Why Immunotherapy Fails in Multiple Myeloma

 , and
, and 
Abstract
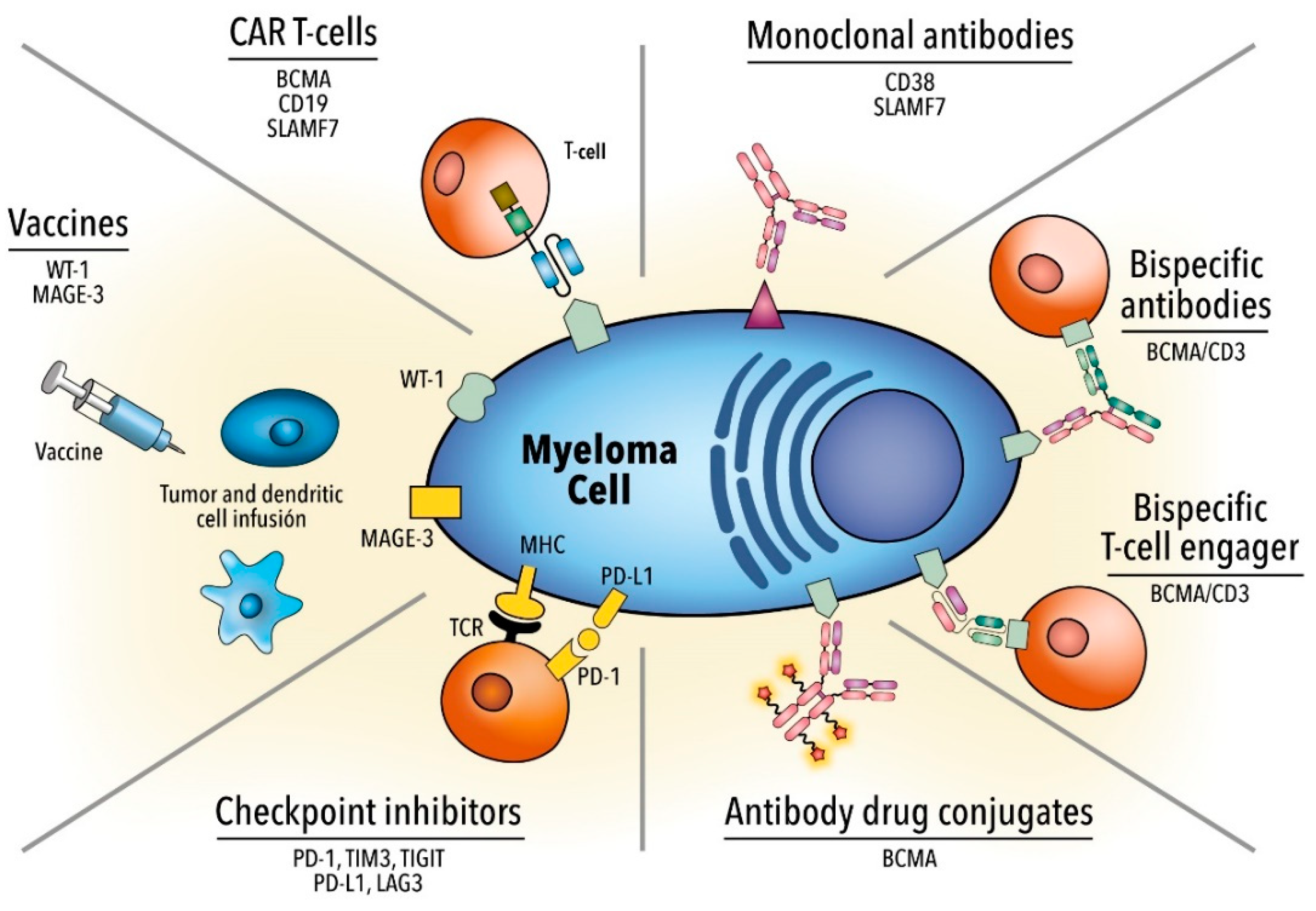
:1. Introduction
2. Monoclonal Antibodies
2.1. Anti-CD38
- Complement-dependent cytotoxicity (CDC): Binding between the Fc tail of the antibody and C1q activates the complement cascade to end with the formation of the membrane attack complex (MAC) [72];
- Antibody-dependent cell-mediated cytotoxicity (ADCC): Binding between FC-gamma receptors on effector cells (T and NK cells) and the Fc tail of daratumumab releases cytotoxic molecules, leading to MM cell death [65];
- Antibody-dependent cellular phagocytosis (ADCP): Opsonization of the tumor cell occurs when the Fc tail of the CD38 antibody binds to the Fc-gamma receptor of phagocytic cells such as monocytes or macrophages [73];
- CD38 expression: Tests performed on modified MM cell lines that express different levels of CD38 have shown greater CDC and ADCC in cells expressing high levels of CD38 compared to cells with low expression. In MM plasma cells, expression is heterogenic and daratumumab activity is correlated with such expression levels [78]. Analysis performed on samples of patients who had been enrolled in daratumumab clinical trials showed a quick and marked decrease in CD38 levels after treatment in all patients; a decrease in CDC and ADCC was also observed in ex vivo tests. Downregulation of CD38 of this type also occurs in cell subsets other than MM cells and mechanisms are not fully understood. Some strategies to overcome such resistance have been proposed and are based on combinations with other drugs capable of increasing CD38 levels such as IMiDs, panobinostat, all-trans retinoic acid (ATRA), and ricolinostat [79,80,81]. The ability of ATRA to resynthesize CD38 is being analyzed in a clinical trial (NCT02751255);
- Complement inhibitory proteins: Tumor cells are known to be capable of increasing soluble and membrane-bound complement regulatory proteins such as C4-binding protein, CD55, or CD59 to protect themselves from complement attacks, similar to the way in which immune checkpoint inhibitor receptors function [82]. Ex vivo analysis using MM cell lines with low expression of CD55 and CD59, and MM cell lines treated with phospholipase-C to remove GPI-anchored proteins (CD55 and CD59) showed increased daratumumab CDC. These observations were not confirmed with MM cells obtained from daratumumab-naïve patients. In addition, an increase in CD55 and CD59 expression was detected in MM cells obtained from patients who were progressing under monotherapy treatment. In this case, ATRA combination may also decrease upregulation of complement inhibitors [78]. Panobinostat, which has shown to increase CD38 levels, also increases CD55 and CD59 levels, possibly explaining the lack of benefit in terms of CDC, although ADCC improved [83];
- CD47-SIRPα interaction: CD47 expressed in tumor cells of solid tumors and hematological malignancies interacts with regulatory transmembrane protein SIRPα that is expressed on dendritic cells and macrophages, decreasing their phagocytic function [84]. Upregulation of CD47 has been observed in drug-resistant MM cells and blocking the interaction between SIRPα and CD47 restores phagocytosis [85]. Anti-CD47 therapies are under evaluation in other lymphoid malignancies and low-dose cyclophosphamide may decrease CD47 expression to improve ADCP [86,87,88];
- Polymorphisms on Fc-gamma receptors: Mechanism of action of daratumumab ADCC and ADCP depend on the activation of Fc-gamma receptors on effector cells [89]. Affinity may differ based on allelic variants of these receptors. Fc-gamma receptors were genotyped in samples of patients with MM included in daratumumab clinical trials, demonstrating a positive correlation between polymorphisms 3A and 2B and outcome in terms of PFS, albeit not OS [90];
- The way in which the microenvironment plays a crucial role in MM has been well studied. Bone marrow stromal cells (BMSC) protect MM cells from drugs and effector cells such as cytotoxic T cells [91]. Interaction between BMSC and MM cells may upregulate anti-apoptotic molecules like survivin, which could contribute to resistance against daratumumab;
- Soluble CD38 (sCD38) may have a draining effect on daratumumab function and diminish efficacy; however, the presence of sCD38 has been observed in only a few patients and in such cases, did not correlate with response. There are no published data about other CD38 antibodies and the impact of sCD38 [82];
- NK cells play a crucial role in ADCC. Some studies have shown a correlation between daratumumab-induced ADCC and NK cell-to-MM cell ratio [78]. Due to their capacity to activate NK cells, IMiDs could improve NK function and ADCC, even in patients with IMiD-refractory MM [65,92]. An increase in ADCC was observed in ex vivo experiments when interaction between NK inhibitory receptors KIR (KIR2DL-1, -2, -3) and their respective ligands was blocked. Similarly, ADCC was reported to improve synergistically with the addition of lenalidomide to the experiment. As NK cells express CD38 on their surface, fratricide and a diminished effector function can arise. When studied in patients, the reduction in NK levels was similar in responders and non-responders to daratumumab and no correlation with outcome was observed. Some measures have nonetheless been proposed to diminish this eventual effect, including the administration of ex vivo-expanded autologous NK cells to increase the count, and pretreatment of such cells with F(ab’)2 fragments of daratumumab to avoid fratricide [93,94].

{kind=link}
{kind=link}
| Mechanisms of Action | Mechanisms of Resistance |
|---|---|
|
|
2.2. Anti-SLAMF7
3. Antibody-Drug Conjugate
4. Bispecific Monoclonal Antibodies
5. Immune Checkpoint Inhibitors
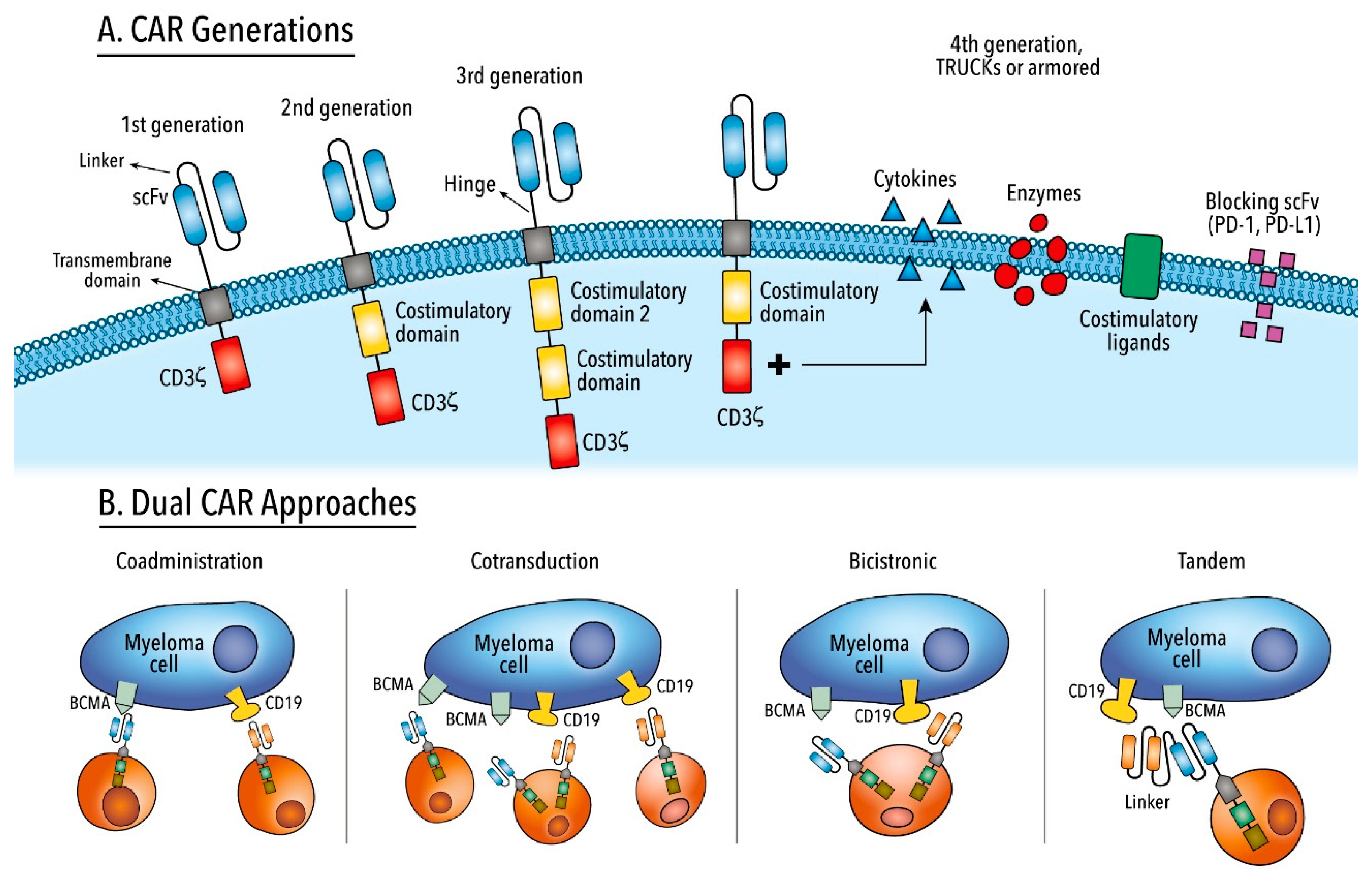
6. Chimeric Antigen Receptor T Cell Therapy
6.1. Mechanisms or Relapse
6.1.1. Antigen-Positive Escape
6.1.2. Antigen-Negative Escape
6.2. CAR T Cell-Related Toxicities
6.3. Product Manufacturing, Access and Economic Challenges
7. Vaccines
8. Personal Perspective
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Cowan, A.J.; Allen, C.; Barac, A.; Basaleem, H.; Bensenor, I.; Curado, M.P.; Foreman, K.; Gupta, R.; Harvey, J.; Hosgood, H.D.; et al. Global Burden of Multiple Myeloma: A Systematic Analysis for the Global Burden of Disease Study 2016. JAMA Oncol. 2018, 4, 1221–1227. [Google Scholar] [CrossRef] [Green Version]
- Sant, M.; Allemani, C.; Tereanu, C.; De Angelis, R.; Capocaccia, R.; Visser, O.; Marcos-Gragera, R.; Maynadié, M.; Simonetti, A.; Lutz, J.M.; et al. Incidence of hematologic malignancies in Europe by morphologic subtype: Results of the HAEMACARE project. Blood 2010, 116, 3724–3734. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, S.V. The multiple myelomas - current concepts in cytogenetic classification and therapy. Nat. Rev. Clin. Oncol. 2018, 15, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, A.; Paul, S.; Rajkumar, S.V.; Ketterling, R.P.; Greipp, P.T.; Dispenzieri, A.; Gertz, M.A.; Buadi, F.K.; Lacy, M.Q.; Dingli, D.; et al. Prognostic significance of interphase FISH in monoclonal gammopathy of undetermined significance. Leukemia 2018, 32, 1811–1815. [Google Scholar] [CrossRef] [PubMed]
- Merz, M.; Hielscher, T.; Hoffmann, K.; Seckinger, A.; Hose, D.; Raab, M.S.; Hillengass, J.; Jauch, A.; Goldschmidt, H. Cytogenetic abnormalities in monoclonal gammopathy of undetermined significance. Leukemia 2018, 32, 2717–2719. [Google Scholar] [CrossRef]
- Mikulasova, A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Jackson, G.H.; Smetana, J.; Kufova, Z.; Pour, L.; Sandecka, V.; Almasi, M.; et al. The spectrum of somatic mutations in monoclonal gammopathy of undetermined significance indicates a less complex genomic landscape than that in multiple myeloma. Haematologica 2017, 102, 1617–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, B.; Zhang, X.G.; Lu, Z.Y.; Bataille, R. Interleukin-6 in human multiple myeloma. Blood 1995, 85, 863–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunn, W.G.; Conley, A.; Deininger, L.; Olson, S.D.; Prockop, D.J.; Gregory, C.A. A crosstalk between myeloma cells and marrow stromal cells stimulates production of DKK1 and interleukin-6: A potential role in the development of lytic bone disease and tumor progression in multiple myeloma. Stem Cells 2006, 24, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Hengeveld, P.J.; Kersten, M.J. B-cell activating factor in the pathophysiology of multiple myeloma: A target for therapy? Blood Cancer J. 2015, 5, e282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprynski, A.C.; Hose, D.; Caillot, L.; Réme, T.; Shaughnessy, J.D.; Barlogie, B.; Seckinger, A.; Moreaux, J.; Hundemer, M.; Jourdan, M.; et al. The role of IGF-1 as a major growth factor for myeloma cell lines and the prognostic relevance of the expression of its receptor. Blood 2009, 113, 4614–4626. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; McMillin, D.W.; Klippel, S.; Hideshima, T.; Chauhan, D.; Richardson, P.G.; Munshi, N.C.; Anderson, K.C. The role of the bone marrow microenvironment in the pathophysiology of myeloma and its significance in the development of more effective therapies. Hematol. Oncol. Clin. N. Am. 2007, 21, 1007–1034. [Google Scholar] [CrossRef] [PubMed]
- Asaoku, H.; Kawano, M.; Iwato, K.; Tanabe, O.; Tanaka, H.; Hirano, T.; Kishimoto, T.; Kuramoto, A. Decrease in BSF-2/IL-6 response in advanced cases of multiple myeloma. Blood 1988, 72, 429–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Görgün, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, D.M.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Minnie, S.A.; Kuns, R.D.; Gartlan, K.H.; Zhang, P.; Wilkinson, A.N.; Samson, L.; Guillerey, C.; Engwerda, C.; MacDonald, K.; Smyth, M.J.; et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood 2018, 132, 1675–1688. [Google Scholar] [CrossRef]
- Lozano, E.; Mena, M.P.; Díaz, T.; Martin-Antonio, B.; Leon, S.; Rodríguez-Lobato, L.G.; Oliver-Caldés, A.; Cibeira, M.T.; Bladé, J.; Prat, A.; et al. Nectin-2 Expression on Malignant Plasma Cells Is Associated with Better Response to TIGIT Blockade in Multiple Myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 4688–4698. [Google Scholar] [CrossRef]
- Prabhala, R.H.; Pelluru, D.; Fulciniti, M.; Prabhala, H.K.; Nanjappa, P.; Song, W.; Pai, C.; Amin, S.; Tai, Y.T.; Richardson, P.G.; et al. Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood 2010, 115, 5385–5392. [Google Scholar] [CrossRef]
- Richardson, P.G.; Weller, E.; Lonial, S.; Jakubowiak, A.J.; Jagannath, S.; Raje, N.S.; Avigan, D.E.; Xie, W.; Ghobrial, I.M.; Schlossman, R.L.; et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood 2010, 116, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Rosiñol, L.; Oriol, A.; Rios, R.; Sureda, A.; Blanchard, M.J.; Hernández, M.T.; Martínez-Martínez, R.; Moraleda, J.M.; Jarque, I.; Bargay, J.; et al. Bortezomib, lenalidomide, and dexamethasone as induction therapy prior to autologous transplant in multiple myeloma. Blood 2019, 134, 1337–1345. [Google Scholar] [CrossRef] [Green Version]
- Leblay, N.; Maity, R.; Hasan, F.; Neri, P. Deregulation of Adaptive T Cell Immunity in Multiple Myeloma: Insights Into Mechanisms and Therapeutic Opportunities. Front. Oncol. 2020, 10, 636. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Stadtmauer, E.A. Cellular and vaccine immunotherapy for multiple myeloma. Hematol. Am. Soc. Hematol. Educ. Program. 2016, 2016, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greil, C.; Engelhardt, M.; Ihorst, G.; Schoeller, K.; Bertz, H.; Marks, R.; Zeiser, R.; Duyster, J.; Einsele, H.; Finke, J.; et al. Allogeneic transplantation of multiple myeloma patients may allow long-term survival in carefully selected patients with acceptable toxicity and preserved quality of life. Haematologica 2019, 104, 370–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Weiss, B.M.; Plesner, T.; Bahlis, N.J.; Belch, A.; Lonial, S.; Lokhorst, H.M.; Voorhees, P.M.; Richardson, P.G.; Chari, A.; et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood 2016, 128, 37–44. [Google Scholar] [CrossRef]
- Plesner, T.; Arkenau, H.T.; Lokhorst, H.M.; Gimsing, P.; Krejcik, J.; Lemech, C.; Minnema, M.; Lassen, U.N.; Laubach, J.P.; Ahmadi, T.; et al. Safety and Efficacy of Daratumumab with Lenalidomide and Dexamethasone in Relapsed or Relapsed, Refractory Multiple Myeloma. Blood 2014, 124, 84. [Google Scholar] [CrossRef]
- Plesner, T.; Arkenau, H.T.; Gimsing, P.; Krejcik, J.; Lemech, C.; Minnema, M.C.; Lassen, U.; Laubach, J.P.; Palumbo, A.; Lisby, S.; et al. Daratumumab in Combination with Lenalidomide and Dexamethasone in Patients with Relapsed or Relapsed and Refractory Multiple Myeloma: Updated Results of a Phase 1/2 Study (GEN503). Blood 2015, 126, 507. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Weisel, K.C.; San Miguel, J.; Cook, G.; Leiba, M.; Suzuki, K.; Kumar, S.; Cavo, M.; Avet-Loiseau, H.; Quach, H.; Hungria, V.; et al. Efficacy of daratumumab in combination with lenalidomide plus dexamethasone (DRd) or bortezomib plus dexamethasone (DVd) in relapsed or refractory multiple myeloma (RRMM) based on cytogenetic risk status. J. Clin. Oncol. 2017, 35 (Suppl. 15), 8006. [Google Scholar] [CrossRef]
- Zonder, J.A.; Mohrbacher, A.F.; Singhal, S.; Van Rhee, F.; Bensinger, W.I.; Ding, H.; Fry, J.; Afar, D.E.; Singhal, A.K. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood 2012, 120, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Jakubowiak, A.; Offidani, M.; Pégourie, B.; De La Rubia, J.; Garderet, L.; Laribi, K.; Bosi, A.; Marasca, R.; Laubach, J.; Mohrbacher, A.; et al. Randomized phase 2 study: Elotuzumab plus bortezomib/dexamethasone vs bortezomib/dexamethasone for relapsed/refractory MM. Blood 2016, 127, 2833–2840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimopoulos, M.A.; Lonial, S.; White, D.; Moreau, P.; Palumbo, A.; San-Miguel, J.; Shpilberg, O.; Anderson, K.; Grosicki, S.; Spicka, I.; et al. Elotuzumab plus lenalidomide/dexamethasone for relapsed or refractory multiple myeloma: ELOQUENT-2 follow-up and post-hoc analyses on progression-free survival and tumour growth. Br. J. Haematol. 2017, 178, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Hoos, A.; Gupta, I.; Bragulat, V.; et al. Antibody-drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: An update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Schönfeld, K.; Zuber, C.; Pinkas, J.; Häder, T.; Bernöster, K.; Uherek, C. Indatuximab ravtansine (BT062) combination treatment in multiple myeloma: Pre-clinical studies. J. Hematol. Oncol. 2017, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Jagannath, S.; Heffner Jr, L.T.; Ailawadhi, S.; Munshi, N.C.; Zimmerman, T.M.; Rosenblatt, J.; Lonial, S.; Chanan-Khan, A.; Ruehle, M.; Rharbaoui, F.; et al. Indatuximab Ravtansine (BT062) Monotherapy in Patients With Relapsed and/or Refractory Multiple Myeloma. Clin. Lymphoma. Myeloma. Leuk. 2019, 19, 372–380. [Google Scholar] [CrossRef]
- Ailawadhi, S.; Kelly, K.R.; Vescio, R.A.; Jagannath, S.; Wolf, J.; Gharibo, M.; Sher, T.; Bojanini, L.; Kirby, M.; Chanan-Khan, A. A Phase I Study to Assess the Safety and Pharmacokinetics of Single-agent Lorvotuzumab Mertansine (IMGN901) in Patients with Relapsed and/or Refractory CD-56-positive Multiple Myeloma. Clin. Lymphoma. Myeloma. Leuk. 2019, 19, 29–34. [Google Scholar] [CrossRef]
- Kaufman, J.L.; Niesvizky, R.; Stadtmauer, E.A.; Chanan-Khan, A.; Siegel, D.; Horne, H.; Wegener, W.A.; Goldenberg, D.M. Phase I, multicentre, dose-escalation trial of monotherapy with milatuzumab (humanized anti-CD74 monoclonal antibody) in relapsed or refractory multiple myeloma. Br. J. Haematol. 2013, 163, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Hipp, S.; Tai, Y.T.; Blanset, D.; Deegen, P.; Wahl, J.; Thomas, O.; Rattel, B.; Adam, P.J.; Anderson, K.C.; Friedrich, M. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia 2017, 31, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Mateos, M.V.; Nahi, H.; Krishnan, A.Y.; van de Donk, N.W.; San Miguel, J.; Oriol, A.; Rosiñol, L.; Chari, A.; Adams, H.; et al. Phase I study of teclistamab, a humanized B-cell maturation antigen (BCMA) x CD3 bispecific antibody, in relapsed/refractory multiple myeloma (R/R MM). J. Clin. Oncol. 2020, 38 (Suppl. 15), 100. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateos, M.V.; Blacklock, H.; Schjesvold, F.; Oriol, A.; Simpson, D.; George, A.; Goldschmidt, H.; Larocca, A.; Chanan-Khan, A.; Sherbenou, D.; et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e459–e469. [Google Scholar] [CrossRef]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.Y.; Zhao, W.H.; Liu, J.; Chen, Y.X.; Cao, X.M.; Yang, Y.; Zhang, Y.L.; Wang, F.X.; Zhang, P.Y.; Lei, B.; et al. Long-Term Follow-up of a Phase 1, First-in-Human Open-Label Study of LCAR-B38M, a Structurally Differentiated Chimeric Antigen Receptor T (CAR-T) Cell Therapy Targeting B-Cell Maturation Antigen (BCMA), in Patients (pts) with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2019, 134 (Suppl. 1), 579. [Google Scholar]
- Zhao, W.H.; Liu, J.; Wang, B.Y.; Chen, Y.X.; Cao, X.M.; Yang, Y.; Zhang, Y.L.; Wang, F.X.; Zhang, P.Y.; Lei, B.; et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2018, 11, 141. [Google Scholar] [CrossRef]
- Xu, J.; Chen, L.J.; Yang, S.S.; Sun, Y.; Wu, W.; Liu, Y.F.; Xu, J.; Zhuang, Y.; Zhang, W.; Weng, X.Q.; et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 9543–9551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madduri, D.; Berdeja, J.G.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D. CARTITUDE-1: Phase 1b/2 Study of Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T Cell Therapy, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136 (Suppl. 1), 22–25. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Jagannath, S.; Berdeja, J.G.; Lonial, S.; Raje, N.S.; Siegel, D.S.; Lin, Y.; Oriol, A.; et al. Idecabtagene vicleucel (ide-cel; bb2121), a BCMA-targeted CAR T-cell therapy, in patients with relapsed and refractory multiple myeloma (RRMM): Initial KarMMa results. J. Clin. Oncol. 2020, 38 (Suppl. 15), 8503. [Google Scholar] [CrossRef]
- Mailankody, S.; Jakubowiak, A.J.; Htut, M.; Costa, L.J.; Lee, K.; Ganguly, S.; Kaufman, J.L.; Siegel, D.S.; Bensinger, W.; Cota, M.; et al. Orvacabtagene autoleucel (orva-cel), a B-cell maturation antigen (BCMA)-directed CAR T cell therapy for patients (pts) with relapsed/refractory multiple myeloma (RRMM): Update of the phase 1/2 EVOLVE study (NCT03430011). J. Clin. Oncol. 2020, 38 (Suppl. 15), 8504. [Google Scholar] [CrossRef]
- Rosenblatt, J.; Vasir, B.; Uhl, L.; Blotta, S.; MacNamara, C.; Somaiya, P.; Wu, Z.; Joyce, R.; Levine, J.D.; Dombagoda, D.; et al. Vaccination with dendritic cell/tumor fusion cells results in cellular and humoral antitumor immune responses in patients with multiple myeloma. Blood 2011, 117, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, A.P.; Aqui, N.A.; Stadtmauer, E.A.; Vogl, D.T.; Fang, H.B.; Cai, L.; Janofsky, S.; Chew, A.; Storek, J.; Akpek, G.; et al. Combination immunotherapy using adoptive T-cell transfer and tumor antigen vaccination on the basis of hTERT and survivin after ASCT for myeloma. Blood 2011, 117, 788–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenblatt, J.; Avivi, I.; Vasir, B.; Uhl, L.; Munshi, N.C.; Katz, T.; Dey, B.R.; Somaiya, P.; Mills, H.; Campigotto, F.; et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 3640–3648. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, A.P.; Aqui, N.A.; Stadtmauer, E.A.; Vogl, D.T.; Xu, Y.Y.; Kalos, M.; Cai, L.; Fang, H.B.; Weiss, B.M.; Badros, A.; et al. Combination immunotherapy after ASCT for multiple myeloma using MAGE-A3/Poly-ICLC immunizations followed by adoptive transfer of vaccine-primed and costimulated autologous T cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 1355–1365. [Google Scholar] [CrossRef] [Green Version]
- Nooka, A.K.; Wang, M.L.; Yee, A.J.; Kaufman, J.L.; Bae, J.; Peterkin, D.; Richardson, P.G.; Raje, N.S. Assessment of Safety and Immunogenicity of PVX-410 Vaccine With or Without Lenalidomide in Patients With Smoldering Multiple Myeloma: A Nonrandomized Clinical Trial. JAMA Oncol. 2018, 4, e183267. [Google Scholar] [CrossRef]
- Cohen, A.D.; Lendvai, N.; Nataraj, S.; Imai, N.; Jungbluth, A.A.; Tsakos, I.; Rahman, A.; Mei, A.H.; Singh, H.; Zarychta, K.; et al. Autologous Lymphocyte Infusion Supports Tumor Antigen Vaccine-Induced Immunity in Autologous Stem Cell Transplant for Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 658–669. [Google Scholar] [CrossRef]
- Deaglio, S.; Mehta, K.; Malavasi, F. Human CD38: A (r)evolutionary story of enzymes and receptors. Leuk Res. 2001, 25, 1–12. [Google Scholar] [CrossRef]
- Deaglio, S.; Vaisitti, T.; Billington, R.; Bergui, L.; Omede, P.; Genazzani, A.A.; Malavasi, F. CD38/CD19: A lipid raft-dependent signaling complex in human B cells. Blood 2007, 109, 5390–5398. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Donk, N.W.C.J.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Groen, R.W.; Noort, W.A.; van Kessel, B.; de Jong-Korlaar, R.; Bakker, J.; Van Bueren, J.J.; Parren, P.W.; Lokhorst, H.M.; Van De Donk, N.W.; et al. Preclinical Evidence for the Therapeutic Potential of CD38-Targeted Immuno-Chemotherapy in Multiple Myeloma Patients Refractory to Lenalidomide and Bortezomib. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2802–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Mateos, M.V.; Cavo, M.; Blade, J.; Dimopoulos, M.A.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): A randomised, open-label, phase 3 trial. Lancet 2020, 395, 132–141. [Google Scholar] [CrossRef]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef]
- Chari, A.; Suvannasankha, A.; Fay, J.W.; Arnulf, B.; Kaufman, J.L.; Ifthikharuddin, J.J.; Weiss, B.M.; Krishnan, A.; Lentzsch, S.; Comenzo, R.; et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood 2017, 130, 974–981. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Quach, H.; Mateos, M.V.; Landgren, O.; Leleu, X.; Siegel, D.; Weisel, K.; Yang, H.; Klippel, Z.; Zahlten-Kumeli, A.; et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): Results from a randomised, multicentre, open-label, phase 3 study. Lancet 2020, 396, 186–197. [Google Scholar] [CrossRef]
- Attal, M.; Richardson, P.G.; Rajkumar, S.V.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): A randomised, multicentre, open-label, phase 3 study. Lancet 2019, 394, 2096–2107. [Google Scholar] [CrossRef]
- De Weers, M.; Tai, Y.T.; Van Der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Verploegen, S.; Bögels, M.; van Egmond, M.; van Bueren, J.J.; Mutis, T.; Groen, R.W.; Breij, E.; Martens, A.C.; Bleeker, W.K.; et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. mAbs 2015, 7, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Franssen, L.E.; Stege, C.A.M.; Zweegman, S.; van de Donk, N.W.C.J.; Nijhof, I.S. Resistance Mechanisms Towards CD38-Directed Antibody Therapy in Multiple Myeloma. J. Clin. Med. 2020, 9, 1195. [Google Scholar] [CrossRef] [PubMed]
- Overdijk, M.B.; Jansen, J.M.; Nederend, M.; van Bueren, J.J.; Groen, R.W.; Parren, P.W.; Leusen, J.H.; Boross, P. The Therapeutic CD38 Monoclonal Antibody Daratumumab Induces Programmed Cell Death via Fcγ Receptor-Mediated Cross-Linking. J. Immunol. 2016, 197, 807–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitadate, A.; Kobayashi, H.; Abe, Y.; Narita, K.; Miura, D.; Takeuchi, M.; Matsue, K. Pre-treatment CD38-positive regulatory T cells affect the durable response to daratumumab in relapsed/refractory multiple myeloma patients. Haematologica 2020, 105, e37–e40. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Groen, R.W.; Lokhorst, H.M.; Van Kessel, B.; Bloem, A.C.; Van Velzen, J.; de Jong-Korlaar, R.; Yuan, H.; Noort, W.A.; Klein, S.K.; et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia 2015, 29, 2039–2049. [Google Scholar] [CrossRef]
- Fedele, P.L.; Willis, S.N.; Liao, Y.; Low, M.S.; Rautela, J.; Segal, D.H.; Gong, J.N.; Huntington, N.D.; Shi, W.; Huang, D.; et al. IMiDs prime myeloma cells for daratumumab-mediated cytotoxicity through loss of Ikaros and Aiolos. Blood 2018, 132, 2166–2178. [Google Scholar] [CrossRef] [Green Version]
- García-Guerrero, E.; Gogishvili, T.; Danhof, S.; Schreder, M.; Pallaud, C.; Pérez-Simón, J.A.; Einsele, H.; Hudecek, M. Panobinostat induces CD38 upregulation and augments the antimyeloma efficacy of daratumumab. Blood 2017, 129, 3386–3388. [Google Scholar] [CrossRef]
- García-Guerrero, E.; Götz, R.; Doose, S.; Sauer, M.; Rodríguez-Gil, A.; Nerreter, T.; Kortüm, K.M.; Pérez-Simón, J.A.; Einsele, H.; Hudecek, M.; et al. Upregulation of CD38 expression on multiple myeloma cells by novel HDAC6 inhibitors is a class effect and augments the efficacy of daratumumab. Leukemia 2020, 29, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Casneuf, T.; Van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barclay, A.N.; Van den Berg, T.K. The interaction between signal regulatory protein alpha (SIRPα) and CD47: Structure, function, and therapeutic target. Annu. Rev. Immunol. 2014, 32, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Iwamura, H.; Kaneko, T.; Ohnishi, H.; Murata, Y.; Okazawa, H.; Kanazawa, Y.; Sato-Hashimoto, M.; Kobayashi, H.; Oldenborg, P.A.; et al. Regulation by SIRPα of dendritic cell homeostasis in lymphoid tissues. Blood 2010, 116, 3517–3525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrisqueta, P.; Sancho, J.M.; Cordoba, R.; Persky, D.O.; Andreadis, C.; Huntington, S.F.; Carpio, C.; Morillo Giles, D.; Wei, X.; Li, Y.F.; et al. Anti-CD47 Antibody, CC-90002, in Combination with Rituximab in Subjects with Relapsed and/or Refractory Non-Hodgkin Lymphoma (R/R NHL). Blood 2019, 134 (Suppl. 1), 4089. [Google Scholar] [CrossRef]
- Naicker, S.; Rigalou, A.; McEllistrim, C.; Natoni, A.; Chiu, C.; Sasser, K.; Ryan, A.; O’Dwyer, M. Patient Data Supports the Rationale of Low Dose Cyclophosphamide to Potentiate the Anti-Myeloma Activity of Daratumumab through Augmentation of Macrophage-Induced ADCP. Blood 2017, 130 (Suppl. 1), 121. [Google Scholar]
- Rigalou, A.; Ryan, A.; Natoni, A.; Chiu, C.; Sasser, K.; O’Dwyer, M.E. Potentiation of Anti-Myeloma Activity of Daratumumab with Combination of Cyclophosphamide, Lenalidomide or Bortezomib Via a Tumor Secretory Response That Greatly Augments Macrophage-Induced ADCP. Blood 2016, 128, 2101. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- van de Donk, N.W.; Casneuf, T.; Di Cara, A.; Parren, P.W.; Zweegman, S.; van Kessel, B.; Lokhorst, H.M.; Usmani, S.Z.; Lonial, S.; Richardson, P.G.; et al. Impact of Fc gamma receptor polymorphisms on efficacy and safety of daratumumab in relapsed/refractory multiple myeloma. Br. J. Haematol. 2019, 184, 475–479. [Google Scholar] [CrossRef] [Green Version]
- Sanne, J.; van de Donk, N.W.; Minnema, M.C.; Huang, J.H.; Aarts-Riemens, T.; Bovenschen, N.; Yuan, H.; Groen, R.W.; McMillin, D.W.; Jakubikova, J.; et al. Accessory cells of the microenvironment protect multiple myeloma from T-cell cytotoxicity through cell adhesion-mediated immune resistance. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 5591–5601. [Google Scholar]
- Van Der Veer, M.S.; de Weers, M.; van Kessel, B.; Bakker, J.M.; Wittebol, S.; Parren, P.W.; Lokhorst, H.M.; Mutis, T. Towards effective immunotherapy of myeloma: Enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab. Haematologica 2011, 96, 284–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijhof, I.S.; van Bueren, J.J.; van Kessel, B.; Andre, P.; Morel, Y.; Lokhorst, H.M.; van de Donk, N.W.; Parren, P.W.; Mutis, T. Daratumumab-mediated lysis of primary multiple myeloma cells is enhanced in combination with the human anti-KIR antibody IPH2102 and lenalidomide. Haematologica 2015, 100, 263–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, Y.; Hughes, T.; Zhang, J.; Caligiuri, M.A.; Benson, D.M.; Yu, J. Fratricide of NK Cells in Daratumumab Therapy for Multiple Myeloma Overcome by Ex Vivo-Expanded Autologous NK Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4006–4017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannons, J.L.; Tangye, S.G.; Schwartzberg, P.L. SLAM family receptors and SAP adaptors in immunity. Annu. Rev. Immunol. 2011, 29, 665–705. [Google Scholar] [CrossRef]
- Collins, S.M.; Bakan, C.E.; Swartzel, G.D.; Hofmeister, C.C.; Efebera, Y.A.; Kwon, H.; Starling, G.C.; Ciarlariello, D.; Bhaskar, S.; Briercheck, E.L.; et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: Evidence for augmented NK cell function complementing ADCC. Cancer Immunol. Immunother. 2013, 62, 1841–1849. [Google Scholar] [CrossRef] [Green Version]
- Hartmut, G.; Mai, E.K.; Hans, S.; Uta, B.; Kaya, H.; Christina, K. Bortezomib, Lenalidomide and Dexamethasone with or Without Elotuzumab as Induction Therapy for Newly-Diagnosed, Transplant-Eligible Multiple Myeloma. 2020. Available online: https://library.ehaweb.org/eha/2020/eha25th/295023/hartmut.goldschmidt.bortezomib.lenalidomide.and.dexamethasone.with.or.without (accessed on 14 December 2020).
- Usmani, S.Z.; Ailawadhi, S.; Sexton, R.; Hoering, A.; Lipe, B.; Hita, S.; Durie, B.G.; Zonder, J.A.; Dhodapkar, M.V.; Callander, N.S.; et al. Primary analysis of the randomized phase II trial of bortezomib, lenalidomide, dexamthasone with/without elotuzumab for newly diagnosed, high-risk multiple myeloma (SWOG-1211). J. Clin. Oncol. 2020, 38 (Suppl. 15), 8507. [Google Scholar] [CrossRef]
- Bristol Myers Squibb Reports Primary Results of ELOQUENT-1 Study Evaluating Empliciti (Elotuzumab) Plus Revlimid (lenalidomide) and Dexamethasone in Patients with Newly Diagnosed, Untreated Multiple Myeloma. Available online: https://news.bms.com/news/corporate-financial/2020/Bristol-Myers-Squibb-Reports-Primary-Results-of-ELOQUENT-1-Study-Evaluating-Empliciti-elotuzumab-Plus-Revlimid-lenalidomide-and-Dexamethasone-in-Patients-with-Newly-Diagnosed-Untreated-Multiple-Myeloma/default.aspx (accessed on 14 December 2020).
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Lendvai, N.; Sborov, D. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Farooq, A.V.; Degli Esposti, S.; Popat, R.; Thulasi, P.; Lonial, S.; Nooka, A.K.; Jakubowiak, A.; Sborov, D.; Zaugg, B.E.; Badros, A.Z.; et al. Corneal Epithelial Findings in Patients with Multiple Myeloma Treated with Antibody-Drug Conjugate Belantamab Mafodotin in the Pivotal, Randomized, DREAMM-2 Study. Ophthalmol. Ther. 2020, 9, 889–911. [Google Scholar] [CrossRef]
- Sanderson, R.D.; Lalor, P.; Bernfield, M. B lymphocytes express and lose syndecan at specific stages of differentiation. Cell Regul. 1989, 1, 27–35. [Google Scholar] [CrossRef]
- Ishitsuka, K.; Jimi, S.; Goldmacher, V.S.; Ab, O.; Tamura, K. Targeting CD56 by the maytansinoid immunoconjugate IMGN901 (huN901-DM1): A potential therapeutic modality implication against natural killer/T cell malignancy. Br. J. Haematol. 2008, 141, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.; Mattes, M.J.; Cardillo, T.M.; Hansen, H.J.; Chang, C.H.; Burton, J.; Govindan, S.; Goldenberg, D.M. CD74: A new candidate target for the immunotherapy of B-cell neoplasms. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13 Pt 2, 5556s–5563s. [Google Scholar] [CrossRef] [Green Version]
- Zanwar, S.; Nandakumar, B.; Kumar, S. Immune-based therapies in the management of multiple myeloma. Blood Cancer, J. 2020, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Kufer, P.; Bargou, R. BiTE: Teaching antibodies to engage T-cells for cancer therapy. Curr. Opin. Mol. Ther. 2009, 11, 22–30. [Google Scholar] [PubMed]
- Velasquez, M.P.; Bonifant, C.L.; Gottschalk SRedirecting, T. cells to hematological malignancies with bispecific antibodies. Blood 2018, 131, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, N. Cancer therapy using bispecific antibodies. Rinsho Ketsueki. 2018, 59, 1942–1947. [Google Scholar]
- Loffler, A.; Kufer, P.; Lutterbuse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmuller, G.; Dorken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar] [CrossRef]
- Gökbuget, N.; Dombret, H.; Bonifacio, M.; Reichle, A.; Graux, C.; Faul, C.; Diedrich, H.; Topp, M.S.; Brüggemann, M.; Horst, H.A.; et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 2018, 131, 1522–1531. [Google Scholar] [CrossRef] [Green Version]
- Goyos, A.; Li, C.M.; Deegen, P.; Bogner, P.; Thomas, O.; Wahl, J.; Goldstein, R.; Friedrich, M.; Coxon, A.; Balazs, M.; et al. Abstract LB-299: Cynomolgus monkey plasma cell gene signature to quantify the in vivo activity of a half-life extended anti-BCMA BiTE® for the treatment of multiple myeloma. Cancer Res. 2018, 78 (Suppl. 13). [Google Scholar] [CrossRef]
- Cho, S.F.; Lin, L.; Xing, L.; Liu, J.; Yu, T.; Wen, K.; Hsieh, P.; Munshi, N.; Anderson, K.; Tai, Y.T. Anti-BCMA BiTE® AMG 701 Potently Induces Specific T Cell Lysis of Human Multiple Myeloma (MM) Cells and Immunomodulation in the Bone Marrow Microenvironment. Blood 2018, 132 (Suppl. 1), 592. [Google Scholar] [CrossRef]
- Pillarisetti, K.; Powers, G.; Luistro, L.; Babich, A.; Baldwin, E.; Li, Y.; Zhang, X.; Mendonça, M.; Majewski, N.; Nanjunda, R.; et al. Teclistamab is an active T cell-redirecting bispecific antibody against B-cell maturation antigen for multiple myeloma. Blood Adv. 2020, 4, 4538–4549. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.J.; Wong, S.W.; Bermúdez, A.; de la Rubia, J.; Mateos, M.V.; Ocio, E.M.; Rodríguez-Otero, P.; San-Miguel, J.; Li, S.; Sarmiento, R.; et al. Interim Results from the First Phase 1 Clinical Study of the B-cell Maturation Antigen (BCMA) 2+1 T Cell Engager (TCE) CC-93269 in Patients (PTS) with Relapsed/Refractory Multiple Myeloma (RRMM). Available online: https://library.ehaweb.org/eha/2020/eha25th/295025/luciano.j.costa.interim.results.from.the.first.phase.1.clinical.study.of.the (accessed on 14 December 2020).
- Richter, J.R.; Landgren, C.O.; Kauh, J.S.; Back, J.; Salhi, Y.; Reddy, V.; Bayever, E.; Berdej, A. Phase 1, multicenter, open-label study of single-agent bispecific antibody t-cell engager GBR 1342 in relapsed/refractory multiple myeloma. J. Clin. Oncol. 2018, 36 (Suppl. 15), TPS3132. [Google Scholar] [CrossRef]
- Cohen, A.D.; Harrison, S.J.; Krishnan, A.; Fonseca, R.; Forsberg, P.A.; Spencer, A. Initial Clinical Activity and Safety of BFCR4350A, a FcRH5/CD3 T-Cell-Engaging Bispecific Antibody, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136 (Suppl. 1), 42–43. [Google Scholar] [CrossRef]
- Chari, A.; Berdeja, J.G.; Oriol, A.; van de Donk, N.W.C.J.; Rodriguez, P.; Askari, E. A Phase 1, First-in-Human Study of Talquetamab, a G Protein-Coupled Receptor Family C Group 5 Member D (GPRC5D) x CD3 Bispecific Antibody, in Patients with Relapsed and/or Refractory Multiple Myeloma (RRMM). Blood 2020, 136 (Suppl. 1), 40–41. [Google Scholar] [CrossRef]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer 2020, 1, 86–98. [Google Scholar] [CrossRef]
- Lancman, G.; Richter, J.; Chari, A. Bispecifics, trispecifics, and other novel immune treatments in myeloma. Hematology 2020, 2020, 264–271. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [Green Version]
- Salmaninejad, A.; Valilou, S.F.; Shabgah, A.G.; Aslani, S.; Alimardani, M.; Pasdar, A.; Sahebkar, A. PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy. J. Cell Physiol. 2019, 234, 16824–16837. [Google Scholar] [CrossRef]
- Costa, F.; Das, R.; Kini Bailur, J.; Dhodapkar, K.; Dhodapkar, M.V. Checkpoint Inhibition in Myeloma: Opportunities and Challenges. Front. Immunol. 2018, 9, 2204. [Google Scholar] [CrossRef] [Green Version]
- Dyck, L.; Mills, K.H.G. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur. J. Immunol. 2017, 47, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Pratt, G.; Goodyear, O.; Moss, P. Immunodeficiency and immunotherapy in multiple myeloma. Br. J. Haematol. 2007, 138, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Ratta, M.; Fagnoni, F.; Curti, A.; Vescovini, R.; Sansoni, P.; Oliviero, B.; Fogli, M.; Ferri, E.; Della Cuna, G.R.; Tura, S.; et al. cells are functionally defective in multiple myeloma: The role of interleukin-6. Blood 2002, 100, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Bahlis, N.J.; King, A.M.; Kolonias, D.; Carlson, L.M.; Liu, H.Y.; Hussein, M.A.; Terebelo, H.R.; Byrne, G.E.; Levine, B.L.; Boise, L.H.; et al. CD28-mediated regulation of multiple myeloma cell proliferation and survival. Blood 2007, 109, 5002–5010. [Google Scholar] [CrossRef] [Green Version]
- Rosenblatt, J.; Avigan, D. Targeting the PD-1/PD-L1 axis in multiple myeloma: A dream or a reality? Blood 2017, 129, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Paiva, B.; Azpilikueta, A.; Puig, N.; Ocio, E.M.; Sharma, R.; Oyajobi, B.O.; Labiano, S.; San-Segundo, L.; Rodriguez, A.; Aires-Mejia, I.; et al. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia 2015, 29, 2110–2113. [Google Scholar] [CrossRef]
- Lozano, E.; Díaz, T.; Mena, M.P.; Suñe, G.; Calvo, X.; Calderón, M.; Pérez-Amill, L.; Rodríguez, V.; Pérez-Galán, P.; Roué, G.; et al. Loss of the Immune Checkpoint CD85j/LILRB1 on Malignant Plasma Cells Contributes to Immune Escape in Multiple Myeloma. J. Immunol. 2018, 200, 2581–2591. [Google Scholar] [CrossRef]
- Guillerey, C.; Harjunpää, H.; Carrié, N.; Kassem, S.; Teo, T.; Miles, K.; Krumeich, S.; Weulersse, M.; Cuisinier, M.; Stannard, K.; et al. TIGIT immune checkpoint blockade restores CD8+ T-cell immunity against multiple myeloma. Blood 2018, 132, 1689–1694. [Google Scholar] [CrossRef] [Green Version]
- Ribrag, V.; Avigan, D.E.; Green, D.J.; Wise-Draper, T.; Posada, J.G.; Vij, R.; Zhu, Y.; Farooqui, M.Z.; Marinello, P.; Siegel, D.S. Phase 1b trial of pembrolizumab monotherapy for relapsed/refractory multiple myeloma: KEYNOTE-013. Br. J. Haematol. 2019, 186, e41–e44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usmani, S.Z.; Schjesvold, F.; Oriol, A.; Karlin, L.; Cavo, M.; Rifkin, R.M.; Yimer, H.A.; LeBlanc, R.; Takezako, N.; McCroskey, R.D.; et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e448–e458. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelinek, T.; Paiva, B.; Hajek, R. Update on PD-1/PD-L1 Inhibitors in Multiple Myeloma. Front. Immunol. 2018, 9, 2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Sadelain, M.; Rivière, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. Yescarta. European Medicines Agency. 2018. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/yescarta (accessed on 22 July 2020).
- FDA. Research C for BE and YESCARTA (Axicabtagene Ciloleucel). Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/yescarta-axicabtagene-ciloleucel (accessed on 22 July 2020).
- Anonymous. Kymriah. European Medicines Agency. 2018. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/kymriah (accessed on 22 July 2020).
- FDA. Research C for BE and KYMRIAH (Tisagenlecleucel). Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/kymriah-tisagenlecleucel (accessed on 22 July 2020).
- Gagelmann, N.; Riecken, K.; Wolschke, C.; Berger, C.; Ayuk, F.A.; Fehse, B.; Kröger, N. Development of CAR-T cell therapies for multiple myeloma. Leukemia 2020. [Google Scholar] [CrossRef] [PubMed]
- Sellner, L.; Fan, F.; Giesen, N.; Schubert, M.L.; Goldschmidt, H.; Müller-Tidow, C.; Dreger, P.; Raab, M.S.; Schmitt, M. B-cell maturation antigen-specific chimeric antigen receptor T cells for multiple myeloma: Clinical experience and future perspectives. Int. J. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Sidana, S.; Shah, N. CAR T-cell therapy: Is it prime time in myeloma? Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 260–265. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, M.; Raje, N. Anti-BCMA CAR T-cell therapy in multiple myeloma: Can we do better? Leukemia 2020, 34, 21–34. [Google Scholar] [CrossRef]
- Rodríguez-Lobato, L.G.; Ganzetti, M.; Fernández de Larrea, C.; Hudecek, M.; Einsele, H.; Danhof, S. CAR T-Cells in Multiple Myeloma: State of the Art and Future Directions. Front. Oncol. Available online: https://www.frontiersin.org/articles/10.3389/fonc.2020.01243/full (accessed on 30 July 2020).
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [Green Version]
- Madry, C.; Laabi, Y.; Callebaut, I.; Roussel, J.; Hatzoglou, A.; Le Coniat, M.; Mornon, J.P.; Berger, R.; Tsapis, A. The characterization of murine BCMA gene defines it as a new member of the tumor necrosis factor receptor superfamily. Int. Immunol. 1998, 10, 1693–1702. [Google Scholar] [CrossRef]
- Ng, L.G.; Mackay, C.R.; Mackay, F. The BAFF/APRIL system: Life beyond B lymphocytes. Mol. Immunol. 2005, 42, 763–772. [Google Scholar] [CrossRef]
- Moreaux, J.; Legouffe, E.; Jourdan, E.; Quittet, P.; Rème, T.; Lugagne, C.; Moine, P.; Rossi, J.F.; Klein, B.; Tarte, K. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood 2004, 103, 3148–3157. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.Y.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; van Eenennaam, H.; et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016, 127, 3225–3236. [Google Scholar] [CrossRef] [Green Version]
- Novak, A.J.; Darce, J.R.; Arendt, B.K.; Harder, B.; Henderson, K.; Kindsvogel, W.; Gross, J.A.; Greipp, P.R.; Jelinek, D.F. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: A mechanism for growth and survival. Blood 2004, 103, 689–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective Targeting of Multiple B-Cell Maturation Antigen-Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdeja, J.G.; Alsina, M.; Shah, N.D.; Siegel, D.S.; Jagannath, S.; Madduri, D.; Kaufman, J.L.; Munshi, N.C.; Rosenblatt, J.; Jasielec, J.K.; et al. Updated Results from an Ongoing Phase 1 Clinical Study of bb21217 Anti-Bcma CAR T Cell Therapy. Blood 2019, 134 (Suppl. 1), 927. [Google Scholar] [CrossRef]
- Mailankody, S.; Htut, M.; Lee, K.P.; Bensinger, W.; Devries, T.; Piasecki, J.; Ziyad, S.; Blake, M.; Byon, J.; Jakubowiak, A. JCARH125, Anti-BCMA CAR T-cell Therapy for Relapsed/Refractory Multiple Myeloma: Initial Proof of Concept Results from a Phase 1/2 Multicenter Study (EVOLVE). Blood 2018, 132 (Suppl. 1), 957. [Google Scholar] [CrossRef]
- Costello, C.L.; Gregory, T.K.; Ali, S.A.; Berdeja, J.G.; Patel, K.K.; Shah, N.D.; Ostertag, E.; Martin, C.; Ghoddusi, M.; Shedlock, D.J.; et al. Phase 2 Study of the Response and Safety of P-Bcma-101 CAR-T Cells in Patients with Relapsed/Refractory (r/r) Multiple Myeloma (MM) (PRIME). Blood 2019, 134 (Suppl. 1), 3184. [Google Scholar] [CrossRef]
- Green, D.J.; Pont, M.; Sather, B.D.; Cowan, A.J.; Turtle, C.J.; Till, B.G.; Nagengast, A.M.; Libby, E.N.; Becker, P.S.; Coffey, D.G.; et al. Fully Human Bcma Targeted Chimeric Antigen Receptor T Cells Administered in a Defined Composition Demonstrate Potency at Low Doses in Advanced Stage High Risk Multiple Myeloma. Blood 2018, 132 (Suppl. 1), 1011. [Google Scholar] [CrossRef]
- Mailankody, S.; Ghosh, A.; Staehr, M.; Purdon, T.J.; Roshal, M.; Halton, E.; Diamonte, C.; Pineda, J.; Anant, P.; Bernal, Y.; et al. Clinical Responses and Pharmacokinetics of MCARH171, a Human-Derived Bcma Targeted CAR T Cell Therapy in Relapsed/Refractory Multiple Myeloma: Final Results of a Phase I Clinical Trial. Blood 2018, 132 (Suppl. 1), 959. [Google Scholar] [CrossRef]
- Gagelmann, N.; Ayuk, F.; Atanackovic, D.; Kröger, N. B cell maturation antigen-specific chimeric antigen receptor T cells for relapsed or refractory multiple myeloma: A meta-analysis. Eur. J. Haematol. 2020, 104, 318–327. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Cao, J.; Wang, G.; Cheng, H.; Wei, C.; Qi, K.; Sang, W.; Zhenyu, L.; Shi, M.; Li, H.; Qiao, J.; et al. Potent anti-leukemia activities of humanized CD19-targeted Chimeric antigen receptor T (CAR-T) cells in patients with relapsed/refractory acute lymphoblastic leukemia. Am. J. Hematol. 2018, 93, 851–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, N.; Trinklein, N.D.; Buelow, B.; Patterson, G.H.; Ojha, N.; Kochenderfer, J.N. Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nat. Commun. 2020, 11, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wang, J.; Wang, D.; Hu, G.; Yang, Y.; Zhou, X.; Meng, L.; Hong, Z.; Chen, L.; Mao, X.; et al. Efficacy and Safety of Fully Human Bcma Targeting CAR T Cell Therapy in Relapsed/Refractory Multiple Myeloma. Blood 2019, 134 (Suppl. 1), 929. [Google Scholar] [CrossRef]
- Jie, J.; Hao, S.; Jiang, S.; Li, Z.; Yang, M.; Zhang, W.; Yu, K.; Xiao, J.; Meng, H.; Ma, L.; et al. Phase 1 Trial of the Safety and Efficacy of Fully Human Anti-Bcma CAR T Cells in Relapsed/Refractory Multiple Myeloma. Blood 2019, 134 (Suppl. 1), 4435. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. 2018, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Brocker, T.; Karjalainen, K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J. Exp. Med. 1995, 181, 1653–1659. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 712. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Palomba, M.L.; Batlevi, C.L.; Riviere, I.; Wang, X.; Senechal, B.; Furman, R.R.; Bernal, Y.; Hall, M.; Pineda, J.; et al. A Phase I First-in-Human Clinical Trial of CD19-Targeted 19-28z/4-1BBL Armored CAR T Cells in Patients with Relapsed or Refractory NHL and CLL Including Richter’s Transformation. Blood 2018, 132 (Suppl.1), 224. [Google Scholar] [CrossRef]
- Boucher, J.C.; Li, G.; Shrestha, B.; Zhang, Y.; Vishwasrao, P.; Cabral, M.L.; Guan, L.; Davila, M.L. Mutation of the CD28 costimulatory domain confers increased CAR T cell persistence and decreased exhaustion. J. Immunol. 2018, 200 (Suppl. 1), 57.28. [Google Scholar]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Madar, A.; Casado-Medrano, V.; Shaw, C.; Wing, A.; Liu, F.; Young, R.M.; June, C.H.; Posey, A.D. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J. Clin. Investig. 2020, 130, 3087–3097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, D.-G.; Ye, Q.; Poussin, M.; Harms, G.M.; Figini, M.; Powell, D.J. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 2012, 119, 696–706. [Google Scholar] [CrossRef]
- Guedan, S.; Chen, X.; Madar, A.; Carpenito, C.; McGettigan, S.E.; Frigault, M.J.; Lee, J.; Posey, A.D.; Scholler, J.; Scholler, N.; et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 2014, 124, 1070–1080. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Pruteanu, I.; Cohen, A.D.; Garfall, A.L.; Milone, M.C.; Tian, L.; Gonzalez, V.E.; Gill, S.; Frey, N.V.; Barrett, D.M.; et al. Identification and Validation of Predictive Biomarkers to CD19- and BCMA-Specific CAR T-Cell Responses in CAR T-Cell Precursors. Blood 2019, 134 (Suppl. 1), 622. [Google Scholar] [CrossRef]
- Busch, D.H.; Fräßle, S.P.; Sommermeyer, D.; Buchholz, V.R.; Riddell, S.R. Role of memory T cell subsets for adoptive immunotherapy. Semin Immunol. 2016, 28, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Kotani, H.; Li, G.; Yao, J.; Mesa, T.E.; Chen, J.; Boucher, J.C.; Yoder, S.J.; Zhou, J.; Davila, M.L. Aged CAR T Cells Exhibit Enhanced Cytotoxicity and Effector Function but Shorter Persistence and Less Memory-like Phenotypes. Blood 2018, 132 (Suppl. 1), 2047. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Perazzelli, J.; Grupp, S.A.; Barrett, D.M. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci. Transl. Med. 2016, 8, 320ra3. [Google Scholar] [CrossRef]
- Schubert, M.-L.; Hoffmann, J.-M.; Dreger, P.; Müller-Tidow, C.; Schmitt, M. Chimeric antigen receptor transduced T cells: Tuning up for the next generation. Int. J. Cancer 2018, 142, 1738–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suen, H.; Brown, R.; Yang, S.; Weatherburn, C.; Ho, P.J.; Woodland, N.; Nassif, N.; Barbaro, P.; Bryant, C.; Hart, D.; et al. Multiple myeloma causes clonal T-cell immunosenescence: Identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia 2016, 30, 1716–1724. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Dancy, E.K.; Cohen, A.D.; Hwang, W.T.; Fraietta, J.A.; Davis, M.M.; Levine, B.L.; Siegel, D.L.; Stadtmauer, E.A.; Vogl, D.T.; et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv. 2019, 3, 2812–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Carol, E.O.; Alli, R.; Basham, J.H.; Abdelsamed, H.A.; Palmer, L.E.; Jones, L.L.; Youngblood, B.; Geiger, T.L. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia 2018, 32, 1157–1167. [Google Scholar] [CrossRef]
- Zhou, J.; Jin, L.; Wang, F.; Zhang, Y.; Liu, B.; Zhao, T. Chimeric antigen receptor T (CAR-T) cells expanded with IL-7/IL-15 mediate superior antitumor effects. Protein Cell 2019, 10, 764–769. [Google Scholar] [CrossRef] [Green Version]
- Ajina, A.; Maher, J. Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol. Cancer Ther. 2018, 17, 1795–1815. [Google Scholar] [CrossRef] [Green Version]
- Calderon, H.; Mamonkin, M.; Guedan, S. Analysis of CAR-Mediated Tonic Signaling. In Chimeric Antigen Receptor T Cells; Humana: New York, NY, USA, 2020. [Google Scholar]
- Hermanson, D.L.; Barnett, B.E.; Rengarajan, S.; Codde, R.; Wang, X.; Tan, Y.; Martin, C.E.; Smith, J.B.; He, J.; Mathur, R.; et al. A Novel Bcma-Specific, Centyrin-Based CAR-T Product for the Treatment of Multiple Myeloma. Blood 2016, 128, 2127. [Google Scholar] [CrossRef]
- Watanabe, N.; Bajgain, P.; Sukumaran, S.; Ansari, S.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology 2016, 5, e1253656. [Google Scholar] [CrossRef] [Green Version]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.L.; Staehr, M.; Masakayan, R.; Tatake, I.J.; Purdon, T.J.; Wang, X.; Wang, P.; Liu, H.; Xu, Y.; Garrett-Thomson, S.C.; et al. Development and Evaluation of an Optimal Human Single-Chain Variable Fragment-Derived BCMA-Targeted CAR T Cell Vector. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1447–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining “T cell exhaustion”. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of Immune Checkpoint Blockade into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Hucks, G.E.; Seif, A.E.; Talekar, M.K.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Gonzalez, V.; Nazimuddin, F.; Gupta, M.; et al. The effect of pembrolizumab in combination with CD19-targeted chimeric antigen receptor (CAR) T cells in relapsed acute lymphoblastic leukemia (ALL). J. Clin. Oncol. 2017, 35 (Suppl. 15), 103. [Google Scholar] [CrossRef]
- Li, A.M.; Hucks, G.E.; Dinofia, A.M.; Seif, A.E.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Fasano, C.; McBride, B.; Gonzalez, V.; et al. Checkpoint Inhibitors Augment CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy in Relapsed B-Cell Acute Lymphoblastic. Leuk. Blood 2018, 132 (Suppl. 1), 556. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Chun, J.Y.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosser, M.E.; Brown, C.E.; Shami, A.F.; Forman, S.J.; Jensen, M.C. Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol. Immunol. 2012, 51, 263–272. [Google Scholar] [CrossRef]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Luo, C.; Wang, Y.; Guo, Y.; Dai, H.; Tong, C.; Ti, D.; Wu, Z.; Han, W. PD-1 silencing impairs the anti-tumor function of chimeric antigen receptor modified T cells by inhibiting proliferation activity. J Immunother. Cancer 2019, 7, 209. [Google Scholar] [CrossRef]
- Zah, E.; Nam, E.; Bhuvan, V.; Tran, U.; Ji, B.Y.; Gosliner, S.B.; Wang, X.; Brown, C.E.; Chen, Y.Y. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat. Commun. 2020, 11, 2283. [Google Scholar] [CrossRef]
- Minnie, S.A.; Hill, G.R. Immunotherapy of multiple myeloma. J. Clin. Investig. 2020, 130, 1565–1575. [Google Scholar] [CrossRef]
- García-Guerrero, E.; Sierro-Martínez, B.; Pérez-Simón, J.A. Overcoming Chimeric Antigen Receptor (CAR) Modified T-Cell Therapy Limitations in Multiple Myeloma. Front. Immunol. 2020, 11, 1128. [Google Scholar] [CrossRef]
- Laurent, S.A.; Hoffmann, F.S.; Kuhn, P.H.; Cheng, Q.; Chu, Y.; Schmidt-Supprian, M.; Hauck, S.M.; Schuh, E.; Krumbholz, M.; Rübsamen, H.; et al. γ-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat. Commun. 2015, 6, 7333. [Google Scholar] [CrossRef]
- Sanchez, E.; Li, M.; Kitto, A.; Li, J.; Wang, C.S.; Kirk, D.T.; Yellin, O.; Nichols, C.M.; Dreyer, M.P.; Ahles, C.P.; et al. B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br. J. Haematol. 2012, 158, 727–738. [Google Scholar] [CrossRef]
- Ghermezi, M.; Li, M.; Vardanyan, S.; Harutyunyan, N.M.; Gottlieb, J.; Berenson, A.; Spektor, T.M.; Andreu-Vieyra, C.; Petraki, S.; Sanchez, E.; et al. Serum B-cell maturation antigen: A novel biomarker to predict outcomes for multiple myeloma patients. Haematologica 2017, 102, 785–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujarski, S.; Soof, C.; Chen, H.; Li, M.; Sanchez, E.; Wang, C.S.; Emamy-Sadr, M.; Swift, R.A.; Rahbari, K.J.; Patil, S.; et al. Serum b-cell maturation antigen levels to predict progression free survival and responses among relapsed or refractory multiple myeloma patients treated on the phase I IRUX trial. J. Clin. Oncol. 2018, 36 (Suppl. 15), e24313. [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.; et al. γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef] [PubMed]
- Holthof, L.C.; Mutis, T. Challenges for Immunotherapy in Multiple Myeloma: Bone Marrow Microenvironment-Mediated Immune Suppression and Immune Resistance. Cancers 2020, 12, 988. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.S.Y.; Taylor, J.R.; Farzaneh, F.; Kemeny, D.M.; Dibb, N.J.; Maher, J. Harnessing the tumour-derived cytokine, CSF-1, to co-stimulate T-cell growth and activation. Mol. Immunol. 2008, 45, 1276–1287. [Google Scholar] [CrossRef]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef] [Green Version]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Chinnasamy, D.; Yu, Z.; Morgan, R.A.; Lee, C.C.; Restifo, N.P.; Rosenberg, S.A. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 2013, 210, 1125–1135. [Google Scholar] [CrossRef]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leen, A.M.; Sukumaran, S.; Watanabe, N.; Mohammed, S.; Keirnan, J.; Yanagisawa, R.; Anurathapan, U.; Rendon, D.; Heslop, H.E.; Rooney, C.M.; et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1211–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkie, S.; Burbridge, S.E.; Chiapero-Stanke, L.; Pereira, A.C.; Cleary, S.; van der Stegen, S.J.; Spicer, J.F.; Davies, D.M.; Maher, J. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J. Biol. Chem. 2010, 285, 25538–25544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, S.; Sukumaran, S.; Bajgain, P.; Watanabe, N.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Fisher, W.E.; Leen, A.M.; Vera, J.F. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.N.; Lee, P.H.; Vodnala, S.K.; Gurusamy, D.; Kishton, R.J.; Yu, Z.; Eidizadeh, A.; Eil, R.; Fioravanti, J.; Gattinoni, L.; et al. T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J. Clin. Investig. 2019, 129, 1551–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruella, M.; Klichinsky, M.; Kenderian, S.S.; Shestova, O.; Ziober, A.; Kraft, D.O.; Feldman, M.; Wasik, M.A.; June, C.H.; Gill, S. Overcoming the Immunosuppressive Tumor Microenvironment of Hodgkin Lymphoma Using Chimeric Antigen Receptor T Cells. Cancer Discov. 2017, 7, 1154–1167. [Google Scholar] [CrossRef] [Green Version]
- Parihar, R.; Rivas, C.; Huynh, M.; Omer, B.; Lapteva, N.; Metelitsa, L.S.; Gottschalk, S.M.; Rooney, C.M. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors. Cancer Immunol. Res. 2019, 7, 363–375. [Google Scholar] [CrossRef]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef] [Green Version]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bethigh FoxO1low Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wei, Q.; Brzostek, J.; Gascoigne, N.R.J. Signaling from T cell receptors (TCRs) and chimeric antigen receptors (CARs) on T cells. Cell Mol. Immunol. 2020, 17, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, I.; Olivera, I.; Bolaños, E.; Cirella, A.; Teijeira, Á.; Berraondo, P.; Melero, I. Engineering bionic T cells: Signal 1, signal 2, signal 3, reprogramming and the removal of inhibitory mechanisms. Cell Mol. Immunol. 2020, 17, 576–586. [Google Scholar] [CrossRef]
- Gattinoni, L.; Finkelstein, S.E.; Klebanoff, C.A.; Antony, P.A.; Palmer, D.C.; Spiess, P.J.; Hwang, L.N.; Yu, Z.; Wrzesinski, C.; Heimann, D.M.; et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005, 202, 907–912. [Google Scholar] [CrossRef]
- Corrigan-Curay, J.; Kiem, H.P.; Baltimore, D.; O’reilly, M.; Brentjens, R.J.; Cooper, L.; Forman, S.; Gottschalk, S.; Greenberg, P.; Junghans, R.; et al. T-cell immunotherapy: Looking forward. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1564–1574. [Google Scholar] [CrossRef] [Green Version]
- Antony, P.A.; Piccirillo, C.A.; Akpinarli, A.; Finkelstein, S.E.; Speiss, P.J.; Surman, D.R.; Palmer, D.C.; Chan, C.C.; Klebanoff, C.A.; Overwijk, W.W.; et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J. Immunol. 2005, 174, 2591–2601. [Google Scholar] [CrossRef] [Green Version]
- Neelapu, S.S. CAR-T efficacy: Is conditioning the key? Blood 2019, 133, 1799–1800. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Gauthier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Gooley, T.; Li, D.; Cherian, S.; Chen, X.; Pender, B.S.; et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood 2019, 133, 1876–1887. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Geyer, M.B.; Brentjens, R.J. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: Interpreting clinical outcomes to date. Blood 2016, 127, 3312–3320. [Google Scholar] [CrossRef]
- Zhang, T.; Cao, L.; Xie, J.; Shi, N.; Zhang, Z.; Luo, Z.; Yue, D.; Zhang, Z.; Wang, L.; Han, W.; et al. Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell malignancies in phase I clinical trials: A meta-analysis. Oncotarget 2015, 6, 33961–33971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Martin, N.; Thompson, E.; Dell’Aringa, J.; Paiva, B.; Munshi, N.; San Miguel, J. Correlation of Tumor BCMA Expression with Response and Acquired Resistance to Idecabtagene Vicleucel in the KarMMa Study in Relapsed and Refractory Multiple Myeloma. 2020. Available online: https://library.ehaweb.org/eha/2020/eha25th/294902/nathan.martin.correlation.of.tumor.bcma.expression.with.response.and.acquired.html?f=listing%3D3%2Abrowseby%3D8%2Asortby%3D1%2Amedia%3D1 (accessed on 8 August 2020).
- Da Via, M.C.; Dietrich, O.; Truger, M.; Arampatzi, P.; Duell, J.; Zhou, X.; Tabares, P.; Danhof, S.; Kraus, S.; Meggendorfer, M. Biallelic Deletion of Chromosome 16p Encompassing the BCMA Locus as a Tumor-Intrinsic Resistance Mechanism to BCMA-Directed CAR T in Multiple Myeloma. 2020. Available online: https://library.ehaweb.org/eha/2020/eha25th/294800/leo.rasche.biallelic.deletion.of.chromosome.16p.encompassing.the.bcma.locus.as.html?f=listing%3D0%2Abrowseby%3D8%2Asortby%3D2%2Asearch%3Dcar-t (accessed on 8 August 2020).
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, J.; Naqvi, A.S.; Luo, M.; Wertheim, G.; Paessler, M.; Thomas-Tikhonenko, A.; Rheingold, S.R.; Pillai, V. Heterogeneity of surface CD19 and CD22 expression in B lymphoblastic leukemia. Am. J. Hematol. 2018, 93, E352–E355. [Google Scholar] [CrossRef] [Green Version]
- Schürch, C.M.; Rasche, L.; Frauenfeld, L.; Weinhold, N.; Fend, F. A review on tumor heterogeneity and evolution in multiple myeloma: Pathological, radiological, molecular genetics, and clinical integration. Virchows Arch. Int. J. Pathol. 2020, 476, 337–351. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef]
- Fischer, J.; Paret, C.; El Malki, K.; Alt, F.; Wingerter, A.; Neu, M.A.; Kron, B.; Russo, A.; Lehmann, N.; Roth, L.; et al. CD19 Isoforms Enabling Resistance to CART-19 Immunotherapy Are Expressed in B-ALL Patients at Initial Diagnosis. J. Immunother. 2017, 40, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacoby, E.; Nguyen, S.M.; Fountaine, T.J.; Welp, K.; Gryder, B.; Qin, H.; Yang, Y.; Chien, C.D.; Seif, A.E.; Lei, H.; et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat. Commun. 2016, 7, 12320. [Google Scholar] [CrossRef] [PubMed]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; van der Stegen, S.J.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef]
- Perez-Amill, L.; Suñe, G.; Antoñana-Vildosola, A.; Castella, M.; Najjar, A.; Bonet, J.; Fernández-Fuentes, N.; Inogés, S.; López, A.; Bueno, C.; et al. Preclinical development of a humanized chimeric antigen receptor against B cell maturation antigen for multiple myeloma. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.; Barrett, D.M. Current status of chimeric antigen receptor therapy for haematological malignancies. Br. J. Haematol. 2016, 172, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Tamura, H.; Ishibashi, M.; Sunakawa, M.; Inokuchi, K. Immunotherapy for Multiple Myeloma. Cancers 2019, 11, 2009. [Google Scholar] [CrossRef] [Green Version]
- Abbott, R.C.; Cross, R.S.; Jenkins, M.R. Finding the Keys to the CAR: Identifying Novel Target Antigens for T Cell Redirection Immunotherapies. Int. J. Mol. Sci. 2020, 21, 515. [Google Scholar] [CrossRef] [Green Version]
- Hosen, N. Chimeric Antigen Receptor T-Cell Therapy for Multiple Myeloma. Cancers 2019, 11, 2024. [Google Scholar] [CrossRef] [Green Version]
- Guedan, S.; Calderon, H.; Posey, A.D.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Zhang, J.; Li, M.; Xu, N.; Qi, W.; Tan, J.; Lou, X.; Yu, Z.; Sun, J.; Wang, Z.; et al. Characterization of novel dual tandem CD19/BCMA chimeric antigen receptor T cells to potentially treat multiple myeloma. Biomark Res. 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Wada, M.; Pinz, K.G.; Liu, H.; Shuai, X.; Chen, X.; Yan, L.E.; Petrov, J.C.; Salman, H.; Senzel, L.; et al. A compound chimeric antigen receptor strategy for targeting multiple myeloma. Leukemia 2018, 32, 402–412. [Google Scholar] [CrossRef] [PubMed]
- de Larrea, C.F.; Staehr, M.; Lopez, A.V.; Ng, K.Y.; Chen, Y.; Godfrey, W.D.; Purdon, T.J.; Ponomarev, V.; Wendel, H.G.; Brentjens, R.J.; et al. Defining an Optimal Dual-Targeted CAR T-cell Therapy Approach Simultaneously Targeting BCMA and GPRC5D to Prevent BCMA Escape–Driven Relapse in Multiple Myeloma. Blood Cancer Discov. 2020. Available online: https://bloodcancerdiscov.aacrjournals.org/content/early/2020/06/30/2643-3230.BCD-20-0020 (accessed on 9 August 2020).
- Popat, R.; Zweegman, S.; Cavet, J.; Yong, K.; Lee, L.; Faulkner, J.; Kotsopoulou, E.; Al-Hajj, M.; Thomas, S.; Cordoba, S.P.; et al. Phase 1 First-in-Human Study of AUTO2, the First Chimeric Antigen Receptor (CAR) T Cell Targeting APRIL for Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2019, 134 (Suppl. 1), 3112. [Google Scholar] [CrossRef]
- Li, C.; Mei, H.; Hu, Y.; Guo, T.; Liu, L.; Jiang, H.; Tang, L.; Wu, Y.; Ai, L.; Deng, J.; et al. A Bispecific CAR-T Cell Therapy Targeting Bcma and CD38 for Relapsed/Refractory Multiple Myeloma: Updated Results from a Phase 1 Dose-Climbing Trial. Blood 2019, 134 (Suppl. 1), 930. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, L.; Liu, L.; Wang, J.; Wang, S.; Gao, L.; Zhang, C.; Liu, Y.; Kong, P.; Liu, J.; et al. A Bcma and CD19 Bispecific CAR-T for Relapsed and Refractory Multiple Myeloma. Blood 2019, 134 (Suppl. 1), 3147. [Google Scholar] [CrossRef]
- Yan, L.; Yan, Z.; Shang, J.; Shi, X.; Jin, S.; Kang, L.; Qu, S.; Zhou, J.; Kang, H.; Wang, R.; et al. Sequential CD19- and Bcma-Specific Chimeric Antigen Receptor T Cell Treatment for RRMM: Report from a Single Center Study. Blood 2019, 134 (Suppl. 1), 578. [Google Scholar] [CrossRef]
- Garcia-Guerrero, E.; Rodríguez-Lobato, L.G.; Danhof, S.; Sierro-Martínez, B.; Goetz, R.; Sauer, M. ATRA Augments BCMA Expression on Myeloma Cells and Enhances Recognition By BCMA-CAR T-Cells. Blood 2020, 136 (Suppl. 1), 13–14. [Google Scholar] [CrossRef]
- Cowan, A.J.; Pont, M.; Sather, B.D.; Turtle, C.J.; Till, B.G.; Nagengast, A.M.; Libby, I.I.I.E.N.; Becker, P.S.; Coffey, D.G.; Tuazon, S.A.; et al. Efficacy and Safety of Fully Human Bcma CAR T Cells in Combination with a Gamma Secretase Inhibitor to Increase Bcma Surface Expression in Patients with Relapsed or Refractory Multiple Myeloma. Blood 2019, 134 (Suppl. 1), 204. [Google Scholar] [CrossRef]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basak, G.W.; Carrier, E. The Search for Multiple Myeloma Stem Cells: The Long and Winding Road. Biol Blood Marrow Transpl. 2010, 16, 587–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, W.; Wang, Q.; Barber, J.P.; Brennan, S.; Smith, B.D.; Borrello, I.; McNiece, I.; Lin, L.; Ambinder, R.F.; Peacock, C.; et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008, 68, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, B.; Puig, N.; Cedena, M.T.; de Jong, B.G.; Ruiz, Y.; Rapado, I.; Martinez-Lopez, J.; Cordon, L.; Alignani, D.; Delgado, J.A.; et al. Differentiation stage of myeloma plasma cells: Biological and clinical significance. Leukemia 2017, 31, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, M.; Radhakrishnan, S.V.; Luetkens, T.; Atanackovic, D. The role of surface molecule CD229 in Multiple Myeloma. Clin. Immunol. 2019, 204, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Stadtmauer, E.A.; Hwang, W.T.; Lacey, S.F.; Melenhorst, J.J.; Krevvata, M.; Carroll, M.P.; Matsui, W.H.; Wang, Q.; Dhodapkar, M.V.; et al. Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI Insight 2019, 21, 4. [Google Scholar] [CrossRef]
- Atanackovic, D.; Panse, J.; Hildebrandt, Y.; Jadczak, A.; Kobold, S.; Cao, Y.; Templin, J.; Meyer, S.; Reinhard, H.; Bartels, K.; et al. Surface molecule CD229 as a novel target for the diagnosis and treatment of multiple myeloma. Haematologica 2011, 96, 1512–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nerreter, T.; Letschert, S.; Götz, R.; Doose, S.; Danhof, S.; Einsele, H.; Sauer, M.; Hudecek, M. Super-resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimination by CD19 CAR-T. Nat. Commun. 2019, 10, 3137. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelapu, S.S. Managing the toxicities of CAR T-cell therapy. Hematol. Oncol. 2019, 37 (Suppl. 1), 48–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yáñez, L.; Sánchez-Escamilla, M.; Perales, M.-A. CAR T Cell Toxicity: Current Management and Future Directions. HemaSphere 2019, 3, e186. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [Green Version]
- Taraseviciute, A.; Tkachev, V.; Ponce, R.; Turtle, C.J.; Snyder, J.M.; Liggitt, H.D.; Myerson, D.; Gonzalez-Cuyar, L.; Baldessari, A.; English, C.; et al. Chimeric Antigen Receptor T Cell-Mediated Neurotoxicity in Nonhuman Primates. Cancer Discov. 2018, 8, 750–763. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transpl. 2019, 25, 625–638. [Google Scholar] [CrossRef] [Green Version]
- Mahadeo, K.M.; Khazal, S.J.; Abdel-Azim, H.; Fitzgerald, J.C.; Taraseviciute, A.; Bollard, C.M.; Tewari, P.; Duncan, C.; Traube, C.; McCall, D.; et al. Management guidelines for paediatric patients receiving chimeric antigen receptor T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 45–63. [Google Scholar] [CrossRef] [Green Version]
- Yakoub-Agha, I.; Chabannon, C.; Bader, P.; Basak, G.W.; Bonig, H.; Ciceri, F.; Corbacioglu, S.; Duarte, R.F.; Einsele, H.; Hudecek, M.; et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: Best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica 2020, 105, 297–316. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; Van Steenbergen, S.; Van Elzakker, P.; Van Krimpen, B.; Groot, C.; Vulto, A.; Den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Thistlethwaite, F.C.; Gilham, D.E.; Guest, R.D.; Rothwell, D.G.; Pillai, M.; Burt, D.J.; Byatte, A.J.; Kirillova, N.; Valle, J.W.; Sharma, S.K.; et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017, 66, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Singh, I.; Zudaire, E.; Yeh, T.M.; Allred, A.J.; Olyslager, Y.; Banerjee, A.; Goldberg, J.D.; et al. Update of CARTITUDE-1: A phase Ib/II study of JNJ-4528, a B-cell maturation antigen (BCMA)-directed CAR-T-cell therapy, in relapsed/refractory multiple myeloma. J. Clin. Oncol. 2020, 38 (Suppl. 15), 8505. [Google Scholar] [CrossRef]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Chang, W.C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef]
- Caruso, H.G.; Hurton, L.V.; Najjar, A.; Rushworth, D.; Ang, S.; Olivares, S.; Mi, T.; Switzer, K.; Singh, H.; Huls, H.; et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 2015, 75, 3505–3518. [Google Scholar] [CrossRef] [Green Version]
- Arcangeli, S.; Rotiroti, M.C.; Bardelli, M.; Simonelli, L.; Magnani, C.F.; Biondi, A.; Biagi, E.; Tettamanti, S.; Varani, L. Balance of Anti-CD123 Chimeric Antigen Receptor Binding Affinity and Density for the Targeting of Acute Myeloid Leukemia. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 1933–1945. [Google Scholar] [CrossRef]
- Han, C.; Sim, S.J.; Kim, S.H.; Singh, R.; Hwang, S.; Kim, Y.I.; Park, S.H.; Kim, K.H.; Lee, D.G.; Oh, H.S.; et al. Desensitized chimeric antigen receptor T cells selectively recognize target cells with enhanced antigen expression. Nat. Commun. 2018, 9, 468. [Google Scholar] [CrossRef] [Green Version]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015, 350, aab4077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanitis, E.; Poussin, M.; Klattenhoff, A.W.; Song, D.; Sandaltzopoulos, R.; June, C.H.; Powell, D.J. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 2013, 1, 43–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Feng, Z.; Ma, J.; Ling, S.; Cao, Y.; Gurung, B.; Wu, Y.; Katona, B.W.; O’Dwyer, K.P.; Siegel, D.L.; et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood 2020, 135, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. “Off-the-shelf” allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Somerville, R.P.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 1112–1121. [Google Scholar] [CrossRef] [Green Version]
- Kochenderfer, J.N.; Dudley, M.E.; Carpenter, R.O.; Kassim, S.H.; Rose, J.J.; Telford, W.G.; Hakim, F.T.; Halverson, D.C.; Fowler, D.H.; Hardy, N.M.; et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013, 122, 4129–4139. [Google Scholar] [CrossRef]
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K.; et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014, 28, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Liu, P.Q.; Zhou, Y.; Zhang, L.; Maiti, S.; Huls, H.; Miller, J.C.; Kebriaei, P.; Rabinovitch, B.; et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 2012, 119, 5697–5705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, C.J.; Peacock, S.; Chaudhry, A.N.; Bradley, J.A.; Bolton, E.M. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell 2012, 11, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Quan, Y.; Yan, Q.; Morales, J.E.; Wetsel, R.A. Targeted Disruption of the β2-Microglobulin Gene Minimizes the Immunogenicity of Human Embryonic Stem Cells. Stem Cells Transl. Med. 2015, 4, 1234–1245. [Google Scholar] [CrossRef]
- Sommer, C.; Boldajipour, B.; Valton, J.; Galetto, R.; Bentley, T.; Sutton, J.; Ni, Y.; Leonard, M.; Van Blarcom, T.; Smith, J.; et al. ALLO-715, an Allogeneic BCMA CAR T Therapy Possessing an Off-Switch for the Treatment of Multiple Myeloma. Blood 2018, 132 (Suppl. 1), 591. [Google Scholar] [CrossRef]
- Cranert, S.A.; Richter, M.; Tong, M.; Weiss, L.; Tan, Y.; Ostertag, E.M.; Coronella, J.; Shedlock, D.J. Manufacture of an Allogeneic CAR-T Stem Cell Memory Product Candidate for Multiple Myeloma, P-Bcma-ALLO1, Is Robust, Reproducible and Highly Scalable. Blood 2019, 134 (Suppl. 1), 4445. [Google Scholar] [CrossRef]
- Mathur, R.; Zhang, Z.; He, J.; Galetto, R.; Gouble, A.; Chion-Sotinel, I.; Filipe, S.; Gariboldi, A.; Veeramachaneni, T.; Manasanch, E.E.; et al. Universal SLAMF7-Specific CAR T-Cells As Treatment for Multiple Myeloma. Blood 2017, 130 (Suppl. 1), 502. [Google Scholar]
- Mailankody, S.; Matous, J.V.; Liedtke, M.; Sidana, S.; Malik, S.; Nath, R. Universal: An Allogeneic First-in-Human Study of the Anti-Bcma ALLO-715 and the Anti-CD52 ALLO-647 in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136 (Suppl. 1), 24–25. [Google Scholar] [CrossRef]
- Santomasso, B.; Bachier, C.; Westin, J.; Rezvani, K.; Shpall, E.J. The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 433–444. [Google Scholar] [CrossRef]
- Nam, S.; Smith, J.; Yang, G. Driving the next wave of innovation in CAR T-cell Therapies|McKinsey. 2019. Available online: https://www.mckinsey.com/industries/pharmaceuticals-and-medical-products/our-insights/driving-the-next-wave-of-innovation-in-car-t-cell-therapies (accessed on 15 August 2020).
- ICER (Institute for Clinical and Economic Review); CTAF (California Technology Assessment Forum). Chimeric Antigen Receptor T-Cell Therapy for B-Cell Cancers: Effectiveness and Value. In Final Evidence Report; ICER: Boston, MA, USA, 2018. [Google Scholar]
- Lin, J.K.; Lerman, B.J.; Barnes, J.I.; Boursiquot, B.C.; Tan, Y.J.; Robinson, A.Q.; Davis, K.L.; Owens, D.K.; Goldhaber-Fiebert, J.D. Cost Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Relapsed or Refractory Pediatric B-Cell Acute Lymphoblastic. Leuk. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 13, JCO2018790642. [Google Scholar] [CrossRef] [Green Version]
- Kebriaei, P.; Izsvák, Z.; Narayanavari, S.A.; Singh, H.; Ivics, Z. Gene Therapy with the Sleeping Beauty Transposon System. Trends Genet TIG 2017, 33, 852–870. [Google Scholar] [CrossRef] [PubMed]
- Ramanayake, S.; Bilmon, I.; Bishop, D.; Dubosq, M.C.; Blyth, E.; Clancy, L.; Gottlieb, D.; Micklethwaite, K. Low-cost generation of Good Manufacturing Practice-grade CD19-specific chimeric antigen receptor-expressing T cells using piggyBac gene transfer and patient-derived materials. Cytotherapy 2015, 17, 1251–1267. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, J.; Avivi, I.; Binyamini, N.; Uhl, L.; Somaiya, P.; Stroopinsky, D.; Palmer, K.A.; Coll, M.D.; Katz, T.; Bisharat, L.; et al. Blockade of PD-1 in Combination with Dendritic Cell/Myeloma Fusion Cell Vaccination Following Autologous Stem Cell Transplantation Is Well Tolerated, Induces Anti-Tumor Immunity and May Lead to Eradication of Measureable Disease. Blood 2015, 126, 4218. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef] [Green Version]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- Pardoll, D. Cancer and the Immune System: Basic Concepts and Targets for Intervention. Semin Oncol. 2015, 42, 523–538. [Google Scholar] [CrossRef] [Green Version]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [Green Version]
- Pawlyn, C.; Davies, F.E. Toward personalized treatment in multiple myeloma based on molecular characteristics. Blood 2019, 133, 660–675. [Google Scholar] [CrossRef] [Green Version]
- Krzyszczyk, P.; Acevedo, A.; Davidoff, E.J.; Timmins, L.M.; Marrero-Berrios, I.; Patel, M.; White, C.; Lowe, C.; Sherba, J.J.; Hartmanshenn, C.; et al. The growing role of precision and personalized medicine for cancer treatment. Technology 2018, 6, 79–100. [Google Scholar] [CrossRef] [Green Version]
- Le Tourneau, C.; Borcoman, E.; Kamal, M. Molecular profiling in precision medicine oncology. Nat. Med. 2019, 25, 711–712. [Google Scholar] [CrossRef]
- Rothwell, D.G.; Ayub, M.; Cook, N.; Thistlethwaite, F.; Carter, L.; Dean, E.; Smith, N.; Villa, S.; Dransfield, J.; Clipson, A.; et al. Utility of ctDNA to support patient selection for early phase clinical trials: The TARGET study. Nat. Med. 2019, 25, 738–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Soria, J.C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Braña, I.; et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat. Med. 2019, 25, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Marquart, J.; Chen, E.Y.; Prasad, V. Estimation of the Percentage of US Patients with Cancer Who Benefit From Genome-Driven Oncology. JAMA Oncol. 2018, 4, 1093–1098. [Google Scholar] [CrossRef]
- Hoos, A.; Eggermont, A.M.; Janetzki, S.; Hodi, F.S.; Ibrahim, R.; Anderson, A.; Humphrey, R.; Blumenstein, B.; Old, L.; Wolchok, J. Improved endpoints for cancer immunotherapy trials. J. Natl. Cancer Inst. 2010, 102, 1388–1397. [Google Scholar] [CrossRef] [Green Version]
- Park, J.J.H.; Hsu, G.; Siden, E.G.; Thorlund, K.; Mills, E.J. An overview of precision oncology basket and umbrella trials for clinicians. CA Cancer J. Clin. 2020, 70, 125–137. [Google Scholar] [CrossRef]
- Jørgensen, J.; Hanna, E.; Kefalas, P. Outcomes-based reimbursement for gene therapies in practice: The experience of recently launched CAR-T cell therapies in major European countries. J. Mark Access Health Policy 2020, 8, 1715536. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, U.H.; Cornell, R.F.; Lakshman, A.; Gahvari, Z.J.; McGehee, E.; Jagosky, M.H.; Gupta, R.; Varnado, W.; Fiala, M.A.; Chhabra, S.; et al. Outcomes of Patients with Multiple Myeloma Refractory to CD38-Targeted Monoclonal Antibody Therapy. Leukemia 2019, 33, 2266–2275. [Google Scholar] [CrossRef]
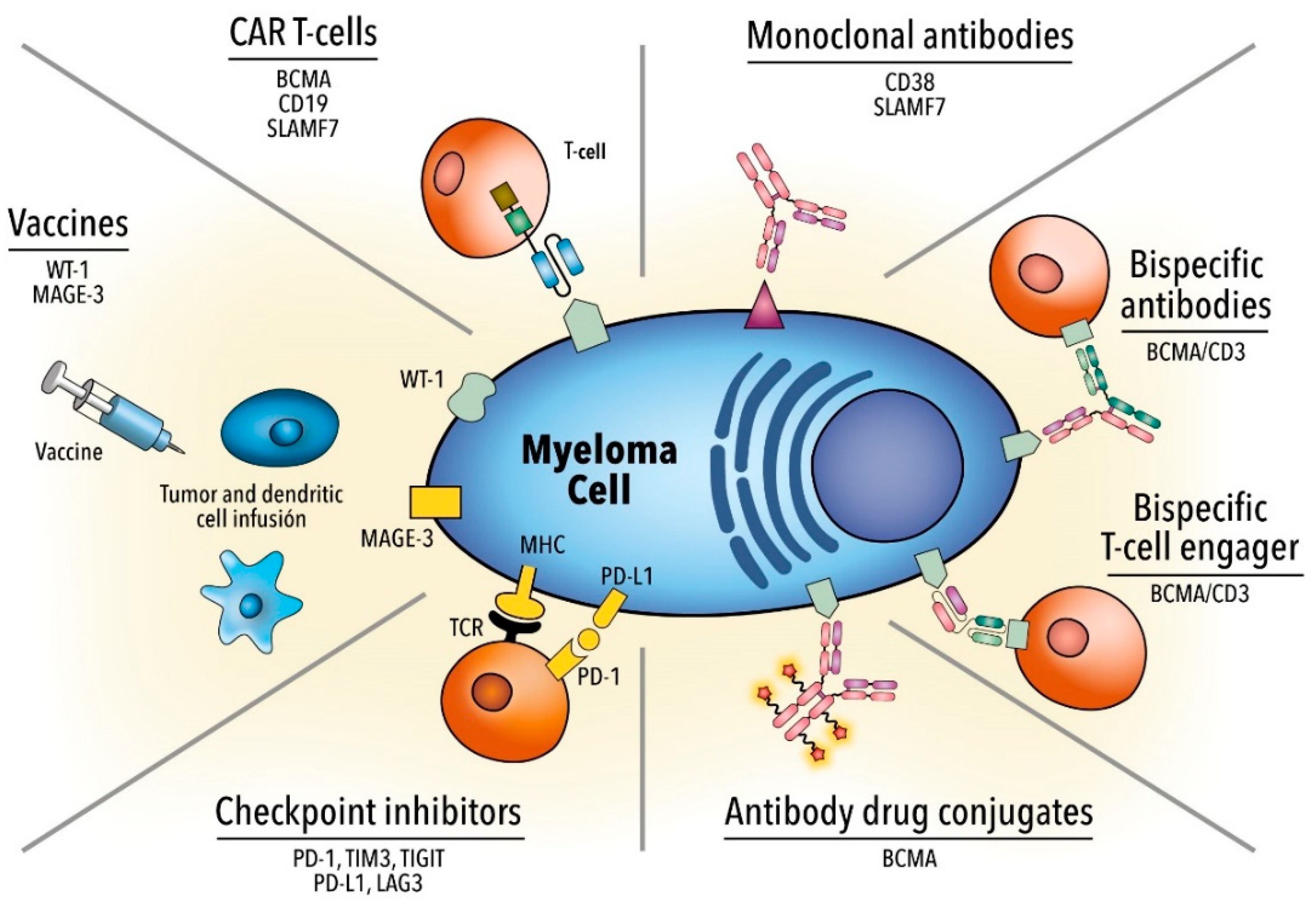
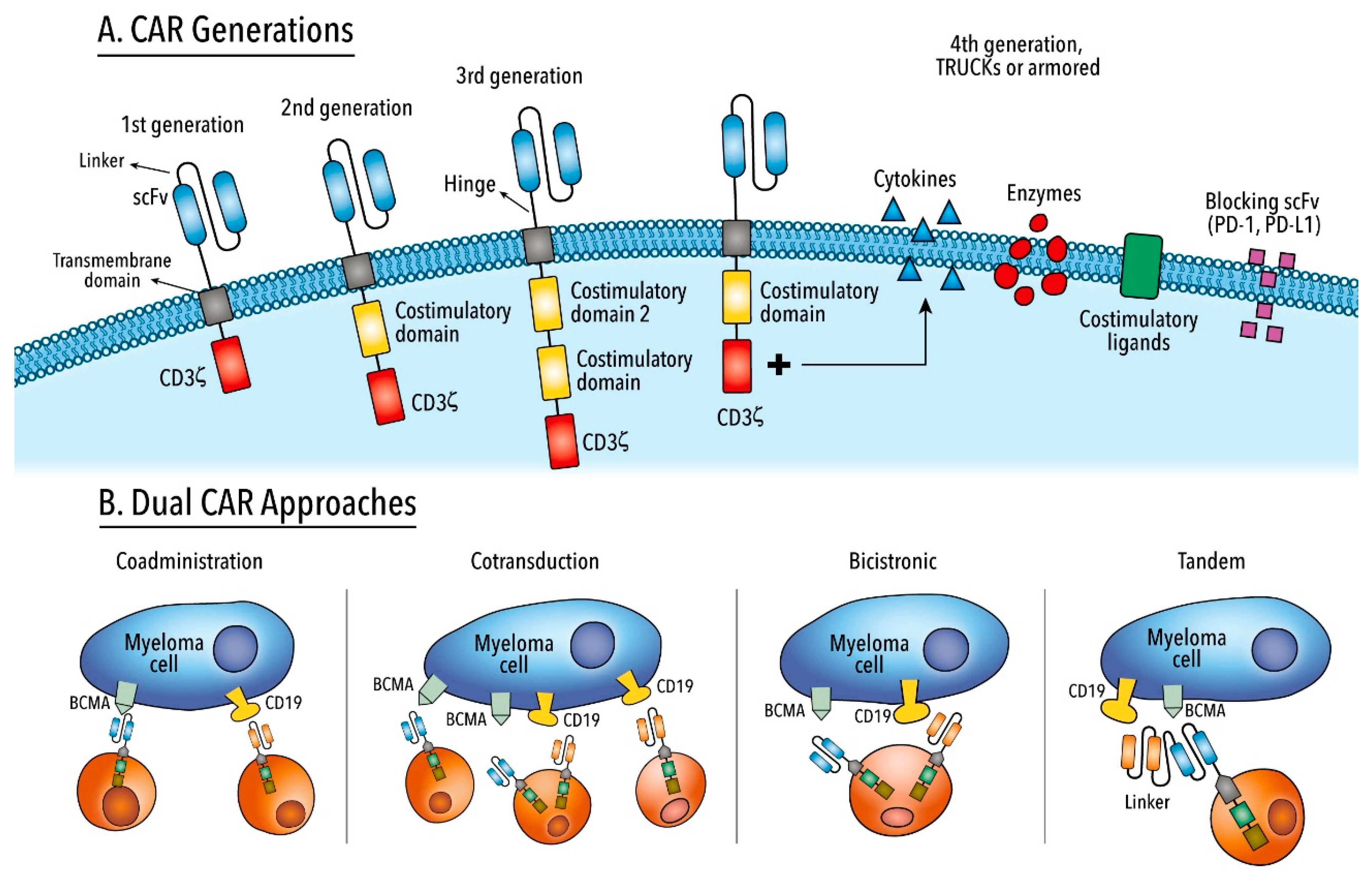
| Agent | Target | Specification | Prior Lines | Response | Prognosis | Toxicity |
|---|---|---|---|---|---|---|
| Monoclonal antibodies | CD38 | First-in-human, phase I/II. Monotherapy 16 mg/kg [25,26] | ≥3 | ORR 31.1% sCR 4.7% | PFS 4 mo OS 20.1 mo | IRR 48% (2.7% ≥ grade 3) |
| GEN 503. Part 2: dose expansion with DRd [27,28] | 2 | ORR 81% sCR 25% | PFS 72% OS 90% | IRR 56% (6.3% ≥ grade 3) | ||
| POLLUX phase III DRd vs. Rd. R refractory were excluded [29] | 1 | CR 43.1 vs. 19.2% (p < 0.001) sCR 22.4 vs. 4.6% (p < 0.001) (DRd vs. Rd) | 12 m PFS 83.2 vs. 60.1% OS 91.2 vs. 76.4% (p < 0.001) | IRR 47.7% (6.3% ≥ grade 3); 92% occurring during the first infusion | ||
| CASTOR phase III DVd [30,31] | 2 | ORR 83.8 vs. 63.2% (p < 0.0001) CR or better 28.8 vs. 9.8% (p < 0.0001) sCR 8.8 vs. 2.6% (DVd vs. Vd) | 18 m PFS 48 vs. 7.9% In high-risk cytogenetics PFS 11.2 vs. 7.2% | IRR 45.3% (8.3% ≥ grade 3) | ||
| SLAMF7/CS-1 | E monotherapy. Phase I, dose escalation 0.5–20 mg/kg [32] | ≥2 | No maximum tolerated dose ORR 0% SD 26.5% | NA | IRR 52% before the initiation of prophylaxis | |
| Vd +/− E, randomized phase II [33] | ≥1 | ORR 65 vs. 63% CR 4 vs. 4% (EVd vs. Vd) | PFS 9.7 vs. 6.9 mo OS 85 vs. 74% | IRR 7% (0% ≥ grade 3) | ||
| ELOQUENT-2 Rd +/− E, randomized phase III [34] | 1–3 | ORR 79 vs. 66% (p = 0.0002) (ERd vs. Rd) | 3 y PFS (3y) 26 vs. 18% 3 y-OS 60 vs. 53% (p = 0.026) | Comparable between groups | ||
| Pd +/− E, randomized phase II [35] | ≥2 | ORR 53 vs. 26% (EPd vs. Pd) | PFS 10.3 vs. 4.7 mo | IRR 5% (0% ≥ grade 3) | ||
| ADC | BCMA | GSK-2857916 conjugated to MMAF; phase I [36,37] | ≥3 | ORR 60% CR 9% sCR 6% | PFS 12 mo | Thrombocytopenia 35% Eye-related events: Blurry vision 52%, dry eyes 37%, photophobia 29% |
| CD138 | Indatuximab ravtansine linked to maytansinoid; phases I/II [38,39] | ≥2 | ORR 5.9% CR 0% SD 42.9% | PFS 3 mo | Fatigue 47% Diarrhea 43% | |
| CD56 | Lorvotuzumab-mertansine; phase I [40] | ≥1 | ORR 5.7% CR 0% SD 42.9% | PFS 26.1 weeks in evaluable | Peripheral neuropathy 5.3% | |
| CD74 | Milatuzumab doxorubicin; phase I [41] | ≥2 | No objective responses. SD 5/19 (26%) for 3 mo | NA | n = 1 grade 3 IRR | |
| Bispecific antibodies | BCMA/CD3 | AMG 420: First-in-human, phase I, dose escalation: maximum tolerated 400 µg/day. No extramedullary disease [42,43] | ≥2 | Dose 400 µg/day ORR 70% sCR 50% | Dose 400 µg/day PFS 9 mo | CRS 38.1% (grade ≥ 3 7.1%) Dose-limiting peripheral neuropathy n = 2 |
| Teclistamab; phase I; dose range: 0.3–270 µg/kg [44] | 6 | ORR 78% in patients receiving highest dose | - | CRS 56% (all grade 1/2) Neurotoxicity 8% (3% grade ≥ 3) IRR 9% | ||
| Immune checkpoint inhibitors | PD-1 | Nivolumab monotherapy; phase I including several neoplasms [45] | ≥1 | ORR 4% SD 63% | - | Drug-related AEs 52% any grade, 19% ≥ grade 3 |
| KEYNOTE-183; phase III, randomized Pd +/− Pembrolizumab [46] | ≥2 | ORR 34 vs. 40% (Pembrolizumab + Pd vs. Pd) | PFS 5.6 vs. 8.4 (median time to progression 8.1 vs. 8.7 mo) (Pembrolizumab + Pd vs. Pd) | Serious AE 63 vs. 46% (Pembrolizumab + Pd vs. Pd) TRM n = 4: unknown cause, neutropenic sepsis, myocarditis, Stevens–Johnson syndrome | ||
| KEYNOTE-185; phase III, randomized Rd +/− Pembrolizumab [46] | Newly-diagnosed ASCT ineligible | ORR 64 vs. 62% (Pembrolizumab + Rd vs. Rd) | PFS not reached | Serious AE 54 vs. 39% (Pembrolizumab + Rd vs. Rd) Terminated because of the uneven number of deaths between groups | ||
| CAR T cell | BCMA | NCI scFv murine/CD28 [47] | 9.5 | ORR 81% (≥CR 13%) | mEFS 7.2 mo | CRS 94% (grade ≥ 3 38%) ICANS NA (grade ≥ 3 19%) |
| UPenn/CART-BCMA scFv human/4-1BB [48] | 7 | ORR 64% (≥CR 11%) | mPFS 4.2 mo | CRS 88% (grade ≥ 3 32%) ICANS 32 (grade ≥ 3 12%) | ||
| LCAR-B38M VHH llama/4-1BB [49,50] | 3 | ORR 88% (≥CR 74%) | mPFS 20 mo18 m OS 68% | CRS 89% (grade ≥ 3 7%) ICANS 2 (grade ≥ 3 0%) | ||
| LCAR-B38M VHH llama/4-1BB [51] | 4 | ORR 88% (≥CR 77%) | 1 y PFS 53%1 y OS 82% | CRS 100% (grade ≥ 3 41%) ICANS NA (grade ≥ 3 NA) | ||
| Ciltacabtagene Autolecuel (LCAR-B38M/JNJ68284528) CARTITUDE-1 [52] | 6 | ORR 97% (sCR 67%) | 1 y PFS 76.6% 1 y OS 88.5% | CRS 95% (grade ≥ 3 4%) ICANS 21% (grade ≥ 3 10%) | ||
| Idecabtagene Vicleucel (bb2121)/scFv murine/4-1BB [24] | 7–8 | ORR 85% (≥CR 45%) | mPFS 11.8 mo | CRS 76% (grade ≥ 3 6%) ICANS 42% (grade ≥ 3 3%) | ||
| Idecabtagene Vicleucel (bb2121)/scFv murine/4-1BB KarMMA [53] | 6 | ORR 73% (≥CR 33%) | mPFS 8.8 mo mOS 19.4 mo | CRS 84% (grade ≥ 3 6%) ICANS 18% (grade ≥ 3 3%) | ||
| Orvacabtagene Autoleucel (JCARH125)/scFv human 4-1BB EVOLVE [54] | 6 | ORR 92% (≥CR 36%) | mPFS 9.3 mo | CRS 89% (grade ≥ 3 3%) ICANS 13% (grade ≥ 3 3%) | ||
| Vaccines | Dendritic cells/tumor fusions | Vaccine composed of autologous dendritic cells and patient-derived myeloma cells; 16 patients included [55] | 4 | SD: 11 | - | Site reaction (grade 1) |
| hTERT/Survivin | NCT00499577 [56] | 1 | IR 36% | mEFS 20 mo | Chills 57% Rash > 85% (grades 1–2) | |
| Dendritic cells/tumor fusions | Two cohorts: 24 patients vaccinated post-ASCT 12 patients vaccinated pre- and post- ASCT [57] | - | ORR 78% (CR 47%) | 2 y PFS 57% | Site reaction (grade 1) Myalgia (grade 1) | |
| MAGE-A3 | NCT01245673 [58] | 1–5 | IR 88% | 2 y OS 74% 2 y EFS 56% | Site reaction >90 % | |
| XBP1 CD138 CS1 | NCT01718899: SMM patients; two cohorts: Monotherapy Combination with IMiDs [59] | 1 | IR 95% | mTTP: 36 w mTTP: not reached | Site reaction 58–100% (grades 1–2) | |
| MAGE-A3 | NCT01380145: vaccinated post ASCT [60] | 1–2 | IR 100% | mPFS 27 mo mOS not reached | Site reaction 54% (grade 1)Myalgia 33% (grades 1–2) |
| Limitation | Rationale | Approach |
|---|---|---|
| Antigen-positive escape | Impaired T cell persistence | Optimize CAR design (human scFv, hinge, costimulatory domains) to avoid antigen-independent tonic signaling and reduce antigenicity Younger T cell donors, transduction to stem cell memory T cell and central memory T cells, block T cell differentiation signaling, or use of non-viral transduction systems Genomic knock-in of the CAR sequence to the TRAC locus |
| Impaired T cell potency | Fine-tuning CAR design (human scFv, hinge, costimulatory domains) Avoid antigen-independent tonic signaling Genomic knock-in of the CAR sequence to the TRAC locus Avoid T cell exhaustion (combine with immune check-point inhibitors or disrupt the checkpoint pathway) Reduce the amount of soluble target antigen Optimize lymphodepletion protocol | |
| Tumor microenvironment-induced immunosuppression | Boost T cell trafficking and migration Overcome inhibitory signals by blocking immune check-point pathways or switching inhibitory signals present in the TME into pro-inflammatory signals Targeting immunosuppressive immune cells (regulatory T cells, tumor-associated macrophages, myeloid-derived suppressor cells) Armored CAR T cells or TRUCKs | |
| Antigen-negative escape | Immune selection pressure Gene mutations Lineage switching Trogocytosis Antigen masking | Identification and selection of the most suitable tumor antigen Fine-tuning antigen binding affinity Targeting multiple tumor antigens (sequential or co-administration of single-target CAR products, dual CARs, or tandem CARs) Upregulate surface density of the target antigen Targeting myeloma stem cells |
| Toxicities | CRS and ICANS | Optimizing reduction in the number of CAR T cells infused or dividing doses on different days Prompt recognition with the use of predictive biomarkers Use of tocilizumab or corticosteroids in early stages of the disease Tailored modifications of the construct, optimizing the costimulatory domain CAR T cells with suicide genes or “OFF-switches” |
| On-target, off-tumor | Affinity tuning of the scFv Advanced CAR engineering: “AND” logic-gate, “ON-switch”, “SPLIT”, or inhibitory CARs | |
| Manufacturing | Amount and quality of T cells Vein-to-vein time Production failure | Allogeneic CAR T cells (major concerns: GvHD and CAR T cell rejection) |
| Access and economic | Infrastructure, workflows, processes, regulatory requirements, and economic burden | Cooperation among multiple stakeholders Use of non-viral gene delivery with transposon/transposase systems Creation of community CAR T cell therapy centers Promote the outpatient setting Shift from centralized to decentralized manufacturing, namely “bedside manufacturing” Cost-effectiveness, cost-benefit, and quality-adjusted life-year analyses Outcome-based reimbursement or staged payment models Legitimate value of immunotherapy as shown by real-world evidence and longer follow-ups |
| Bispecific Antibodies | CAR T Cell | |
|---|---|---|
| Production | “Off-the-shelf”: No need for manufacturing time, allowing for immediate treatment of the patient | Individual manufacturing for each patient, starting with autologous lymphapheresis Approach: Allogeneic CAR T cells under development |
| Administration | Continuous intravenous infusion Approach: extended half-life bispecific antibodies | Punctual infusion of the product (dose is sometimes split up into several days to reduce AEs) |
| T cell phenotype and effector function | Binding of endogenous CD8+ and CD4+ T cells, which have a superior cytotoxic function than naïve T cells | The product is mostly composed of naïve CD8+ and CD4+ T cells; these cells have higher self-renewal, survival, and penetration in lymphoid tissues |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Lobato, L.G.; Oliver-Caldés, A.; Moreno, D.F.; Fernández de Larrea, C.; Bladé, J. Why Immunotherapy Fails in Multiple Myeloma. Hemato 2021, 2, 1-42. https://doi.org/10.3390/hemato2010001
Rodríguez-Lobato LG, Oliver-Caldés A, Moreno DF, Fernández de Larrea C, Bladé J. Why Immunotherapy Fails in Multiple Myeloma. Hemato. 2021; 2(1):1-42. https://doi.org/10.3390/hemato2010001
Chicago/Turabian StyleRodríguez-Lobato, Luis Gerardo, Aina Oliver-Caldés, David F. Moreno, Carlos Fernández de Larrea, and Joan Bladé. 2021. "Why Immunotherapy Fails in Multiple Myeloma" Hemato 2, no. 1: 1-42. https://doi.org/10.3390/hemato2010001
APA StyleRodríguez-Lobato, L. G., Oliver-Caldés, A., Moreno, D. F., Fernández de Larrea, C., & Bladé, J. (2021). Why Immunotherapy Fails in Multiple Myeloma. Hemato, 2(1), 1-42. https://doi.org/10.3390/hemato2010001