1. Introduction
Immune checkpoint inhibitors (ICIs) augment existing anti-tumor T-cell responses that have been rendered ineffective by inhibitory pathways, including the upregulation of inhibitory receptors referred to as immune checkpoints. Programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) are the most well-known targetable immune checkpoints. CTLA-4 serves to limit initiation of T-cell activation by competing with CD28 in binding with the co-stimulatory T-cell ligands, B7-1 (CD80) or B7-2 (CD86), preventing overactivation [
1,
2]. Monoclonal antibodies that block the binding of CTLA-4 to CD28 ligands enhance the strength and duration of initial T-cell activation [
3]. PD-1, on the other hand, functions to dampen T-cell receptor signaling following persistent antigen stimulation within the tumor microenvironment [
4]. Such chronic stimulation induces T-cell exhaustion, a process of T-cell reprogramming. This process includes upregulation of PD-1, which, upon binding to its ligands, programmed death-ligand 1 (PD-L1) or programmed death-ligand 2 (PD-L2), inhibits T-cell functioning in peripheral tissues [
4]. Although PD-1 ligands are normally expressed on many tissues, some cancer cells also highly express PD-L1 [
1,
5,
6]. Monoclonal antibodies designed to disrupt the interaction between PD-1 and PD-L1, by binding to either molecule, lead to amplified T-cell receptor signaling [
2]. This, in turn, revitalizes the proliferation and functionality of exhausted T cells, enabling them to once again initiate an effective anti-tumor response [
2]. It is essential to note that PD-1 operates through mechanisms different from CTLA-4, providing a biological rationale for the synergistic use of immune checkpoint inhibitors targeting both pathways.
These ICIs have transformed the treatment of several types of cancer in adults. There are currently six Food and Drug Administration (FDA)-approved ICIs targeting components of the PD-1/PD-L1 interaction and one FDA-approved ICI targeting CTLA-4: nivolumab (anti–PD-1); pembrolizumab (anti–PD-1); cemiplimab (anti–PD-1); atezolizumab (anti–PD-L1); durvalumab (anti–PD-L1); avelumab (anti–PD-L1); and ipilimumab (anti-CTLA-4). Notably, therapies targeting the PD-1 pathway have been granted approval by the FDA for the treatment of eighteen different types of cancer in adults that affect the skin, lung, head and neck, genitourinary, gastrointestinal, and lymphoid systems [
2]. Their utilization often results in a 10% to 30% improvement in overall response rates compared to conventional therapies [
7]. Furthermore, ICIs are being integrated into initial treatment approaches for adult cancers, such as metastatic non-small cell lung cancer, where the addition of pembrolizumab to standard-of-care chemotherapy has led to an additional 10% to 25% objective response rate compared to chemotherapy alone [
8].
With few exceptions, the application of ICIs in the realm of pediatric oncology has not experienced the same magnitude of success. This likely stems from an inherent difference in the immunogenicity of pediatric cancers, which are known to have a far lower tumor mutational burden (TMB) and fewer neoantigens than adult cancers [
9]. Despite this, certain types of pediatric malignancies have benefited from the use of ICIs, namely, Hodgkin lymphoma, metastatic melanoma, and the malignancies that afflict patients with constitutional mismatch repair deficiency syndrome (CMMRD).
The underlying biology of Hodgkin lymphoma is a known critical factor influencing susceptibility to immune checkpoint inhibition. The pathologic Reed–Sternberg cells of Hodgkin lymphoma exhibit genetic alterations in chromosome 9p24.1, leading to the overexpression of PD-L1 and PD-L2. A study by Karim et al. revealed a significantly high expression of PD-L1 in all examined Hodgkin lymphoma cases (10/10) [
10]. This genetic profile makes classic Hodgkin lymphoma particularly susceptible to checkpoint blockade. The introduction of ICIs has demonstrated significant efficacy, with an objective response rate of 60% in cases of relapsed or refractory Hodgkin lymphoma [
5]. Pembrolizumab is currently approved for this indication.
In 2017, the FDA granted approval for the use of ipilimumab in the treatment of unresectable or metastatic melanoma in pediatric patients aged twelve years or older following the success of two early-phase clinical trials [
11,
12]. More recently, the combination of nivolumab and ipilimumab has demonstrated considerable promise, with 58% of patients surviving to the three-year mark. Encouraging results have also been demonstrated with this combination therapy in patients with intracranial metastases who had previously failed conventional treatments, those with leptomeningeal disease, or those exhibiting neurological symptoms. In this challenging cohort, 46% of patients demonstrated an intracranial response at the seventeen-month mark [
13].
As previously discussed, pediatric cancers typically exhibit a quiet mutational landscape with low expression of neoantigens, resulting in a limited ability to activate an immune response. Tumors in patients with CMMRD, however, are an exception due to their extraordinarily elevated TMB. This heightened mutational burden arises from a germline biallelic loss of mismatch repair function, leading to a devastating cancer predisposition syndrome with childhood onset, characterized by aggressive and challenging cancers. The use of ICIs in CMMRD was first reported in 2016 [
14]. A subsequent report of therapy with either nivolumab or pembrolizumab in 45 progressive or recurrent tumors from 38 patients with CMMRD demonstrated an objective response and/or stable disease in 55.5% of malignancies. Furthermore, the overall survival at three years for the entire cohort was 41.4%, indicating a dramatic improvement in survival for a patient population that historically has faced dismal outcomes [
15].
While advances in chemotherapy protocols, radiation therapy, and surgical techniques have dramatically improved overall survival, relapsed pediatric malignancies remain a formidable challenge [
16]. These treatments often leave patients grappling with a host of treatment-related side effects, which can profoundly impact their quality of life. In response to this dilemma and the limitations of current salvage therapies, ICIs have emerged as a promising therapeutic approach. Designed to reengage a patient’s own immune system, ICIs offer a unique advantage by eradicating cancer cells while concurrently mitigating many of the unfortunate sequelae associated with conventional therapies. The notable success of ICIs in treating both adult malignancies and specific pediatric cancers has not only transformed the landscape of cancer care but has also ignited efforts in pediatric oncology to explore their application across various tumor types. Ongoing research is dedicated to accurately identifying pediatric patients who stand to benefit most from the targeted and less debilitating effects of these innovative therapies.
At our large but non-academic institution, PD-L1 testing is most often performed in heavily pretreated patients who have failed multiple lines of traditional treatment, including chemotherapy, surgery, and radiation, with the hope of offering immunotherapy as a salvage treatment option. PD-L1 expression is tested through all available methods, often yielding complex results, characterized by a variety of PD-L1 clones and measurements of expression. Nevertheless, this approach is undertaken with the hope of detecting indications that the malignancy may respond favorably to immunotherapy. This is the challenging reality confronting pediatric oncologists as they navigate the search for viable treatment options for refractory and often rare malignancies. In the following sections, we aim to describe a single institution’s experience with immune checkpoint inhibition in seven pediatric patients with a variety of malignancies, and hope to advance the exploration of these treatment modalities in the treatment of children with cancer.
2. Case Descriptions
At our institution, we performed a retrospective chart review and identified patients who received ICIs and/or underwent immunohistochemistry (IHC) testing for PD-L1, quantification of TMB, and classification of microsatellite instability (MSI) status via two commercial companies, Foundation Medicine and Tempus. In total, we identified seven patients who received therapy with ICIs, including nivolumab and ipilimumab. All but three of these patients demonstrated positive PD-L1 expression, high TMB, and/or MSI-high status.
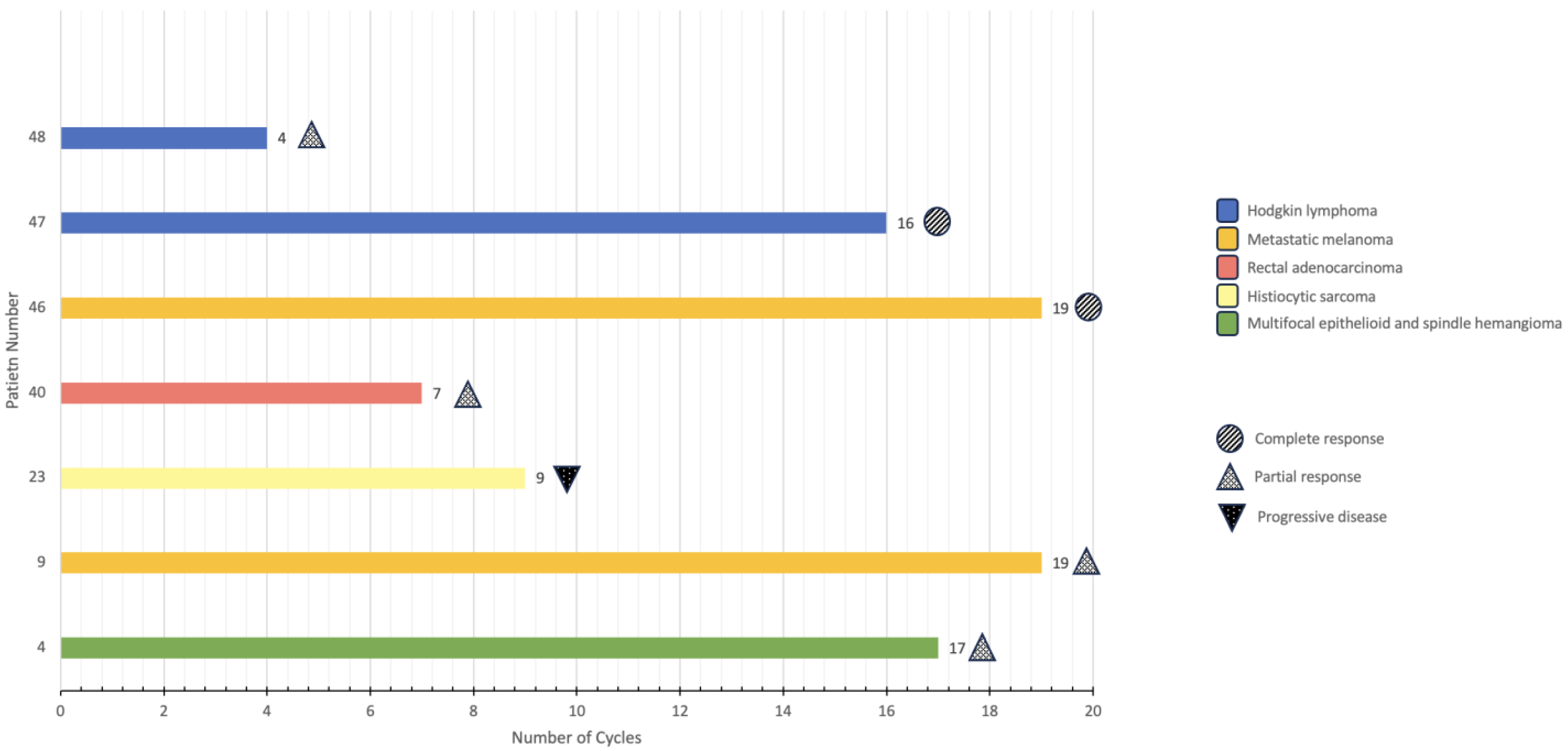
Of the seven patients identified, three were females, and four were males. Age at diagnosis ranged from eleven to seventeen with a median age of thirteen. The diagnoses included: multifocal epithelioid and spindle cell hemangioma (n = 1); metastatic melanoma (n = 2); histiocytic sarcoma (n = 1); rectal adenocarcinoma in the setting of CMMRD (n = 1); and Hodgkin lymphoma (n = 2). Two patients underwent genomic testing via Foundation Medicine, and two patients underwent genomic testing via Tempus. Three patients did not undergo genomic testing but received ICIs based on their malignancy (patients 46, 47, and 48).
The patients described received between four and nineteen cycles of immunotherapy as of November 2023 (
Figure 1). The different immunotherapy regimens employed included: nivolumab exclusively; nivolumab plus ipilimumab; and nivolumab plus brentuximab. Immunotherapy-related symptoms included: diffuse itching (n = 1); headache (n = 1); allergic reaction (n = 1); nausea/vomiting (n = 1); transaminitis (n = 1); flu-like symptoms (n = 1); self-limited neuritis (n = 1); low thyroid-stimulating hormone (TSH) (n = 1); back pain (n = 2); anemia (n = 1); neutropenia (n = 1); mild rash (n = 1); decreased appetite/anorexia (n = 2); and weight loss (n = 1). Overall, all patients tolerated the infusions well without significant adverse events that would require the discontinuation of immunotherapy. Of these seven patients, three are still being treated with immunotherapy, one is being treated with other modalities, two have achieved remission, and one has relapsed.
The clinical courses of these seven patients, including age at diagnosis, immunotherapy biomarkers (if genomic testing was performed), current status or therapeutic outcome, and adverse effects experienced with immunotherapy (if any), are outlined below.
2.1. Patient 4: Multifocal Epithelioid and Spindle Cell Hemangioma
Patient 4 is a fourteen-year-old male who was diagnosed with a multifocal epithelioid and spindle cell hemangioma of the left hand and wrist at age thirteen. Magnetic resonance imaging (MRI) of the left hand revealed multiple enhancing osseous lesions involving the second, third, fourth, and fifth metacarpals; the third proximal phalanx; the hamate; the capitate; and the trapezoid. Diagnostic biopsy and curettage of the dominant lesion in the fourth metacarpal revealed an epithelioid and spindle cell hemangioma.
Less than a month after the biopsy was performed, the patient proceeded to surgery for excision and curettage of the lesions. The initial treatment strategy after surgery was observation. The tumor exhibited rapid progression, leading to significantly increased swelling and the development of neurological symptoms. His surgeon opined that with this rapid progression, another debulking procedure would not benefit the patient and that the only option for definitive control was amputation.
IHC testing using the Dako® PD-L1 22C3 clone revealed a tumor proportion score (TPS) of 10%. Based on this result and to avoid or delay amputation of the distal left upper extremity, the patient was initiated on a regimen of nivolumab (3 milligrams (mg)/kilogram (kg) intravenous (IV) every three weeks) and ipilimumab (1 mg/kg IV every three weeks). He received four cycles of this combination, then he was switched to a regimen of nivolumab (3 mg/kg IV every two weeks) exclusively. As of October 2023, he has received thirteen cycles of nivolumab alone and seventeen cycles of immunotherapy in total. Overall, he has tolerated the infusions well without significant adverse effects. His only reported symptom was diffuse itching post-infusion, but he did not develop a rash. With immunotherapy, several of the lesions have mildly decreased in size, and the patient is scheduled to undergo another debulking procedure. In the meantime, he will continue receiving nivolumab.
2.2. Patient 9: Metastatic Melanoma
Patient 9 is a twenty-year-old female with a history of several congenital nevi who was diagnosed with melanoma of the upper forehead via a shave biopsy at age seventeen. She promptly underwent wide local excision of the lesion and sentinel lymph node biopsy. Excision of the lesion demonstrated residual melanoma, pathologic stage 1A with negative but close margins. The sentinel lymph node biopsy was negative for metastatic disease.
Shortly after this procedure, the patient was lost to follow-up. Approximately a year after the procedure, the patient presented with an enlarged lymph node on the right side of the neck. Excision of this submandibular lymph node revealed metastatic melanoma. She then underwent a modified radical dissection of the right neck, which demonstrated no evidence of metastatic melanoma in thirty lymph nodes. There was, however, a nodular focus of melanoma within the subcutis of her previous incision. Thus, she was re-staged and diagnosed with nodular recurrence of melanoma, pathologic stage IIIB.
TMB was 16.3 mutations (m)/megabase (MB), in the 93rd percentile. She also demonstrated a BRAF V600E mutation. Based on these results, the patient proceeded with adjuvant immunotherapy consisting of nivolumab (3 mg/kg IV every two weeks). To date, she has received nineteen cycles of nivolumab. In total, fifty-two weeks of therapy, or twenty-seven cycles of nivolumab, are planned. Overall, she has tolerated the infusions well without significant adverse effects. Her only reported symptom was a headache post-infusion. Restaging scans have demonstrated no evidence of disease in the brain, neck, chest, abdomen, or pelvis.
2.3. Patient 23: Histiocytic Sarcoma
Patient 23 is a sixteen-year-old male with a history of B-cell acute lymphoblastic leukemia (B-ALL) diagnosed with histiocytic sarcoma of the left thorax at age thirteen. Imaging revealed a mass involving the left parietal and visceral pleura, a large left-sided pleural effusion, and an anterior mediastinal mass.
Initially, clinicians were concerned that the patient’s B-ALL had relapsed, but sampling of the left-sided pleural effusion and bilateral bone marrow aspirates and biopsies demonstrated no evidence of malignancy or atypia. The diagnosis was confirmed when surgeons biopsied the pleura on the left side. Biopsy demonstrated non-LCH, with the neoplastic cells staining positive for CD68, CD163, fascin, and factor XIIIa and negative for S100, CD1a, Pax-5, ALK, CD3, and CD20.
Initial treatment consisted of prednisone. After the patient tapered and discontinued the steroids, the left-sided pleural effusion, which was drained during his initial hospitalization, reaccumulated. Additionally, the pleural-based lesions and anterior mediastinal mass progressed. Thus, a more aggressive treatment approach was taken, and the patient began therapy with clofarabine. Initially, imaging demonstrated a positive treatment response after two cycles of clofarabine, but repeat imaging after the fourth cycle of clofarabine demonstrated no significant change. Thus, the patient underwent a fifth and final but truncated cycle of clofarabine.
IHC testing using the Dako® PD-L1 22C3 clone revealed a TPS of 20%. Based on this result and the lack of further response to clofarabine, the patient was switched to therapy with nivolumab (3 mg/kg IV weekly). He had a reaction to his second infusion of nivolumab, developing facial flushing and reporting symptoms of dyspnea, throat tightness, abdominal pain, and facial numbness. The infusion was stopped, and single doses of intravenous (IV) diphenhydramine (1 mg/kg) and IV hydrocortisone (1 mg/kg) were administered. The facial flushing and symptoms resolved, and he was able to restart and complete the infusion at a slower rate. From then on, he was pre-medicated with diphenhydramine prior to an immunotherapy infusion.
He received two three-week cycles of nivolumab, then ipilimumab was added to the regimen. With this modified regimen, he received nivolumab (3 mg/kg IV) every two weeks and ipilimumab (1 mg/kg IV) every six weeks. Additionally, adjuvant therapy with radiation was initiated. Early in this regimen, he presented multiple times with reaccumulation of the left-sided pleural effusion, ultimately requiring left-sided pleurodesis. In total, he received nine six-week cycles of combination immunotherapy, ending therapy in June 2022 after imaging continued to demonstrate no evidence of progression. With the administration of diphenhydramine prior to each infusion, he tolerated the infusions well without significant adverse effects.
Ten months off immunotherapy, a positron emission tomography (PET) scan demonstrated nodular uptake in the duodenum and the right hemipelvis, concerning for progression. Imaging findings in the left thorax, the initial site of disease, have remained stable. Biopsy of the duodenal lesion demonstrated non-LCH. Thus, the patient started therapy with trametinib (2 mg by mouth daily). Reported side effects include the development of facial and back acne and dandruff. Otherwise, the patient has tolerated trametinib without significant adverse effects. We have described this patient’s unique diagnosis and clinical course in detail in a case report [
17].
2.4. Patient 40: Rectal Adenocarcinoma
Patient 40 is a twelve-year-old male recently diagnosed with rectal adenocarcinoma. Imaging demonstrated perirectal soft tissue infiltration and prominent perirectal lymph nodes, indicating local metastasis, but there was no evidence of distant metastasis. The patient began chemotherapy with folinic acid, 5-fluorouracil, and oxaliplatin (FOLFOX), scheduled for administration every 2 weeks.
Then, testing revealed a TMB of 86.3 m/MB, in the 100th percentile. His MSI status was categorized as high, and germline testing revealed a base substitution in the PMS2 gene, leading to constitutional mismatch repair deficiency syndrome (CMMRD). Notably, no pathogenic alterations were identified in the KRAS, NRAS, or BRAF genes.
Based on these results, nivolumab (3 mg/kg IV) was added to the third and fourth cycles of FOLFOX. Although repeat imaging continued to demonstrate a progressive decrease in the size of the mass, after the fourth cycle of FOLFOX (and second cycle with nivolumab added), the patient was switched to combination immunotherapy with nivolumab (3 mg/kg IV every three weeks) and ipilimumab (1 mg/kg IV every three weeks) after consultation with experts. After receiving four cycles of combination immunotherapy, as per the National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines, the patient was switched to nivolumab exclusively (3 mg/kg IV every two weeks). Recent surveillance imaging continues to demonstrate a positive treatment response.
During his second infusion of nivolumab, the patient experienced severe back pain. The infusion was stopped, ibuprofen was administered, and he was able to resume the infusion an hour later. Thus, with every subsequent infusion, the patient was pre-medicated with ibuprofen, and nivolumab was infused at a slower rate. Additionally, the patient experienced immunotherapy-induced anemia and neutropenia, the former of which was managed with transfusions of packed red blood cells for a hemoglobin level of less than 8 grams (g)/deciliter (dL). Otherwise, the patient tolerated the infusions well without significant adverse effects.
2.5. Patient 46: Metastatic Melanoma
Patient 46 is a twenty-one-year-old male who was diagnosed with melanoma of the right neck via a shave biopsy at age seventeen. He promptly underwent full-thickness excision of the lesion, which demonstrated residual melanoma, pathologic stage IIB with positive margins. Sentinel lymph node biopsy was negative for metastatic disease. He then underwent definitive wide excision and reconstruction. Imaging was negative for distant metastasis. Thus, the patient was monitored closely thereafter.
Approximately twenty-two months after the initial melanoma was diagnosed, the patient relapsed, as imaging revealed innumerable metastases in the brain, lungs, lymph nodes, kidneys, vertebrae, and skin. The patient was promptly started on a regimen of combination immunotherapy, consisting of nivolumab (1 mg/kg IV every three weeks) and ipilimumab (3 mg/kg IV every three weeks). He also received whole-brain radiation with boosts directed at the intracranial metastases.
He tolerated the first infusion of immunotherapy fairly well, developing flu-like symptoms immediately after the infusion that resolved without intervention. He also developed sharp back pain during the second infusion that resolved with the administration of diphenhydramine, allowing him to resume and complete the infusion. From then on, he was pre-medicated with diphenhydramine. He also developed nausea, vomiting, and grade 3 transaminitis after the fourth infusion, which required hospitalization. This transaminitis improved to grade 1 with a course of corticosteroids. Repeat imaging demonstrated an excellent treatment response, with significantly improved burden of disease in the brain, neck, chest, abdomen, and pelvis.
Thus, after the fourth cycle of combination immunotherapy and after improvement of the transaminitis, the patient was switched to nivolumab alone (3 mg/kg IV every two weeks). With maintenance immunotherapy, the patient experienced an episode of nausea, anorexia, weight loss, and low TSH (<0.1 mU/L), which again resolved with a course of corticosteroids. He also experienced two brief episodes of numbness and tingling on the right side of his body that resolved without intervention and a third episode that progressed to altered mental status (dysarthria and decreased responsiveness). That episode lasted several hours and required hospitalization but then resolved spontaneously.
The patient ultimately received fifteen six-week cycles of nivolumab alone and nineteen cycles of immunotherapy in total. Based on surveillance imaging that demonstrated no evidence of disease, the patient discontinued immunotherapy and is being monitored closely.
2.6. Patient 47: Hodgkin Lymphoma
Patient 47 is an eighteen-year-old female with a complex past medical history, including cerebral palsy, diagnosed with classic Hodgkin lymphoma, nodular sclerosis type, at age fifteen. A CT scan revealed an anterior mediastinal mass occupying more than one third of the chest and bilateral supraclavicular lymphadenopathy. Thus, the patient’s disease was classified as stage IIA with bulk disease in the mediastinum. She promptly began chemotherapy as per Children’s Oncology Group (COG) AHOD1131. After two cycles of this regimen, repeat imaging demonstrated a partial response with a Deauville score of 4. Thus, she was classified as a slow early responder (SER). The patient received two more cycles of chemotherapy as per COG AHOD1131 and then proceeded to involved-site radiation therapy (ISRT) for consolidation of treatment given her SER status. The end-of-therapy PET scan demonstrated no evidence of active lymphoma.
Twenty-two months off therapy, the patient relapsed, as a PET scan demonstrated several avid lymph nodes above the diaphragm. Thus, the patient began salvage therapy with brentuximab (1.8 mg/kg IV every three weeks) and nivolumab (3 mg/kg IV every three weeks). She developed a mild, erythematous rash and decreased appetite after the first cycle of immunotherapy. Otherwise, she tolerated the infusions well without significant adverse effects. A repeat PET scan after two cycles of immunotherapy demonstrated a remarkable and complete treatment response. In total, the patient received sixteen cycles of combination immunotherapy with brentuximab and nivolumab. Clinicians recommended high-dose chemotherapy with autologous stem cell rescue followed by ISRT to consolidate treatment, but the patient’s guardian did not wish to proceed with these therapies given the patient’s comorbidities. The end-of-therapy PET scan was negative for disease. Currently, the patient is being monitored off therapy and continues to do well.
2.7. Patient 48: Hodgkin Lymphoma
Patient 48 is a twelve-year-old female diagnosed with classic Hodgkin lymphoma, nodular sclerosis type, at age eleven. Imaging revealed disease above and below the diaphragm but no visceral organ involvement. Thus, her disease was classified as stage IIIA, intermediate risk. She promptly began chemotherapy as per COG AHOD0031. After four cycles of this regimen, a PET scan demonstrated several avid lymph nodes, concerning for residual/refractory disease. As the findings were questionable and could have been due to inflammation or infection, the clinicians decided to observe and repeat imaging in four weeks.
Repeat imaging demonstrated definite progression of disease, with hypermetabolic lymphadenopathy in the neck, chest, and upper abdomen. Thus, the patient began salvage therapy with brentuximab (1.8 mg/kg IV every three weeks) and nivolumab (3 mg/kg IV every three weeks), as per COG AHOD1721. Overall, she tolerated the immunotherapy infusions well without significant adverse effects. After completing four cycles of combined immunotherapy with brentuximab and nivolumab, the patient appeared to exhibit progression of disease, with imaging demonstrating increased size and PET avidity of lymph nodes in several regions. Thus, the patient started salvage chemotherapy with ifosfamide, gemcitabine, and vinorelbine.
Biopsy of two of the involved lymph nodes, however, did not demonstrate disease, and repeat imaging demonstrated resolution of the previously enlarged and avid lymph nodes. These imaging findings were thus deemed to be infectious in nature, and the patient proceeded to high-dose chemotherapy followed by autologous stem cell rescue. Repeat imaging was negative for disease. Post-transplant maintenance immunotherapy and ISRT were recommended following the patient’s recovery. Thus, she is scheduled to begin brentuximab (1.8 mg/kg IV every three weeks for up to sixteen cycles) and radiation in the immediate future.
3. Discussion
Immunotherapy represents an exciting and rapidly evolving front of precision, or personalized, medicine. Much of the focus of pediatric oncology research has shifted from survival to minimizing the negative effects of conventional therapies and seeking alternative therapies, such as immunotherapy and agents that target certain mutations, that often do not carry the weight of such toxicities. These alternative therapies also offer hope to patients with malignancies that have relapsed, are refractory to conventional therapies, or are rare and have no established standard of care that clinicians can refer to and follow. Nivolumab, for example, is a human IgG4 monoclonal antibody directed against PD-1 that has been approved by the FDA for the treatment of a rapidly increasing list of malignancies, including metastatic melanoma in patients aged twelve or older (as an adjuvant therapy), relapsed/progressive classic Hodgkin lymphoma in adult patients, and metastatic colorectal cancer in patients with high microsatellite instability or mismatch repair deficiency in patients aged twelve or older [
18,
19]. Ipilimumab, the first ICI approved by the FDA, is a human IgG1 monoclonal antibody directed against CTLA-4 that has also been approved for the treatment of metastatic melanoma in patients aged twelve or older (as an adjuvant therapy) and metastatic colorectal cancer in patients with high microsatellite instability or mismatch repair deficiency in patients aged twelve or older (in combination with nivolumab) [
19,
20].
The most common adverse reactions caused by nivolumab and ipilimumab include fatigue, rash, pruritus, nausea, decreased appetite, back pain, and headache [
18,
20]. Many of these symptoms were observed in our patients but were effectively managed by targeting the symptom (i.e., premedicating the patient with ibuprofen for back pain) and decreasing the rate of infusion, as shown in
Table 1. This allowed many patients to continue receiving immunotherapy. More serious adverse reactions have been observed with the administration of nivolumab and/or ipilimumab, including immune-mediated pneumonitis, colitis, hepatitis, endocrinopathies (adrenal insufficiency, hyperglycemia, hypothyroidism, etc.), nephritis and renal dysfunction, encephalitis, and infusion reactions [
18,
20]. We also observed immune-mediated adverse reactions in our patients, including low TSH and cytopenias, but these effects resolved or were managed with courses of corticosteroids and transfusions, respectively.
Despite these risks, immunotherapy has been established as an effective and viable treatment option, not only for patients with relapsed or refractory malignancies, but also for patients with rare, difficult-to-treat malignancies. It even appears that some ICIs are more effective than others. For example, in a randomized, double-blind, phase 3 clinical trial, researchers established that adjuvant therapy with nivolumab in patients with completely resected melanoma resulted in higher recurrence-free survival rates than adjuvant therapy with ipilimumab (70.5% versus 60.8%, respectively) [
19]. Furthermore, nivolumab was less toxic, causing treatment-related grade 3 or 4 adverse reactions in only 14.4% of patients, whereas ipilimumab caused such events in 45.9% of patients [
19]. These results, especially regarding toxicity, have guided our clinicians, who often choose to treat patients with nivolumab alone and/or incorporate ipilimumab into as few cycles as possible.
Although immunotherapy had been established as an effective treatment option for our two patients with metastatic melanoma, our two patients with classic Hodgkin lymphoma, and our patient with rectal adenocarcinoma due to CMMRD, the utilization of immunotherapy was guided by genomic testing results in our patient with multifocal epithelioid and spindle cell hemangioma and in our patient with histiocytic sarcoma. Epithelioid and spindle cell hemangioma is a morphologic variant of epithelioid hemangioma that can arise in the bone or soft tissues and almost uniformly afflicts the extremities, especially the hands and feet, as it did in our patient [
21]. A recent clinicopathologic analysis of eighteen cases of epithelioid and spindle cell hemangioma observed that thirteen patients underwent surgical intervention (excision and/or curettage) and that seven (54%) of these patients experienced recurrence of the primary tumor [
21]. Follow-up data were not available for all patients, but for those with recent follow-up, nine demonstrated no evidence of disease, and six patients demonstrated stable/persistent disease [
21]. Of these six patients, four patients (80%) demonstrated cessation of disease progression after two years [
21]. Thus, the researchers concluded that epithelioid and spindle cell hemangioma, whether it is the primary tumor or a recurrence, behaves indolently; thus, observation is a viable treatment strategy for patients with multifocal disease not suitable for surgical intervention [
21]. Our patient, however, presented with an unusual clinical course that demanded action. Observation was initially employed after surgical intervention, but his lesions demonstrated rapid progression that caused severe neurological symptoms. Based on genomic testing, which demonstrated positive PD-L1 expression, a regimen consisting of nivolumab and ipilimumab was employed to avoid or delay amputation of the distal left upper extremity. This approach was somewhat successful, not completely resolving the lesions, but shrinking them to a size where surgeons felt comfortable performing another debulking procedure.
The non-Langerhans cell histiocytoses comprise a group of histiocytic disorders/neoplasms that do not meet the phenotypic criteria of LCH (cells that stain positive for CD1a, langerin, and S100) and can be conceptually divided into three groups: cutaneous non-LCH; cutaneous non-LCH with a major systemic component; and systemic non-LCH [
22]. Our patient presented with perplexing histopathology and a past medical history of B-ALL but fits best into the category of systemic non-LCH and was specifically diagnosed with histiocytic sarcoma based on IHC results. Traditional therapies employed against non-LCH include corticosteroids, immunomodulatory agents, chemotherapy, radiation, and bone marrow transplantation, among others; these strategies have demonstrated wildly variable success [
23,
24]. For more than a decade, however, clinicians have been exploring the use of targeted agents to treat these rare malignancies, including trametinib, an
MEK inhibitor, as many of these patients exhibit mutations in the
MAPK pathway [
24].
This patient’s histiocytic sarcoma proved refractory to conventional therapies, including prednisone and clofarabine. Again, based on genomic testing, which demonstrated positive PD-L1 expression, with a high TPS of 20%, a regimen consisting of nivolumab and ipilimumab in combination with targeted radiation therapy was employed to prolong the patient’s survival. Surprisingly, this treatment approach achieved stability of disease, allowing the patient to return to school and participate in extracurricular activities. The cessation of immunotherapy, however, led to relapse at a different anatomic site, illustrating the challenges and uncertainty associated with the use of immunotherapy. The patient has started therapy with trametinib, as several studies and case reports have demonstrated success, including a recent multi-center analysis, which demonstrated a response rate of 71% in seventeen adult patients with non-LCH [
24]. Interestingly, eight out of eleven patients (73%) without a
BRAF mutation demonstrated an objective response—results that are encouraging for our patient, who did not demonstrate an
MAPK pathway mutation on genomic testing [
24].
As these cases demonstrate, the use of immunotherapy in pediatric oncology remains challenging and uncertain. This is in part due to the current dependence on biomarkers that are tailored to the fundamentally different immunobiology of adult cancers. PD-L1 expression, TMB, and MSI status are the biomarkers utilized to identify patients with potential susceptibility to immune checkpoint inhibition. For example, the use of PD-L1 expression as a biomarker is not straightforward, presenting a complex scenario when clinicians must interpret the results. The ability to re-engage exhausted T-cells in anti-tumor defense via immunotherapy is anticipated when tumors exhibit PD-L1 expression on the cell surface, making these malignancies seemingly suitable candidates for anti-PD-1 or anti-PD-L1 immunotherapies. Clinical trials, however, have uncovered a nuanced reality—not all tumors with PD-L1 expression demonstrate sensitivity to ICIs, and, conversely, not all PD-L1-negative tumors are resistant to ICIs [
5,
25]. For example, some patients with urothelial and renal cell carcinoma have demonstrated a positive treatment response to nivolumab or pembrolizumab despite little or negative PD-L1 expression [
26,
27]. Meanwhile, for patients with lung cancer and melanoma, there remains a generally positive association between clinical outcome and degree of PD-L1 expression, although PD-L1 expression is not absolutely required for therapeutic benefit [
25]. These complexities are further potentiated by variations in positivity thresholds for PD-L1 expression, which range from 1% or more of tumor cells to 50% or more, depending on the antibody clone used for immunohistochemical assessment and on the type of malignancy the patient has [
25]. Unlike in adult cancers, where degree of PD-L1 expression has shown some association with therapeutic outcomes, PD-L1 expression in pediatric tumors is sporadic in nature, and its use as a biomarker remains uncertain [
28,
29]. These challenges underscore the limited reliability of PD-L1 expression as a biomarker and indicator of likely success when anti-PD-1 or anti-PD-L1 therapies are being considered. Furthermore, this highlights the importance of integrating the interpretation of PD-L1 expression with the results of other biomarkers, such as TMB and MSI status.
TMB, a measure of mutational load, is affected by a variety of factors, including exposure to mutagens, such as ultraviolet light in melanoma and cigarette smoke in lung cancer; temozolomide-based chemotherapy in gliomas; mutations in the
POLE and
POLD1 genes, which encode the proofreading domains of DNA polymerases; and MSI. The higher neoantigen burden, resulting from a significant accumulation of mutations, intuitively leads to the recognition of the tumor as foreign and eradication by the immune system. In adult oncology, a TMB threshold of 10 or more m/MB has been associated with improved response to ICIs and prolonged progression-free survival, irrespective of PD-L1 expression status [
2,
30]. Clinical evidence in adult solid tumors indicates an association between increased TMB and greater sensitivity to immune checkpoint inhibition with anti-PD-L1, including combination therapy with nivolumab and ipilimumab [
30,
31]. While the search for an appropriate, evidence-based TMB threshold in pediatric oncology is still ongoing and traditionally the mutational landscape has been considered quiet, emerging investigations have reported findings that parallel the adult experience [
9]. For example, among pediatric patients with a central nervous system (CNS) tumor, a high TMB was found to correlate with higher rates of progression and death as compared to low-TMB tumors, indicating that these patients with high-TMB tumors could potentially benefit [
32]. Furthermore, Villani et al. examined a wide variety of pediatric cancers and noted that a significant proportion of high-TMB tumors were observed in relapsed childhood cancers and, notably, were not restricted to patients with CMMRD [
33]. This supports repeating genomic testing of relapsed pediatric cancers after initial therapy. Villani et al. also suggested a lower TMB cutoff of 5 or more m/MB for the pediatric population [
34].
Microsatellite instability (MSI) is a rare condition characterized by hypermutability resulting from defects in the DNA mismatch repair pathway. This instability manifests as changes in microsatellite repeat lengths, leading to the classification of tumors into two categories: MSI-high (MSI-H), indicating defective DNA mismatch repair activity, and microsatellite stable (MSS), where no detectable defects in DNA mismatch repair are present. Clinical evidence suggests a significant correlation between MSI status and the response to anti-PD-1 immune checkpoint inhibitors, such as nivolumab and pembrolizumab, with MSI-H tumors exhibiting a higher likelihood of responding to these inhibitors compared to MSS tumors.
MSI can be directly assessed by panels designed to detect changes in microsatellite repeat lengths. These panels, however, were initially developed for adult cancers and may present challenges in pediatric patients with CMMRD. Chung et al. demonstrated a considerably high false-negative rate with the use of these panels, as only 18% of confirmed CMMRD pediatric patients demonstrated high MSI [
35]. To address these limitations, it is recommended to employ additional methods, such as immunohistochemical staining for DNA mismatch repair proteins, when determining MSI status in pediatric cancers [
2]. This multi-faceted approach enhances the accuracy of MSI assessment and ensures more reliable results, particularly in the context of pediatric oncology.
It is important to recognize that no single biomarker can accurately predict an individual patient’s response to ICIs, likely because of the complex and dynamic nature of the tumor microenvironment and tumor cells’ interactions with host immune cells. Consequently, a comprehensive and evidence-based understanding of PD-L1 expression, TMB, and MSI status may aid in enhancing the accuracy of patient selection and optimizing the success of immune checkpoint inhibition in pediatric malignancies. Due to the mismatch between existing biomarkers and the immunobiological landscape of pediatric cancers, however, future research is trending towards the development of specialized biomarkers tailored to the unique context of pediatric oncology [
36].
The current landscape of clinical trials investigating immune checkpoint inhibition in pediatric oncology has, for the most part, yielded disappointing results [
5,
37]. The efficacy of ICIs in children closely approaches that observed in adult cancers only within specific populations, notably patients with relapsed/refractory Hodgkin lymphoma and patients with germline DNA mismatch repair deficiency, polymerase proofreading deficiency, or CMMRD [
5,
34,
38]. In these patients, there is an upregulation of PD-L1 expression on tumor cells or a mutational burden that either approaches or exceeds that of ICI-responsive adult tumors, respectively. Therefore, as expected, most current ongoing clinical trials are primarily exploring the utilization of ICIs in pediatric lymphomas, as shown in
Table 2. These trials are exclusively phase 1 or 2 studies, and primarily focus on relapsed/refractory solid tumors. In clinical practice, the initiation of ICIs often relies on genetic testing performed at the initial diagnosis, but the information this genomic profiling provides is usually utilized late in the disease course, dictating fourth, fifth, or later lines of treatment. By recognizing these limitations to the application of immunotherapy, we observe that there is a clear opportunity for expansion. This entails broadening the spectrum of diagnoses included in clinical trials, standardizing the timepoints at which genomic testing should be performed, and collaborating with other institutions to stage phase 3 clinical trials that examine the combination of ICIs with conventional chemotherapies on a much larger scale.



 ,
, 

{kind=link}