
Treatment of Lower Risk Myelodysplastic Syndromes


{kind=link}
Abstract
:1. Introduction
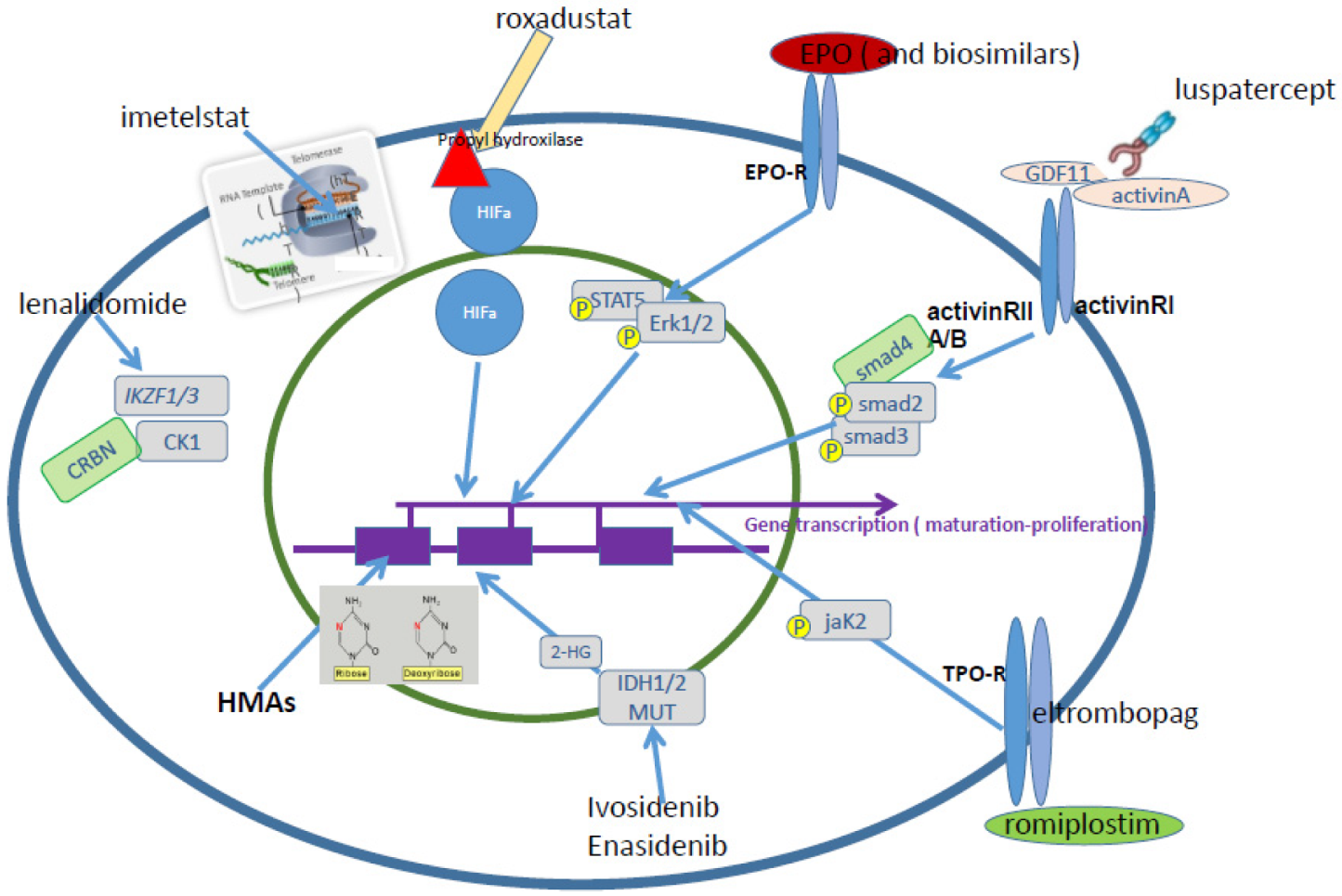
2. Therapy
Funding
Conflicts of Interest
References
- Bersanelli, M.; Travaglino, E.; Meggendorfer, M.; Matteuzzi, T.; Sala, C.; Mosca, E.; Chiereghin, C.; Di Nanni, N.; Gnocchi, M.; Zampini, M.; et al. Classification and Personalized Prognostic Assessment on the Basis of Clinical and Genomic Features in Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Ossa, J.A.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognosis Scoring System for Myelodysplastic Syndromes; American Society of Hematology: Washington, DC, USA, 2021. [Google Scholar]
- Oliva, E.N.; Dimitrov, B.D.; Benedetto, F.; D’Angelo, A.; Nobile, F. Hemoglobin level threshold for cardiac remodeling and quality of life in myelodysplastic syndrome. Leuk. Res. 2005, 29, 1217–1219. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Haase, D.; Santini, V.; Sanz, G.F.; Platzbecker, U.; Mey, U. Myelodysplastic syndromes: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2021, 32, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, E.; Li, J.; Greenberg, P.; Wu, D.; Hou, M.; Figueroa, E.H.M.; Guadalupe Rodriguez, M.; Dong, X.; Ghosh, J.; Izquiero, M.; et al. TELESTO Study Investigators Iron Chelation in Transfusion-Dependent Patients with Low- to Intermediate-1-Risk Myelodysplastic Syndromes: A Randomized Trial. Ann. Intern. Med. 2020, 172, 513–522. [Google Scholar] [CrossRef]
- Park, S.; Kelaidi, C.; Sapena, R.; Vassilieff, D.; Beyne-Rauzy, O.; Coiteux, V.; Vey, N.; Ravoet, C.; Cheze, S.; Rose, C.; et al. Early introduction of ESA in low risk MDS patients may delay the need for RBC transfusion: A retrospective analysis on 112 patients. Leuk. Res. 2010, 34, 1430–1436. [Google Scholar] [CrossRef]
- Johnson & Johnson EPREX® (Epoetin Alfa). Marketing Authorisation Extended to Include Treatment of Symptomatic Anaemia in Patients with Low or Intermediate-1-Risk Myelodysplastic Syndromes. Available online: https://www.jnj.com/media-cen-ter/press-releases/eprex-epoetin-alfa-marketing-authorisation-extended-to-include-treatment-of-symptomat-ic-anaemia-in-patients-with-low-or-intermediate-1-risk-myelodysplastic-syndromes (accessed on 30 August 2021).
- Fenaux, P.; Santini, V.; Spiriti, M.A.A.; Giagounidis, A.; Schlag, R.; Radinoff, A.; Gercheva-Kyuchukova, L.; Anagnostopoulos, A.; Oliva, E.N.; Symeonidis, A. A phase 3 randomized, placebo-controlled study assessing the efficacy and safety of epoetin-α in anemic patients with low-risk MDS. Leukemia 2018, 32, 2648–2658. [Google Scholar] [CrossRef] [Green Version]
- Hellström-Lindberg, E.; Gulbrandsen, N.; Lindberg, G.; Ahlgren, T.; Dahl, I.M.S.; Dybedal, I.; Grimfors, G.; Hesse-Sundin, E.; Hjorth, M.; Kanter-Lewensohn, L.; et al. A validated decision model for treating the anaemia of myelodysplastic syndromes with erythropoietin + granulocyte colony-stimulating factor: Significant effects on quality of life. Br. J. Haematol. 2003, 120, 1037–1046. [Google Scholar] [CrossRef]
- Santini, V.; Schemenau, J.; Levis, A. Can the revised IPSS predict response to erythropoietic-stimulating agents in patients with classical IPSS low or intermediate-1 MDS? Blood 2013, 122, 2286–2288. [Google Scholar] [CrossRef] [Green Version]
- Aloe Spiriti, M.A.; Latagliata, R.; Niscola, P.; Cortelezzi, A.; Francesconi, M.; Ferrari, D.; Volpe, E.; Clavio, M.; Grossi, A.; Tambone Reyes, M.; et al. Impact of a new dosing regimen of epoetin alfa on quality of life and anemia in patients with low-risk myelodysplastic syndrome. Ann. Hematol. 2005, 84, 167–176. [Google Scholar] [CrossRef]
- Balleari, E.; Filiberti, R.A.; Salvetti, C.; Allione, B.; Angelucci, E.; Bruzzone, M.; Calzamiglia, T.; Cavaliere, M.; Cavalleri, M.; Cilloni, D.; et al. Effects of different doses of erythropoietin in patients with myelodysplastic syndromes: A propensity score-matched analysis. Cancer Med. 2019, 8, 7567–7576. [Google Scholar] [CrossRef] [Green Version]
- Kosmider, O.; Passet, M.; Santini, V.; Platzbecker, U.; Andrieu, V.; Zini, G.; Beyne-Rauzy, O.; Guerci, A.; Masala, E.; Balleari, E.; et al. Are somatic mutations predictive of response to erythropoiesis stimulating agents in lower risk myelodysplastic syndromes? Haematologica 2016, 101, e280–e283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Greenberg, P.; Yucel, A.; Farmer, C.; O’Neill, F.; Brandao, C.D.O.; Fenaux, P. Clinical effectiveness and safety of erythropoietin-stimulating agents for the treatment of low- and intermediate-1−risk myelodysplastic syndrome: A systematic literature review. Br. J. Haematol. 2019, 184, 134–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.W.; Sato, M.; Gore, S.D.; Baer, M.R.; Ke, X.; McNally, D.; Davidoff, A. Erythropoiesis-stimulating agents are not associated with increased risk of thrombosis in patients with myelodysplastic syndromes. Haematologica 2012, 97, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basiorka, A.; McGraw, K.L.; Abbas-Aghababazadeh, F.; McLemore, A.; Vincelette, N.D.; Ward, G.A.; Eksioglu, E.A.; Sallman, D.A.; Al Ali, N.; Padron, E.; et al. Assessment of ASC specks as a putative biomarker of pyroptosis in myelodysplastic syndromes: An observational cohort study. Lancet Haematol. 2018, 5, e393–e402. [Google Scholar] [CrossRef]
- Ward, G.A.; McGraw, K.L.; Abbas-Aghababazadeh, F.; Meyer, B.S.; McLemore, A.F.; Vincelette, N.D.; Lam, N.B.; Aldrich, A.L.; Al Ali, N.H.; Padron, E.; et al. Oxidized mitochondrial DNA released after inflammasome activation is a disease biomarker for myelodysplastic syndromes. Blood Adv. 2021, 5, 2216–2228. [Google Scholar] [CrossRef]
- Reza, S.; Shetty, V.; Dar, S.; Qawi, H.; Raza, A. Tumor Necrosis Factor-α Levels Decrease with Anticytokine Therapy in Patients with Myelodysplastic Syndromes. J. Interf. Cytokine Res. 1998, 18, 871–877. [Google Scholar] [CrossRef]
- Santini, V.; Valcárcel, D.; Platzbecker, U.; Komrokji, R.S.; Cleverly, A.L.; Lahn, M.M.; Janssen, J.; Zhao, Y.; Chiang, A.; Giagounidis, A.; et al. Phase II Study of the ALK5 Inhibitor Galunisertib in Very Low-, Low-, and Intermediate-Risk Myelodysplastic Syndromes. Clin. Cancer Res. 2019, 25, 6976–6985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komrokji, R.; Garcia-Manero, G.; Ades, L.; Prebet, T.; Steensma, D.P.; Jurcic, J.G.; Sekeres, M.; Berdeja, J.; Savona, M.R.; Beyne-Rauzy, O.; et al. Sotatercept with long-term extension for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes: A phase 2, dose-ranging trial. Lancet Haematol. 2018, 5, e63–e72. [Google Scholar] [CrossRef]
- Platzbecker, U.; Germing, U.; Götze, K.S.; Kiewe, P.; Mayer, K.; Chromik, J.; Radsak, M.; Wolff, T.; Zhang, X.; Laadem, A.; et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): A multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017, 18, 1338–1347. [Google Scholar] [CrossRef]
- Raftopoulos, H.; Laadem, A.; Hesketh, P.J.; Goldschmidt, J.; Gabrail, N.; Osborne, C.; Ali, M.; Sherman, M.L.; Wang, D.; Glaspy, J.A.; et al. Sotatercept (ACE-011) for the treatment of chemotherapy-induced anemia in patients with metastatic breast cancer or advanced or metastatic solid tumors treated with platinum-based chemotherapeutic regimens: Results from two phase 2 studies. Support. Care Cancer 2016, 24, 1517–1525. [Google Scholar] [CrossRef] [Green Version]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Diez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhani, O.; et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef]
- Krönke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mni, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef]
- Sievers, Q.L.; Gasser, J.A.; Cowley, G.S.; Fischer, E.S.; Ebert, B.L. Genome-wide screen identifies cullin-RING ligase machinery required for lenalido-mide-dependent CRL4CRBN activity. Blood 2018, 132, 1293. [Google Scholar] [CrossRef] [Green Version]
- List, A.; Dewald, G.; Bennett, J.; Giagounidis, A.; Raza, A.; Feldman, E.; Powell, B.; Greenberg, P.; Thomas, D.; Stone, R.; et al. Lenalidomide in the Myelodysplastic Syndrome with Chromosome 5q Deletion. N. Engl. J. Med. 2006, 355, 1456–1465. [Google Scholar] [CrossRef] [Green Version]
- Fenaux, P.; Giagounidis, A.; Selleslag, D.; Beyne-Rauzy, O.; Mufti, G.; Mittleman, M.; Muus, P.; te Boekhorst, P.; Sanz, G.; del Canizo, C.; et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfu-sion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood 2011, 118, 3765–3776. [Google Scholar] [CrossRef] [PubMed]
- Jädersten, M.; Saft, L.; Smith, A.; Kulasekararaj, A.; Pomplun, S.; Göhring, G.; Hedlund, A.; Hast, R.; Schlegelberger, B.; Porwit, A.; et al. TP53 Mutations in Low-Risk Myelodysplastic Syndromes With del(5q) Predict Disease Progression. J. Clin. Oncol. 2011, 29, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- López-Cadenas, F.; Lumbreras, E.; Xicoy, B.; Sanchez-Garcia, J.; Fenaux, P.; Coll, R.; Slama, B.; Hernandez-Rivas, A.; Thepot, S.; Bernal, T.; et al. Phase 3 Study of Lenalidomide (LEN) Vs Placebo in Non-Transfusion Dependent (TD) Low Risk Del(5q) MDS Patients with Del(5q)—Preliminary Blinded Analysis of the European Sintra-REV Trial. Blood 2018, 132, 468. [Google Scholar] [CrossRef]
- Brunner, A.M.; Weng, S.; Cronin, A.; Fathi, A.T.; Habib, A.R.; Stone, R.; Graubert, T.; Steensma, D.P.; Abel, G.A. Impact of lenalidomide use among non-transfusion dependent patients with myelodysplastic syndromes. Am. J. Hematol. 2018, 93, 1119–1126. [Google Scholar] [CrossRef] [Green Version]
- Santini, V.; Almeida, A.; Giagounidis, A.; Gropper, S.; Jonasova, A.; Vey, N.; Mufti, G.J.; Buckstein, R.; Mittelman, M.; Platzbecker, U.; et al. Randomized Phase III Study of Lenalidomide Versus Placebo in RBC Transfu-sion-Dependent Patients with Lower-Risk Non-del(5q) Myelodysplastic Syndromes and Ineligible for or Refractory to Erythropoie-sis-Stimulating Agents. J. Clin. Oncol. 2016, 34, 2988–2996. [Google Scholar] [CrossRef]
- Santini, V.; Fenaux, P.; Giagounidis, A.; Platzbecker, U.; List, A.F.; Haferlach, T.; Zhong, J.; Wu, C.; Mavrommatis, K.; Beach, C.L.; et al. Impact of somatic mutations on response to lenalidomide in lower-risk non-del(5q) myelodysplastic syndromes patients. Leukemia 2021, 35, 897–900. [Google Scholar] [CrossRef] [PubMed]
- List, A.F.; Sun, Z.; Verma, A.; Bennett, J.M.; Komrokji, R.S.; McGraw, K.; Maciejewski, J.; Altman, J.K.; Cheema, P.S.; Claxton, D.F.; et al. Lenalidomide-Epoetin Alfa Versus Lenalidomide Monotherapy in Myelodysplastic Syndromes Re-fractory to Recombinant Erythropoietin. J. Clin. Oncol. 2021, 39, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Fenaux, P.; Van Eygen, K.; Raza, A.; Santini, V.; Germing, U.; Font, P.; Diez-Campelo, M.; Thepot, S.; Vellenga, E.; et al. Imetelstat Achieves Meaningful and Durable Transfusion Independence in High Transfusion–Burden Patients with Lower-Risk Myelodysplastic Syndromes in a Phase II Study. J. Clin. Oncol. 2021, 39, 48–56. [Google Scholar] [CrossRef]
- Santini, V.; Fenaux, P.; Van Eygen, K.; Raza, A.; Germing, U.; Font, P.; Diez-Campelo, M.; Thepot, S.; Vellenga, E.; Patnaik, M.M.; et al. On-Target Activity of Imetelstat Correlates with Clinical Benefits, Including Overall Sur-vival (OS), in Heavily Transfused Non-Del(5q) Lower Risk MDS (LR-MDS) Relapsed/Refractory (R/R) to Erythropoiesis Stimulating Agents (ESAs). Blood 2021, 138, 2598. [Google Scholar] [CrossRef]
- Henry, D.H.; Glaspy, J.; Harrup, R.; Mittelman, M.; Zhou, A.; Carraway, H.E.; Bradley, C.; Saha, G.; Modelska, K.; Bartels, P.; et al. Roxadustat for the treatment of anemia in patients with lower-risk myelodysplastic syndrome: Open-label, dose-selection, lead-in stage of a phase 3 study. Am. J. Hematol. 2021, 92, 174–184. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Santini, V. Targeting ineffective hematopoiesis in myelodysplastic syndromes. Am. J. Hematol. 2021, 97, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Carraway, H.E.; Saygin, C. Therapy for lower-risk MDS. Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 426–433. [Google Scholar] [CrossRef]
- Jabbour, E.; Short, N.J.; Montalban-Bravo, G.; Huang, X.; Bueso-Ramos, C.; Qiao, W.; Yang, H.; Zhao, C.; Kadia, T.; Borthakur, G.; et al. Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood 2017, 130, 1514–1522. [Google Scholar] [CrossRef] [Green Version]
- Musto, P.; Maurillo, L.; Spagnoli, A.; Gozzini, A.; Rivellini, F.; Lunghi, M.; Villani, O.; Aloe-Spiriti, M.A.; Venditti, A.; Santini, V.; et al. Azacitidine for the treatment of lower risk myelodysplastic syndromes: A retrospective study of 74 patients enrolled in an Italian named patient program. Cancer 2010, 116, 1485–1494. [Google Scholar] [CrossRef]
- Dhillon, S. Decitabine/Cedazuridine: First Approval. Drugs 2020, 80, 1373–1378. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; McCloskey, J.K.; Griffiths, E.A.; Yee, K.; Zeidan, A.M.; Al-Kali, A.; Deeg, H.J.; Patel, P.; Sabloff, M.; Keating, M.-M.; et al. Oral Decitabine/Cedazuridine in Patients with Lower Risk Myelodysplastic Syndrome: A Longer-Term Follow-up of from the Ascertain Study. Blood 2021, 138, 66. [Google Scholar] [CrossRef]
- Wei, A.H.; Döhner, H.; Pocock, C.; Montesinos, P.; Afanasyev, B.; Dombret, H.; Ravandi, F.; Sayar, H.; Jang, J.-H.; Porkka, K.; et al. Oral Azacitidine Maintenance Therapy for Acute Myeloid Leukemia in First Remission. N. Engl. J. Med. 2020, 383, 2526–2537. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Santini, V.; Almeida, A.; Platzbecker, U.; Jonasova, A.; Silverman, L.R.; Falantes, J.; Reda, G.; Buccisano, F.; Fenaux, P.; et al. Phase III, Randomized, Placebo-Controlled Trial of CC-486 (Oral Azacitidine) in Patients with Lower-Risk Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Roboz, G.J.; Collins, R.; Sekeres, M.A.; Stone, R.M.; Attar, E.C.; Frattini, M.G.; et al. Enasidenib in patients with mutant IDH2 myelodysplastic syndromes: A phase 1 subgroup analysis of the multicentre, AG221-C-001 trial. Lancet Haematol. 2020, 7, e309–e319. [Google Scholar] [CrossRef]
- Di Nardo, C.D.; Foran, J.M.; Watts, J.M.; Stein, E.M.; de Botton, S.; Fathi, A.T.; Prince, G.T.; Stein, A.S.; Stone, R.M.; Patel, P.A.; et al. Ivosidenib (IVO) in patients with IDH1-mutant relapsed/refractory myelodysplastic syn-drome (R/R MDS): Updated enrollment of a phase 1 dose escalation and expansion study. Clin. Lymphoma Mueloma Leuk. 2020, 20, s321. [Google Scholar] [CrossRef]
- Sebert, M.; Cluzeau, T.; Beyne Rauzy, O.; Bastard, A.S.; Dimicoli-Salazar, S.; Thepot, S.; Peterlin, P.; Park, S.; Gourin, M.-P.; Park, S.; et al. Ivosidenib Monotherapy Is Effective in Patients with IDH1 Mutated Myelodysplastic Syndrome (MDS): The Idiome Phase 2 Study by the GFM Group. Blood 2021, 138, 62. [Google Scholar] [CrossRef]
- Ades, L.; Dimicoli-Salazar, S.; Sebert, M.; Cluzeau, T.; Bastard, A.S.; Laribi, K.; Fossard, G.; Itzykson, R.; Rauzy, O.B.; Garnier, A.; et al. Enasidenib (ENA) Is Effective in Patients with IDH2 Mutated Myelodysplastic Syndrome (MDS): The Ideal Phase 2 Study by the GFM Group. Blood 2021, 138, 63. [Google Scholar] [CrossRef]
- Meng, F.; Chen, X.; Yu, S.; Ren, X.; Liu, Z.; Fu, R.; Li, L. Safety and Efficacy of Eltrombopag and Romiplostim in Myelodysplastic Syndromes: A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 582686. [Google Scholar] [CrossRef]
- Oliva, E.N.; Alati, C.; Santini, V.; Poloni, A.; Molteni, A.; Niscola, P.; Salvi, F.; Sanpaolo, G.; Balleari, E.; Germing, U.; et al. Eltrombopag versus placebo for low-risk myelodysplastic syndromes with thrombocytopenia (EQoL-MDS): Phase 1 results of a single-blind, randomised, controlled, phase 2 superiority trial. Lancet Haematol. 2017, 4, e127–e136. [Google Scholar] [CrossRef]
- Vicente, A.; Patel, B.A.; Gutierrez-Rodrigues, F.; Groarke, E.M.; Giudice, V.; Lotter, J.; Feng, X.; Kajigaya, S.; Weinstein, B.; Barranta, E.; et al. Eltrombopag monotherapy can improve hematopoiesis in patients with low to intermediate risk-1 myelodysplastic syndrome. Haematologica 2020, 105, 2785–2794. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Giles, F.J.; Greenberg, P.L.; Paquette, R.L.; Wang, E.S.; Gabrilove, J.L.; Garcia-Manero, G.; Hu, K.; Franklin, J.L.; Berger, D.P. Phase 2 study of romiplostim in patients with low- or intermediate-risk myelodysplastic syndrome receiving azacitidine therapy. Blood 2010, 116, 3163–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giagounidis, A.; Mufti, G.J.; Fenaux, P.; Sekeres, M.A.; Szer, J.; Platzbecker, U.; Kuendgen, A.; Gaidano, G.; Wiktor-Jedrzejczak, W.; Hu, K.; et al. Results of a randomized, double-blind study of romiplostim versus placebo in patients with low/intermediate-1-risk myelodysplastic syndrome and thrombocytopenia. Cancer 2014, 120, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Fenaux, P.; Sekeres, M.A.; Szer, J.; Platzbecker, U.; Kuendgen, A.; Gaidano, G.; Wiktor-Jedrzejczak, W.; Carpenter, N.; Mehta, B.; et al. Long-term follow-up for up to 5 years on the risk of leukaemic progression in thrombocytopenic patients with lower-risk myelodysplastic syndromes treated with romiplostim or placebo in a randomised dou-ble-blind trial. Lancet Haematol. 2018, 5, e117–e126. [Google Scholar] [CrossRef]
- Stahl, M.; Deveaux, M.; De Witte, T.; Neukirchen, J.; Sekeres, M.A.; Brunner, A.M.; Roboz, G.J.; Steensma, D.P.; Bhatt, V.R.; Platzbecker, U.; et al. The use of immunosuppressive therapy in MDS: Clinical outcomes and their predictors in a large international patient cohort. Blood Adv. 2018, 2, 1765–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunthararajah, Y.; Nakamura, R.; Wesley, R.; Wang, Q.J.; Barrett, A.J. A simple method to predict response to immunosuppressive therapy in patients with myelodysplastic syndrome. Blood 2003, 102, 3025–3027. [Google Scholar] [CrossRef] [Green Version]
- Trowbridge, J.J.; Starczynowski, D.T. Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. J. Exp. Med. 2021, 218, e20201544. [Google Scholar] [CrossRef]
- Kawano, H.; LaMere, M.W.; Byun, D.K.; Gordnier, C.L.; LaMere, E.A.; Ho, T.-C.; Ashton, J.M.; Frisch, B.J.; Bajaj, J.; Liesveld, J.L.; et al. Interleukin-1/Toll-like Receptor Inhibition Can Restore the Disrupted Bone Marrow Microen-vironment in Mouse Model of Myelodysplastic Syndromes. Blood 2021, 138, 1510. [Google Scholar] [CrossRef]
- Gyan, E.; Andrieu, V.; Sanna, A.; Caille, A.; Schemenau, J.; Sudaka, I.; Siguret, V.; Malet, M.; Park, S.; Bordessoule, D.; et al. Myelodysplastic syndromes with single neutropenia or thrombocytopenia are rarely refractory cytopenias with unilineage dysplasia by World Health Organization 2008 criteria and have favorable prognosis. Br. J. Haematol. 2016, 175, 975–979. [Google Scholar] [CrossRef]
- Alessandrino, E.P.; Della Porta, M.G.; Malcovati, L.; Jackson, C.H.; Pascutto, C.; Bacigalupo, A.; Van Lint, M.T.; Falda, M.; Bernardi, M.; Onida, F.; et al. Optimal timing of allogeneic hematopoietic stem cell transplantation in patients with myelodysplastic syndrome. Am. J. Hematol. 2013, 88, 581–588. [Google Scholar] [CrossRef]
- de Witte, T.; Bowen, D.; Robin, M.; Malcovati, L.; Niederwieser, D.; Yakoub-Agha, I.; Mufti, G.J.; Fenaux, P.; Sanz, G.; Martino, R.; et al. Allogeneic hematopoietic stem cell transplantation for MDS and CMML: Recommendations from an international expert panel. Blood 2017, 129, 1753–1762. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santini, V. Treatment of Lower Risk Myelodysplastic Syndromes. Hemato 2022, 3, 153-162. https://doi.org/10.3390/hemato3010013
Santini V. Treatment of Lower Risk Myelodysplastic Syndromes. Hemato. 2022; 3(1):153-162. https://doi.org/10.3390/hemato3010013
Chicago/Turabian StyleSantini, Valeria. 2022. "Treatment of Lower Risk Myelodysplastic Syndromes" Hemato 3, no. 1: 153-162. https://doi.org/10.3390/hemato3010013
APA StyleSantini, V. (2022). Treatment of Lower Risk Myelodysplastic Syndromes. Hemato, 3(1), 153-162. https://doi.org/10.3390/hemato3010013