279–283. [Google Scholar] [CrossRef]
- Haddad, S.; Oktay, L.; Erol, I.; Şahin, K.; Durdagi, S. Utilizing Heteroatom Types and Numbers from Extensive Ligand Libraries to Develop Novel hERG Blocker QSAR Models Using Machine Learning-Based Classifiers. ACS Omega 2023, 8, 40864–40877. [Google Scholar] [CrossRef]
- Venkateshappa, R.; Hunter, D.V.; Muralidharan, P.; Nagalingam, R.S.; Huen, G.; Faizi, S.; Luthra, S.; Lin, E.; Cheng, Y.M.; Hughes, J.; et al. Targeted activation of human ether-à-go-go-related gene channels rescues electrical instability induced by the R56Q+/- long QT syndrome variant. Cardiovasc. Res. 2023, 119, 2522–2535. [Google Scholar] [CrossRef]
- Falah, M.; Rayan, M.; Rayan, A. A Novel Paclitaxel Conjugate with Higher Efficiency and Lower Toxicity: A New Drug Candidate for Cancer Treatment. Int. J. Mol. Sci. 2019, 20, 4965. [Google Scholar] [CrossRef] [PubMed]
- Rayan, M.; Shadafny, S.; Falah, A.; Falah, M.; Abu-Lafi, S.; Asli, S.; Rayan, A. A Novel Docetaxel-Biotin Chemical Conjugate for Prostate Cancer Treatment. Molecules 2022, 27, 961. [Google Scholar] [CrossRef]
- Cai, Y.D.; Liu, X.J.; Xu, X.; Zhou, G.P. Support vector machines for predicting protein structural class. BMC Bioinform. 2001, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.J. Hidden Markov Models and their Applications in Biological Sequence Analysis. Curr. Genom. 2009, 10, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Ferran, E.A.; Ferrara, P.; Pflugfelder, B. Protein classification using neural networks. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1993, 1, 127–135. [Google Scholar]
- Gupta, K.; Sehgal, V.; Levchenko, A. A method for probabilistic mapping between protein structure and function taxonomies through cross training. BMC Struct. Biol. 2008, 8, 40. [Google Scholar] [CrossRef]
- Chemical-Computing-Group MOE. Molecular Operating Environment, version 2011.10; Chemical Computing Group: Montreal, QC, Canada, 2011. [Google Scholar]
- Chou, K.C.; Elrod, D.W. Protein subcellular location prediction. Protein Eng. 1999, 12, 107–118. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, X.; Dai, Z.; Zou, X. Classification of G-protein coupled receptors based on support vector machine with maximum relevance minimum redundancy and genetic algorithm. BMC Bioinform. 2010, 11, 325. [Google Scholar] [CrossRef] [PubMed]
- Rufai, M.; Alabison, R.; Abidemi, A.; Dansu, E. Solution to The Travelling Salesperson Problem Using Simulated Annealing Algorithm. Electron. J. Math. Anal. Appl. 2017, 5, 135–142. [Google Scholar]
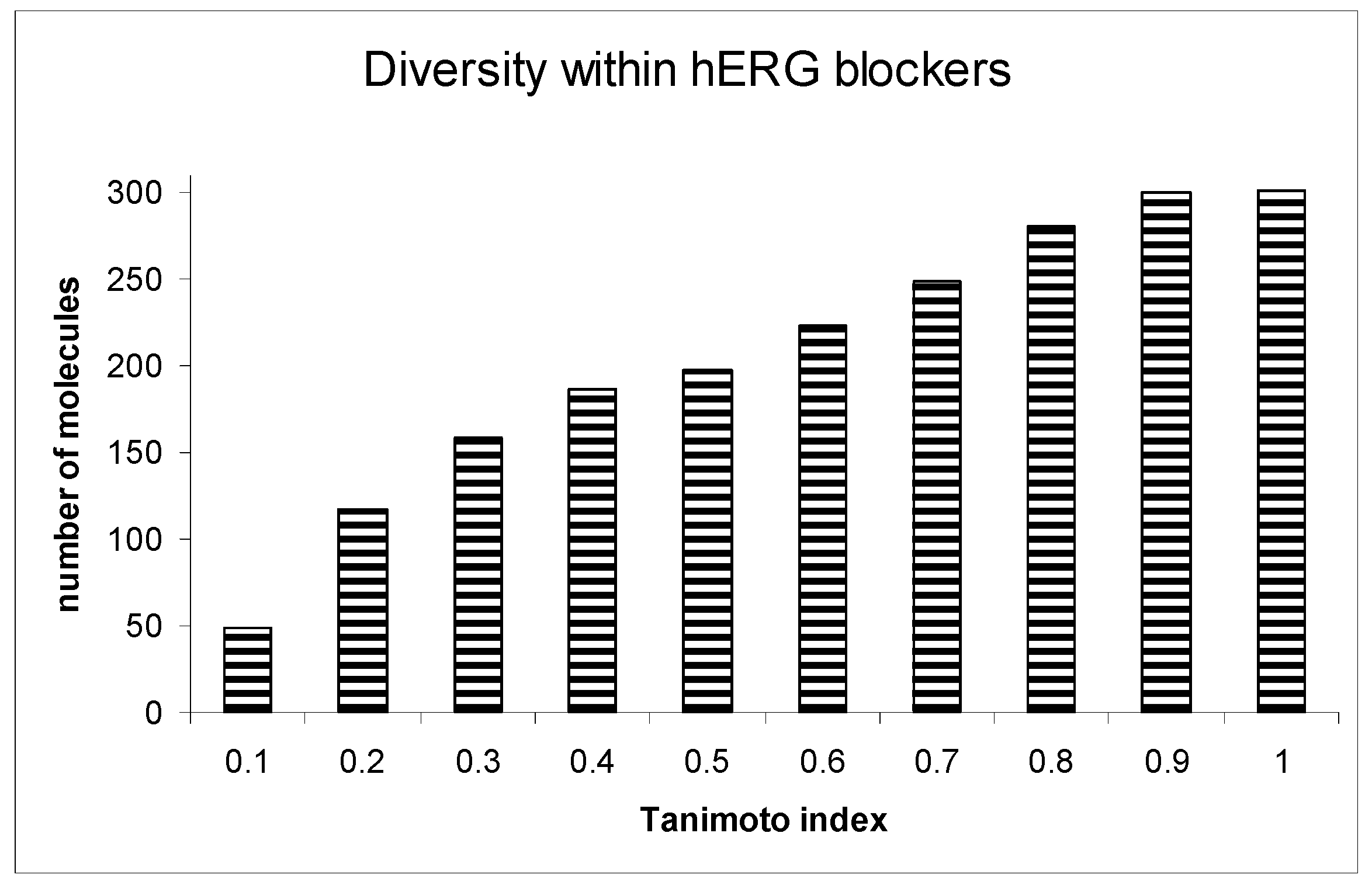


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Prediction Accuracy in % |
|---|---|
| SVM dipeptide-based | 99.5 |
| SVM aa composition-based | 96.5 |
| BLAST | 86.5 |
| PROSITE pattern | 92.0 |
| Pfam profile HMMs | 97.0 |
| PRINTS | 99.0 |
| PROSITE profile using pfscan | 97.0 |
| QFC algorithm | 99.5 |
| Linear discriminant analysis | 98.7 |
| Quadratic discriminant analysis | 98.5 |
| Logistic discriminant analysis | 97.7 |
| K-nearest neighbor method (KNN) | 98.3 |
| ILE | 100.0 |
| Method | Prediction Accuracy in % |
|---|---|
| SVM dipeptide-based | 99.5 |
| SVM aa composition-based | 96.5 |
| BLAST | 86.5 |
| PROSITE pattern | 92.0 |
| Pfam profile HMMs | 97.0 |
| PRINTS | 99.0 |
| PROSITE profile using pfscan | 97.0 |
| QFC algorithm | 99.5 |
| Linear discriminant analysis | 98.7 |
| Quadratic discriminant analysis | 98.5 |
| Logistic discriminant analysis | 97.7 |
| K-nearest neighbor method (KNN) | 98.3 |
| ILE | 100.0 |
| Method | Prediction Accuracy in % |
|---|---|
| SVM | 88.4 |
| BLAST | 83.3 |
| SAM-T2K HMM | 69.9 |
| kernNN | 64.0 |
| Decision tree | 77.3 |
| Naïve Bayes | 93.0 |
| ILE | 99.8 |
| Method | Prediction Accuracy in % |
|---|---|
| SVM | 86.3 |
| SVMtree | 82.9 |
| BLAST | 74.5 |
| SAM-T2K HMM | 70.0 |
| kernNN | 51.0 |
| Decision tree | 70.8 |
| Naïve Bayes | 92.4 |
| Covariant-discriminant | 83.2 |
| ILE | 100.0 |
| Percent Sequence Identity α | Total Number of Models β | Percent π Models with RMSD Lower Than 1 Å | Percent Models with RMSD Lower Than 2 Å | Percent Models with RMSD Lower Than 3 Å |
|---|---|---|---|---|
| 25–29 | 15 | 40 Ω | 100 | 100 |
| 30–39 | 883 | 28 | 98 | 100 |
| 40–49 | 2365 | 50 | 99.9 | 100 |
| 50–59 | 423 | 75 | 100 | 100 |
| 60–69 | 51 | 90 | 100 | 100 |
| 70–79 | 181 | 100 | 100 | 100 |
| 80–89 | 289 | 100 | 100 | 100 |
| 90–95 | 44 | 100 | 100 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raiyn, J.; Rayan, A.; Abu-Lafi, S.; Rayan, A. From Sequence to Solution: Intelligent Learning Engine Optimization in Drug Discovery and Protein Analysis. BioTech 2024, 13, 33. https://doi.org/10.3390/biotech13030033
Raiyn J, Rayan A, Abu-Lafi S, Rayan A. From Sequence to Solution: Intelligent Learning Engine Optimization in Drug Discovery and Protein Analysis. BioTech. 2024; 13(3):33. https://doi.org/10.3390/biotech13030033
Chicago/Turabian StyleRaiyn, Jamal, Adam Rayan, Saleh Abu-Lafi, and Anwar Rayan. 2024. "From Sequence to Solution: Intelligent Learning Engine Optimization in Drug Discovery and Protein Analysis" BioTech 13, no. 3: 33. https://doi.org/10.3390/biotech13030033
APA StyleRaiyn, J., Rayan, A., Abu-Lafi, S., & Rayan, A. (2024). From Sequence to Solution: Intelligent Learning Engine Optimization in Drug Discovery and Protein Analysis. BioTech, 13(3), 33. https://doi.org/10.3390/biotech13030033