
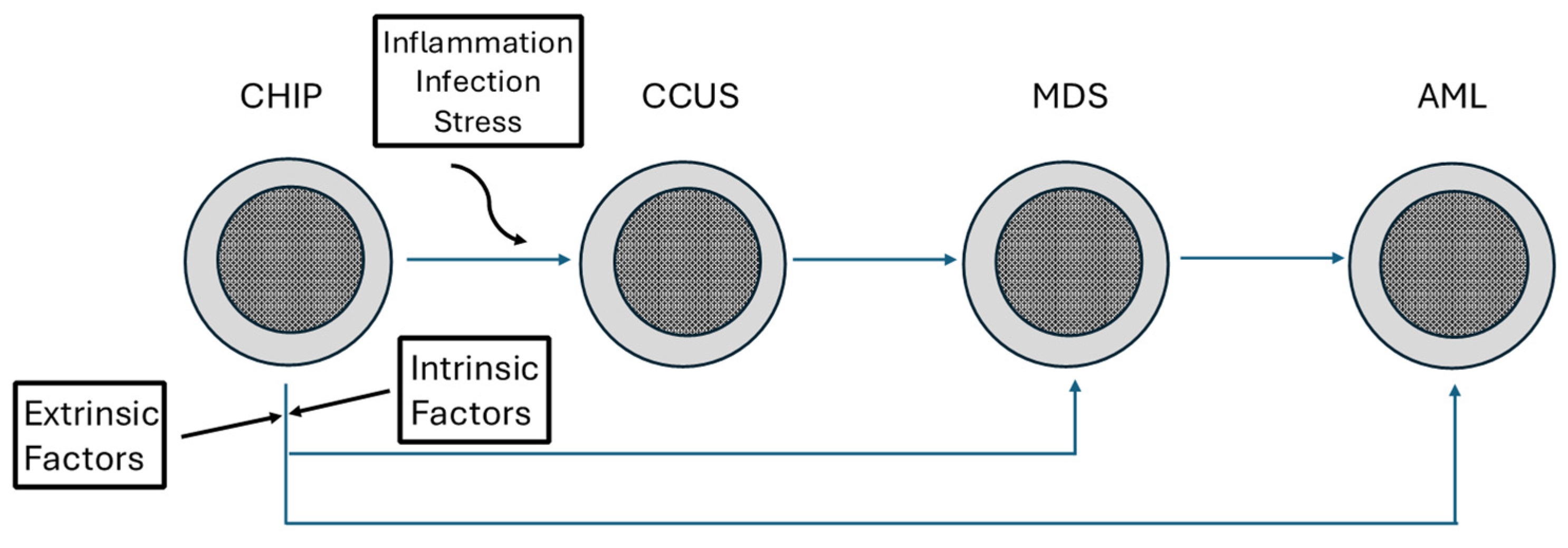
Clonal Hematopoiesis, a Risk Condition for Developing Myeloid Neoplasia



Abstract
:1. Introduction
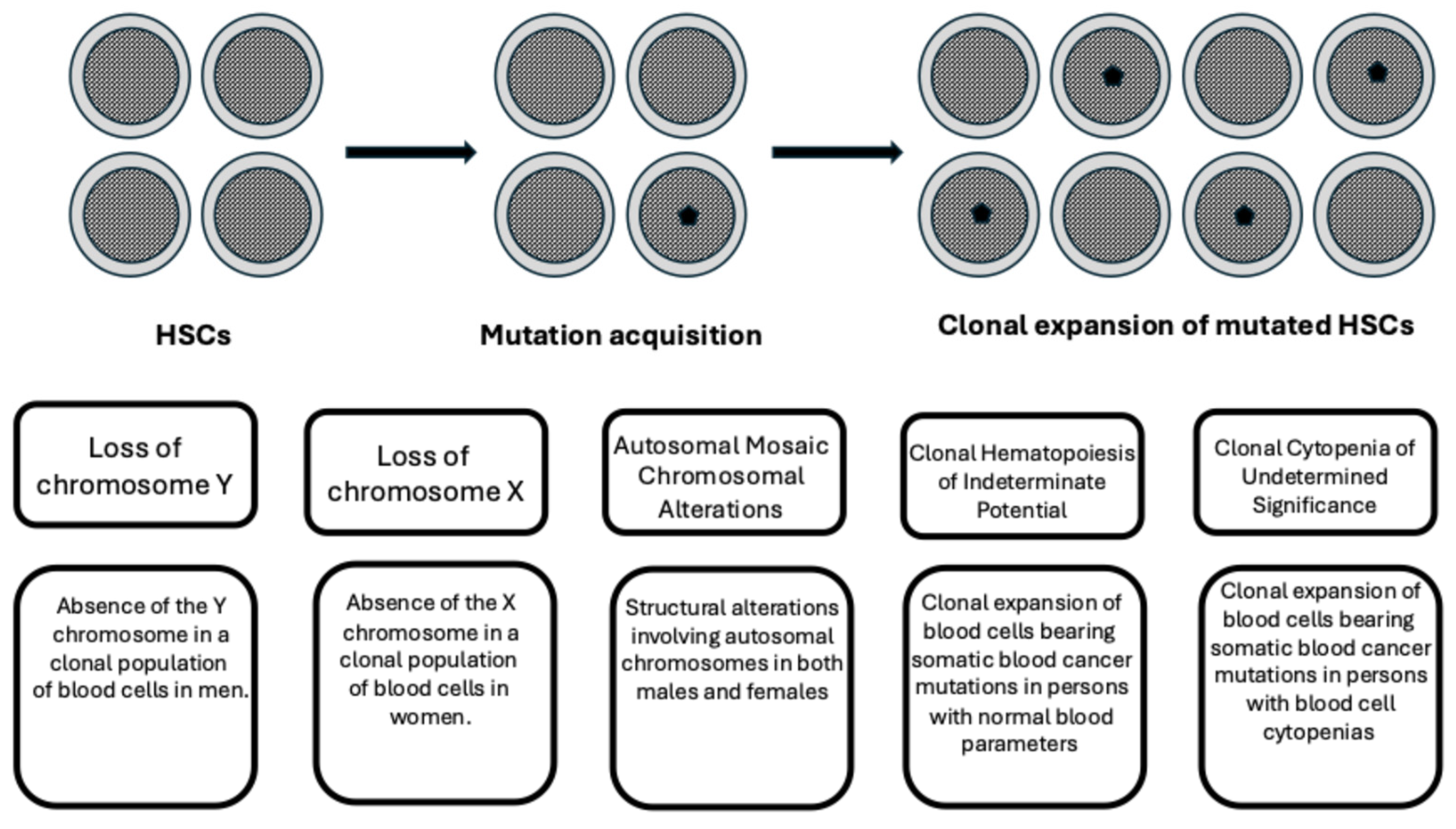
2. Definition of Clonal Hematopoiesis
3. Mutational Spectrum of CHIP
3.1. DNMT3A Mutations
3.2. TET2 Mutations
3.3. ASXL1 Mutations
3.4. Splicing Factor Mutations
3.5. TP53 and PPM1D Mutations
4. Heritability of CHIP
5. Lymphoid CHIP
6. Mosaic Chromosomal Alterations (mCAs)
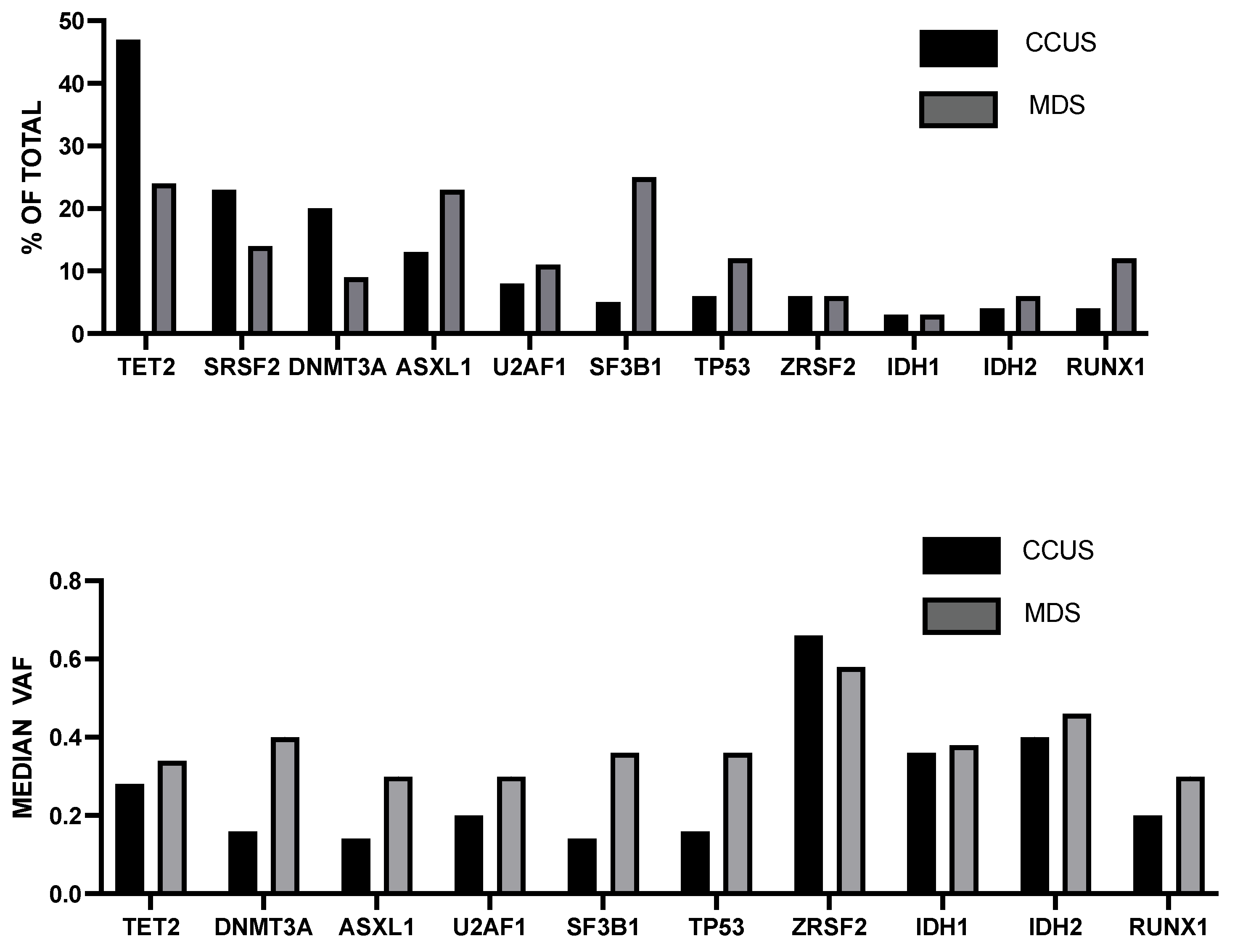
7. Clonal Cytopenia of Undetermined Significance (CCUS)
8. Clonal Monocytosis of Undetermined Significance (CMUS) and Clonal Cytopenia and Monocytosis of Undetermined Significance (CCMUS)
9. Platelet-Restricted Clonal Hematopoiesis (PLT-CH)
10. Clonal Hematopoiesis in Very Old Individuals, Aged >80 Years
11. Longitudinal Dynamics of CH Clones
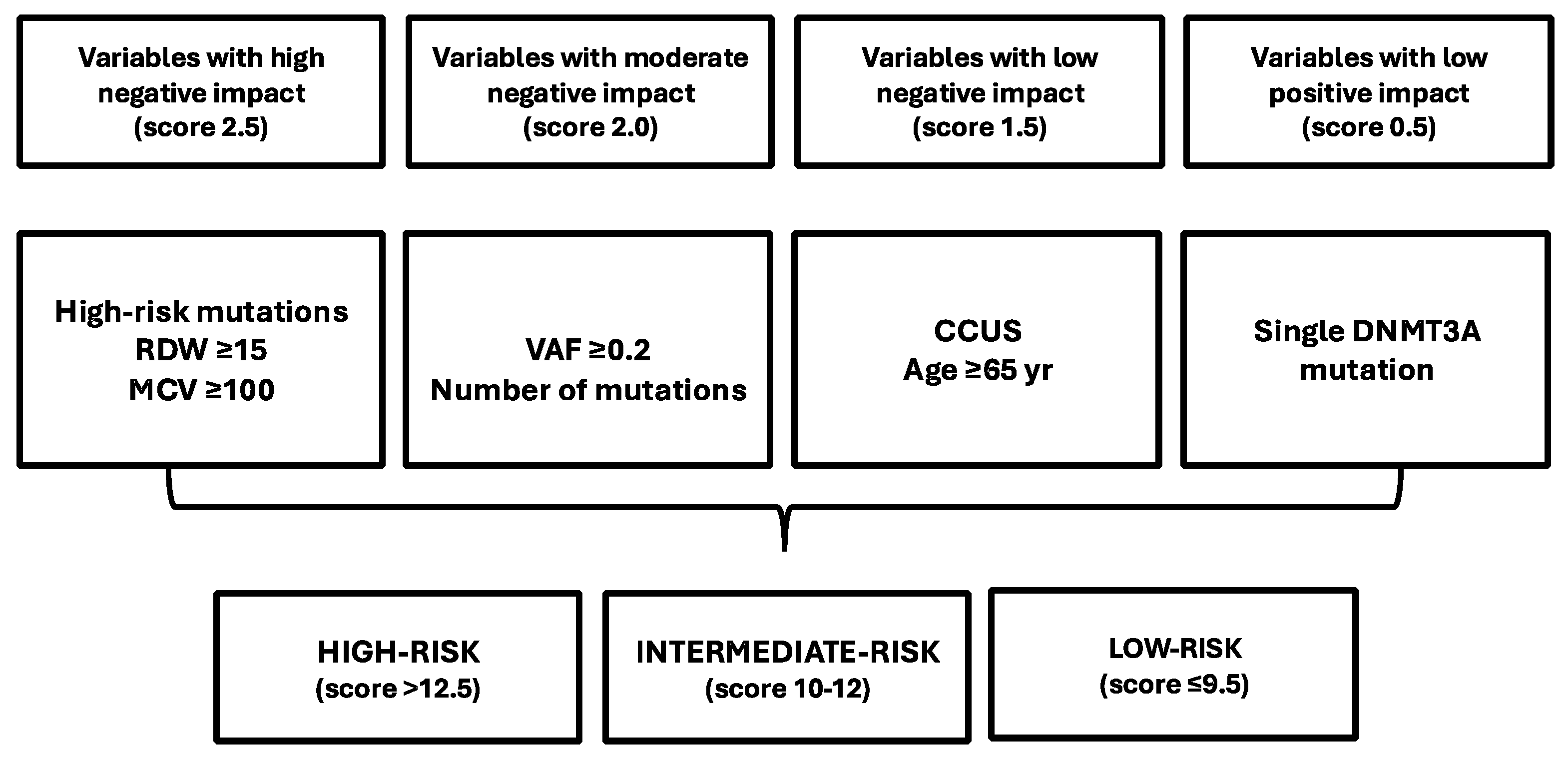
12. Prediction of the Risk of Developing Myeloid Neoplasms in Individuals Bearing CHIP Mutations
13. Effects of Donor-Engrafted Clonal Hematopoiesis in Autologous and Allogeneic Stem Cell Transplantation
14. CH in Bone Marrow Failure Syndromes
14.1. CH in Acquired Aplastic Anemia
14.2. CH in Paroxysmal Nocturnal Hemoglobinuria
14.3. CH in Inherited BMFs
14.4. CH in Fanconi Anemia
14.5. CH in Diamond–Blackfan Anemia
14.6. CH in Schwan-Diamond Syndrome (SDS)
14.7. CH in SAMD9/SAMD9L Syndrome
14.8. CH in Telomere Biology Disorders
15. CH in Germline Predisposition Syndromes
15.1. CH in Patients with RUNX1 Germline Mutations
15.2. CH in Familial Thrombocytopenia Related to ANKRD26 and ETV6 Germline Mutations
15.3. CH in Patients with Germline DDX41 Mutations
15.4. CH in Germline GATA2 Deficiency Syndrome
16. Treatment of CHIP and CCUS
16.1. Anti-IL-1β
16.2. Ascorbic Acid
16.3. IDH Inhibitors
16.4. TGFβ Pathway Inhibitors
16.5. Decitabine/Cedazuridine
16.6. Therapeutic Targeting of Splicing Factor Mutations in CH
17. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moore, L.; Cagan, A.; Coorens, T.H.H.; Neville, M.D.C.; Snahfvi, R.; Sanders, M.A.; Oliver, T.R.W.; Leongamornlert, D.; Ellis, P.; Noorani, A.; et al. The mutational landscape of human somatic and germline cells. Nature 2021, 597, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, E.; Spencer Chapman, M.; Williams, N.; Dawson, K.J.; Mende, N.; Calderbank, E.F.; Jung, H.; Mitchell, T.; Coorens, T.; Spencer, D.H.; et al. Clonal dynamics of hematopoiesis across the human lifespan. Nature 2022, 606, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Lee-Six, H.; Oebro, N.F.; Shepherd, M.S.; Grossmann, S.; Dawson, K.; Belmonte, M.; Hunlky, B.; Martincorena, I.; Anderson, E.; O’Neill, L.; et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 2018, 561, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.G.; Rosendhal Huber, A.; Oka, R.; Vereuln, M.; Patel, S.H.; Hasaart, K.; de la Fonteijne, L.; Varela, I.; Camargo, F.D.; van Broxtel, R. Somatic mutations reveal lineage relationships and age-related mutagenesis in human hematopoiesis. Cell Rep. 2018, 25, 2308–2316. [Google Scholar] [CrossRef]
- Brandsma, A.M.; Bertrums, E.; van Rooismalen, M.J.; Horman, D.A.; Oka, R.; Verheulen, M.; Manders, F.; Ubels, J.; Bbelderbos, M.E.; van Boxtel, R. Mutation signatures of pediatric acute myeloid leukemia and normal blood progenitors associated with differential patient outcomes. Blood Cancer Discov. 2021, 2, 484–499. [Google Scholar] [CrossRef]
- Su, T.Y.; Hauenstein, J.; Somuncular, E.; Dumral, O.; Leonard, E.; Gustafsson, C.; Tzortis, E.; Forlani, A.; Johansson, A.S.; Qian, H.; et al. Aging is associated with functional and molecular changes in distinct hematopoietic stem cell subsets. Nat. Commun. 2024, 15, 7966. [Google Scholar] [CrossRef]
- Poscablo, D.M.; Worthington, A.K.; Smith-Berdan, S.; Rommel, M.; Manso, B.A.; Adili, R.; Mok, L.; Reggiardo, R.E.; Cool, T.; Mogharrab, R.; et al. An age-progressive platelet differentiation path from hematopoietic stem cells causes exacerbated thrombosis. Cell 2024, 187, 3090–3107. [Google Scholar] [CrossRef]
- Mansell, E.; Lin, D.S.; Loghran, S.J.; Milsom, M.D.; Trowbridge, J.J. New insight into the causes, consequences, and correction of hematopoietic stem cell aging. Exp. Hematol. 2023, 125–126, 1–5. [Google Scholar] [CrossRef]
- Chapman, M.S.; Mitchell, E.; Yoshida, K.; Williams, N.; Fabre, M.A.; Ranzoni, A.M.; Robinson, P.S.; Robinson, P.S.; Kregar, L.; Wilk, C.M.; et al. Mutagenic DNA lesions in normal hematopoietic stem cells persist for years causing distinct patterns of mutation. Blood 2024, 144 (Suppl. 1), 2673. [Google Scholar] [CrossRef]
- Busque, L.; Patel, J.P.; Figueroa, M.; Vasanthjakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef]
- Genovese, G.; Kahler, A.K.; Handsaker, R.E. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lu, C.; Wang, J. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- McKerrell, T.; Park, N.; Moreno, T.; Grove, C.S.; Postingl, H.; Stephens, J.; Understanding Society Scientific Group; Crawley, C.; Craig, J.; Scoot, M.A.; et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015, 10, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Young, A.L.; Challen, G.A.; Birmann, B.M.; Druley, T.E. Clonal hematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016, 7, 12484. [Google Scholar] [CrossRef]
- Zirk, F.; Stacey, S.N.; Nordahhl, G.; Frigge, M.L.; Magnusson, O.; Jansdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudnnond, J.; et al. Clonal hematopoiesis with and without candidate driver mutations, is common in elderly. Blood 2017, 130, 742–752. [Google Scholar]
- Stacey, C.N.; Zink, F.; Halidorsson, G.; Stafansdottir, L.; Gudjonsson, S.; Einorsson, G.; Hjörleifsson, G.; Eiriksdottir, T.; Helgadottir, A.; Bjornsdottir, G.; et al. Genetics and epidemiology of mutational barcode-defined clonal hematopoiesis. Nat. Genet. 2023, 55, 2149–2159. [Google Scholar] [CrossRef]
- Bernstein, N.; Chapman, M.S.; Nyamondo, K.; Chen, Z.; Williams, N.; Mitchell, E.; Campbell, P.J.; Cohen, R.L.; Nangalia, J. Analysis of somatic mutations in whole blood from 200,618 individuals identifies pervasive positive selection and novel drivers of clonal hematopoiesis. Nat. Genet. 2024, 56, 1147–1155. [Google Scholar] [CrossRef]
- Feldman, T.; Bercovich, A.; Moskovitz, Y.; Chapal-Ilani, N.; Mitchell, A.; Medeiros, J.; Biezuner, T.; Kaushansky, N.; Minden, M.D.; Gupta, V.; et al. Recurrent deletions in clonal hematopoiesis are driven by microhomology-mediated end joining. Nat. Commun. 2021, 12, 2455. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Mayle, A.; Jeong, M.; Luo, M.; Rodriguez, B.; Mallaney, C.; Celik, H.; Yang, L.; Xia, Z.; et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem. Cell 2014, 15, 350–364. [Google Scholar] [CrossRef]
- Jeong, M.; Park, H.J.; Celik, H.; Ostrander, E.; Reyes, J.; Guzman, A.; Rodriguez, B.; Lei, Y.; Lee, Y.; Ding, L.; et al. Loss of Dnmt3a immortalizes hematopoietic stem cells in vivo. Cell Rep. 2018, 23, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hormachea-Agulkla, D.; Matatall, K.; Le, D.T.; Kain, B.; Long, X.; Kus, P.; Jaksik, R.; Challen, G.A.; Kimmel, M.; King, K.Y. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNγ signaling. Cell Stem. Cell 2021, 28, 1428–1442. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ostrander, E.; Kukhar, O.; Mallaney, C.; Sun, J.; Haussler, E.; Celik, H.; Koh, W.K.; King, K.; Gontarz, P.; et al. Txnip enhances fitness of Dnmt3a-mutant hematopoietic stem cells via p21. Blood Cancer Discov. 2022, 3, 220–239. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Li, X.P.; Liang, Y. Exploring the mechanism of chromatin spatial structure changes and transcriptional regulation in age-related clonal hematopoiesis caused by DNMT3A mutations using chromatin capture technology. Blood 2024, 144 (Suppl. 1), 192–193. [Google Scholar] [CrossRef]
- Oran, B.; Champlin, R.E.; Wang, F.; Tanaka, T.; Saliba, R.M.; Al-Atrash, G.; Garcia-Manero, G.; Kantarjian, H.; Cao, K.; Shpall, E.J.; et al. Donor clonal hematopoiesis increases risk of acute graft versus host disease after matched sibling transplantation. Leukemia 2022, 36, 257–262. [Google Scholar] [CrossRef]
- Frick, M.; Chan, W.; Arends, C.M.; Hablesreiter, R.; Halik, A.; Heuser, M.; Michonneau, D.; Blau, O.; Hoyer, K.; Christen, F.; et al. Role of donor clonal hematopoiesis in allogeneic hematopoietic stem-cell transplantation. J. Clin. Oncol. 2019, 37, 375–385. [Google Scholar] [CrossRef]
- Gibson, C.J.; Kim, H.T.; Zhao, L.; Murdock, H.M.; Hambley, B.; Ogata, A.; Madero-Marroquin, R.; Wang, S.; Green, L.; Fleharty, M.; et al. Donor clonal hematopoiesis and recipient outcomes after transplantation. J. Clin. Oncol. 2022, 40, 189–201. [Google Scholar] [CrossRef]
- Wallace, L.; Engelken, M.; Myers, J.A.; Harper, J.; Clark, B.; Cullins, D.; Zoine, J.T.; Shanmugan, R.; Schattgen, S.; Velasquez, P.; et al. Dnmt3a mutant hematopoietic stem cells produce hyperactive T cells with increased alloimmune and anti-leukemic activity. Blood 2024, 144 (Suppl. 1), 1288–1289. [Google Scholar] [CrossRef]
- Stelmach, P.; Vonfitch, D.; Merbach, A.K.; Lin, D.; Beneyto-Calabuig, S.; Scheller, M.; Leppa, A.M.; Grunschlager, F.; Vassiliou, G.S.; Haas, S.; et al. Mapping genotype. To phenotype. In clonal hematopoiesis uncovers downregulated MHC-II molecules DNMT3AR882-mutant hematopoietic stem cells. Blood 2024, 144 (Suppl. 1), 1292–1293. [Google Scholar] [CrossRef]
- Wang, H.; Divaris, K.; Pan, B.; Li, X.; Lim, J.H.; Sha, G.; Barovic, M.; Giannakou, D.; Korostoff, J.M.; Bing, Y.; et al. Clonal hematopoiesis driven by mutated DNMT3A. promotes inflammatory bone loss. Cell 2023, 187, 3690–3711. [Google Scholar] [CrossRef]
- Kohnke, T.; Karigane, D.; Hilgart, E.; Fan, A.C.; Kayamari, K.; Miyauchi, M.; Collins, C.T.; Suchy, F.P.; Rangavajhula, A.; Feng, Y.; et al. DNMT3AR882H is not required for disease maintenance in primary human AML, but is associated with increased leukemic stem cell frequency. bioRxiv 2024. [Google Scholar] [CrossRef]
- Zou, Z.; Dou, X.; Li, Y.; Zhang, Z.; Wang, J.; Gao, B.; Xiao, Y.; Wang, Y.; Zhao, L.; Sun, C.; et al. RNA m5C oxidation by TET2 regulates chromatin state and leukemogenesis. Nature 2024, 634, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Tuslstrup, M.; Soerensen, M.; Hansen, J.W.; Gillberg, L.; Needhamsen, M.; Kaastrup, K.; Helin, K.; Christensen, K.; Weinschfeldt, J.; Gronbaeck, K. TET2 mutations are associated with hypermethylation at key regulatory enhancers in normal and malignant hematopoiesis. Nat. Commun. 2021, 12, 6061. [Google Scholar] [CrossRef]
- Yang, Y.; Cathelin, S.; Liu, A.; Subedi, A.; Maher, A.; Hasseini, M.; Ayyathan, D.M.; Vanner, R.; Chan, A.M. TET2 deficiency increases the competitive advantage of hematopoietic progenitor and stem cells through upregulation of thrombopoietic receptor signaling. Nat. Commun. 2025, 16, 2384. [Google Scholar] [CrossRef] [PubMed]
- Aivalioti, M.; Bartholdy, B.; Pradhan, K.; Bhagat, T.; Zintiridou, A.; Jeong, J.J.; Tiruthiuvanathan, V.; Pujato, M.; Paranjpe, A.; Chang, C.; et al. PU.1-dependent enhancer inhibition separates Tet2-deficient hematopoiesis from Malignant Transformation. Blood Cancer Discov. 2022, 3, 444–4676. [Google Scholar] [CrossRef]
- De Dominici, M.; Naddaf, L.; Zaberezhnyy, V.; Gooidspeed, A.; Li, S.; Degregori, J. Hematopoietic stem cell aging promotes expansion of Tet2 mutant clones by cell intrinsic mechanisms. Blood 2024, 144 (Suppl. 1), 191. [Google Scholar] [CrossRef]
- Li, S.S.; Karmakar, S.; Motakis, E.; Liu, Y.; Sengupta, K.; Naddaf, L.; Roeder, T.; Vu, B.; Mujica, K.; Pietras, E.M.; et al. Tet2 deficiency mitigates epigenetic aging in clinical hematopoiesis. Blood 2024, 144 (Suppl. 1), 5638–5639. [Google Scholar] [CrossRef]
- Vail, D.J.; Jiang, D.; Chung, K.; Zhang, Y.; Martinez, S.; Guarnera, L.; Parker, Y.; Melenhorst, J.J.; Lindner, D.; Maciejewski, J.; et al. Age-related cellular factors facilitate TET2 mutant clone hematopoiesis. Blood 2024, 144 (Suppl. 1), 1278–1279. [Google Scholar] [CrossRef]
- Encabo, H.H.; Aramburu, I.V.; Garcia-Albornoz, M.; Piganeau, M.; Wood, H.; Song, A.; Ferrelli, A.; Sharma, A.; Minutti, C.M.; Domart, M.C.; et al. Loss of TET2 in human hematopoietic stem cells alters the development and function of neutrophils. Cell Stem. Cell 2023, 30, 781–799. [Google Scholar] [CrossRef]
- Caiado, F.; Kovtonyuk, L.V.; Gonullu, N.G.; Fullin, J.; Boettcher, S.; Manz, M.G. Aging drives Tet2+/− clonal hematopoiesis via IL-1 signaling. Blood 2023, 141, 886–903. [Google Scholar] [CrossRef]
- McClatchy, J.; Strogantsev, R.; Wolfe, E.; Lin, H.Y.; Mohammadhosseini, M.; Davis, B.A.; Eden, C.; Goldman, D.; Fleming, W.H.; Conley, P.; et al. Clonal hematopoiesis related TET2 loss-of-function impedes IL1β-mediated epigenetic reprogramming in hematopoietic stem and progenitor cells. Nat. Commun. 2023, 14, 8102. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, N.A.; Turkalj, S.; Zeng, A.; Stoilova, B.; Metzner, M.; Rahmig, S.; Nagree, M.; Shah, S.; Moore, R.; Usukhbayar, B.; et al. Selective advantage of mutant stem cells in human clonal hematopoiesis is associated with attenuated response to inflammation and aging. Cell Stem. Cell 2024, 31, 1127–1144. [Google Scholar] [CrossRef] [PubMed]
- Marbr, A.R.; Halpin, M.; Corbin, D.; Gordon, B.; Benrashid, S.; Whipp, E.C.; Truxall, J.; Cahn, O.; Mundy-Bosse, B.; Cooper, J.; et al. Targeting inflammation in Tet2 mutant clonal hematopoiesis via inhibition of Brd4 demonstrates activity in both murine and human models. Blood 2024, 144, 2661–2662. [Google Scholar] [CrossRef]
- Kohnke, T.; Nuno, K.A.; Alder, C.C.; Gars, E.J.; Phan, P.; Fan, A.C.; Majeti, R. Human ASXL1-mutant hematopoiesis is driven by a truncated protein associated with aberrant deubiquitination of H2AK119. Blood Cancer Discov. 2024, 5, 202–223. [Google Scholar] [CrossRef]
- Dawoud, A.A.; Tapper, W.J.; Cross, N. Clonal myelopoiesis in the UK biobank cohort: ASXL1 mutations are strongly associated with smoking. Leukemia 2020, 34, 2660–2672. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Goyama, S.; Sugiura, Y.; Inoue, D.; Asada, S.; Yamasaki, S.; Matsumoto, A.; Yamaguchi, K.; Isobe, Y.; Tsuchida, A.; et al. Mutant ASXL1 induces age-related expansion of phenotypic hematopoietic stem cells through activation of Akt/mTOR pathway. Nat. Commun. 2021, 12, 1826. [Google Scholar] [CrossRef]
- Dong, Z.; Sepulveda, H.; Artega-Vazquez, L.J.; Blouin, C.; Fernandez, J.; Binder, M.; Chou, W.C.; Tien, H.F.; Patnaik, M.M.; Faulker, G.J.; et al. A mutant ASXL1-EHMT complex contributes to heterochromatin dysfunction in clonal hematopoiesi and chronic myelomonocytic leukemia. Proc. Natl. Acad. Sci. USA 2025, 122, e241330210121. [Google Scholar] [CrossRef]
- Kamphuis, P.; van Bergen, M.; van Zeventer, I.; deGraal, A.; Dimohamed, A.; Zalzbrum, J.; Schuring, J.J.; van der Rajiden, B.; Huls, G.; Jansen, J.H. Abnormal platelet counts and clonal hematopoiesis in the general population. Hemasphere 2023, 7, e821. [Google Scholar] [CrossRef]
- Yacout, M.; Katamesh, B.; Jabban, Y.; He, R.; Viswanatha, D.; Jevremovic, D.; Greipp, P.; Bosonen, K.; Palmer, J.; Foran, J.; et al. Characterisation and prognostic impact of ZRSF2 mutations in myeloid neoplasms. Leukemia 2024, 38, 2727–2730. [Google Scholar] [CrossRef]
- Desai, P.; Mencia-Trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, E.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef]
- Abelson, S.; Collord, G.; Ng, S.; Weissbrod, O.; Cohen, N.M.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Shirai, C.L.; Ley, J.N.; White, B.S.; Kim, S.; Tibbitts, J.; Shao, J.; Ndonwi, M.; Wadugu, B.; Duncavage, E.J.; Okeyo-Owuor, T.; et al. Mutant U2AF1 expression alters hematopoiesis and pre-mRNA splicing in vivo. Cancer Cell 2015, 27, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Fei, D.L.; Zhen, T.; Durham, B.; Feerone, J.; Zhang, T.; Garrett, L.; Yoshimi, A.; Abdel-Wahab, O.; Bradley, R.K.; Liu, P.; et al. Impaired hematopoiesis and leukemia development in mice with a conditional knock-in allele of a mutant splicing factor gene U2AF1. Proc. Natl. Acad. Sci. USA 2018, 115, E10437–E10446. [Google Scholar] [CrossRef]
- Dutta, A.; Yang, Y.; Le, B.; Zhang, Y.; Abdal-Wahab, O.; Zang, C.; Mahi, G. U2af1 is required for survival and function of hematopoietic stem/progenitor cells. Leukemia 2021, 35, 2382–2398. [Google Scholar] [CrossRef]
- Hoag, B.; De Dominici, M.; Alberti, M.O.; DeGregori, J. Exploring aging dependent adaptations of splicing factor mutations in clonal hematopoiesis. Blood 2023, 142 (Suppl. 1), 5597. [Google Scholar] [CrossRef]
- Bonner, E.A.; Song, A.; Arriaga-Gomez, E.; Gadek, K.; Galvis, R.B.; Linton, J.; Venkataram, R.A.; Sinha, S.; Rongvaux, A.; Evan, M.; et al. MDS-associated SF3B1 mutations promote aberrant hematopoietic cell fate choice by disrupting mediator kinase module component CDK8. Blood 2024, 144 (Suppl. 1), 344–345. [Google Scholar] [CrossRef]
- Sarchi, M.; Clogh, C.A.; Crosse, E.I.; Aydinyan, N.; Wellington, R.; Yang, F.; Gallì, A.; Creamer, P.; Stewart, S.; Bardley, R.K.; et al. Mis splicing of mitotic regulators sensitizes SF3B1-mutated human HSCs to CHK1 inhibition. Blood Cancer J. 2024, 5, 353–370. [Google Scholar] [CrossRef]
- Liang, Y.; Tebaldi, T.; Tejeski, K.; Joshi, P.; Stefani, G.; Taylor, A.; Song, Y.; Vasic, R.; Maziarz, J.; Balasubramian, K.; et al. SRSF2 mutations drive oncogenesis by activating a global program of aberrant alternative splicing in hematopoietic cells. Leukemia 2018, 32, 2659–2671. [Google Scholar] [CrossRef]
- Kon, A.; Yamazaki, S.; Nannya, Y.; Kataoka, K.; Ota, Y.; Nakagawa, M.M.; Yoshida, K.; Shiozawa, Y.; Morita, M.; Yoshizato, T.; et al. Physiological Srsf2 P95H expression causes impaired hematopoietic stem cell functions and aberrant RNA splicing in mice. Blood 2018, 131, 621–635. [Google Scholar] [CrossRef]
- Bapat, A.; Keita, N.; Martelly, W.; Kang, P.; Seet, C.; Jacobsen, J.R.; Stoilov, P.; Hu, C.; Crooks, G.M.; Sharma, S.; et al. Myeloid disease mutations of splicing factor SRSF2 cause G2-M arrest and skewed differentiation of human hematopoietic stem and progenitor cells. Stem. Cells 2018, 36, 1663–1675. [Google Scholar] [CrossRef]
- Bapat, G.; Fobelteder, J.; Schlacher, A.; Auinger, L.; Martinez-Krams, D.; Edrickrema, A.; Kashofer, K.; Beham-Schmid, C.; Greinks, T.; Woelber, A.; et al. Modeling the development of SRSF2mutated myeloid malignancies by CRISPR/Cas9 mediated genome engineering of primary human hematopoietic stem and progenitor cells. Blood 2021, 138 (Suppl. 1), 2180. [Google Scholar]
- Cortes-Lopez, M.; Chamely, P.; Hawkinz, A.G.; Stanley, R.F.; Swett, A.D.; Ganesan, S.; Moulheddine, T.H.; Dai, X.; Kluegel, L.; Chen, C.; et al. Single-cell multi-omics defines impact of splicing aberrations in human hematopoietic clonal outgrowths. Cell Stem. Cell 2023, 30, 1262–1281. [Google Scholar] [CrossRef] [PubMed]
- Inoue, D.; Polaski, J.T.; Taylor, J.; Costal, P.; Chen, S.; Kobayashi, S.; Hogg, S.J.; Hayacvhi, Y.; Pined, J.M.B.; Marabti, E.E.; et al. Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat. Genet. 2021, 33, 707–718. [Google Scholar] [CrossRef]
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivstov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, C.; et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019, 365, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Pourebrahim, R.; Khazaei, S.; Ostermann, L.; Zhao, R.; Muftuoglu, M.; Andreef, M. Age-specific induction of mutant p53 drives clonal hematopoiesis in adult mice leading to acute myeloid leukemia. Blood 2023, 142 (Suppl. 1), 1382–1383. [Google Scholar] [CrossRef]
- Pourebrahim, R.; Montoya, R.H.; Akiyama, H.; Ostermann, L.; Khazaei, S.; Muftuouglu, M.; Baran, N.; Zhao, R.; Lesluyes, T.; Liu, B.; et al. Age-specific induction of mutant p53 drives clonal hematopoiesis and acute myeloid leukemia in adult mice. Cell Rep. Med. 2024, 5, 101558. [Google Scholar] [CrossRef]
- Wong, T.N.; Ramsinghj, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role of TP53 mutations in the priming and evolution of therapy-related acute myeloid leukemia. Nature 2015, 518, 552–555. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Q.; Yu, H.; Capitano, M.L.; Vemula, S.; Nabinger, S.C.; Gao, R.; Yao, C.; Kobayashi, M.; Geng, Z.; et al. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat. Commun. 2019, 10, 5649. [Google Scholar] [CrossRef]
- Adelman, E.R.; Huang, H.T.; Roisman, A.; Olsson, A.; Colaprico, A.; Qin, T.; Lindsley, R.C.; Bejar, R.; Salomonis, N.; Grimes, H.L.; et al. Aging human hematopoietic stem cells manifest profound epigenetic reprogramming of enhancers that may predispose to leukemia. Cancer Discov. 2019, 99, 1080–1101. [Google Scholar] [CrossRef]
- Barajas, S.; Vemula, S.; Chen, S.; Chen, H.; Cai, W.; Xiao, S.; Halene, S.; Abdel-Wahab, O.; Mayo, L.; Savage, S.A.; et al. Chronic inflammation drives p53 mutant clonal hematopoiesis via activating the NLRP1 inflammosome. Blood 2022, 140 (Suppl. 1), 5743–5744. [Google Scholar] [CrossRef]
- Rodriguez-Meira, A.; Norfo, R.; Wen, S.; Chédiville, A.L.; Rahman, H.; O’Sullivan, J.; Wang, G.; Louka, E.; Kretschemar, W.W.; Paterson, A.; et al. Single-cell multi-omics identifies chronic inflammation as a driver of TP53-mutant leukemic evolution. Nat. Genet. 2023, 55, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Lind, K.; Dutta, S.; Cosenza, M.; Eder, T.; Fobeltedere, J.; Pabst, G.; Scholl, W.; Prokesch, A.; Reinisch, A.; Woelfer, A.; et al. Impact of mono- and bi-allelic TP53 aberrations on transformation of human HSPCs. Blood 2024, 144 (Suppl. 1), 5631–5632. [Google Scholar] [CrossRef]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 2020, 52, 1219–1226. [Google Scholar] [CrossRef]
- Nead, K.T.; Kim, T.; Joo, L.; McDowell, T.; Wong, J.; Chan, I.; Brock, E.; Zhao, J.; Xu, T.; Tang, C.; et al. Impact of cancer therapy on clonal hematopoiesis mutations and subsequent clinical outcomes. Blood Adv. 2024, 8, 5215–5224. [Google Scholar] [CrossRef]
- Hsu, J.I.; Dayaram, T.; Tovy, A.; De Brakeleer, E.; Jeong, M.; Wang, F.; Zhang, J.; Hoffernan, T.P.; Gera, S.; Kovacs, J.J.; et al. PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem. Cell 2018, 23, 700–713. [Google Scholar] [CrossRef]
- Miller, P.G.; Sperling, A.S.; Meyerhofer, C.; McConkey, M.E.; Ellegast, J.M.; Da Silva, C.; Cohen, D.N.; Wang, C.; Sharda, A.; Yan, N.; et al. PPM1D modulates. Hematopoietic cell fitness and response to DNA damage and is a therapeutic target in myeloid malignancy. Blood 2023, 142, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.W.; Pedersen, D.A.; Larsen, L.A.; Husby, S.; Clemmensen, S.B.; Hjelmborg, J.; Favero, F.; Weischendfeldt, J.; Christense, K.; Gronbaek, K. Clonal hematopoiesis in elderly twins: Concordance, discordance, and mortality. Blood 2020, 135, 261–268. [Google Scholar] [CrossRef]
- Fabre, M.A.; Mekerrel, T.; Zwiebel, M.; Vijayabaskar, M.S.; Park, N.; Wells, P.M.; Rad, R.; Deloukas, P.; Small, K.; Steves, C.J.; et al. Concordance for clonal hematopoiesis is limited in elderly twins. Blood 2020, 135, 269–273. [Google Scholar] [CrossRef]
- Franco, S.; Godley, L.A. Genetic and environmental risks for clonal hematopoiesis and cancer. J. Exp. Med. 2025, 222, e20230931. [Google Scholar] [CrossRef]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K. Inherited causes of clonal hematopoiesis in 97,691 whole genomes. Nature 2020, 586, 763–768. [Google Scholar] [CrossRef]
- Kar, S.P.; Quiros, P.M.; Gu, M.; Jiang, T.; Mitchell, J.; Langdon, R.; Iyer, V.; Barcena, C.; Vijayabaskar, M.S.; Fabre, M.A.; et al. Genome-wide analyses of 200,453 individuals yield new insights into the causes and consequences of clonal hematopoiesis. Nat. Genet. 2022, 54, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.D.; Damask, A.; O’Kaeffe, S.; Banerjee, N.; Li, D.; Watanabe, K.; Marketta, A.; Van Meter, M.; Semrau, S.; Horowitz, J.; et al. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature 2022, 612, 301–309. [Google Scholar] [CrossRef] [PubMed]
- De Boy, E.A.; Tassia, M.G.; Schrantz, K.E.; Yan, S.M.; Ciosner, Z.L.; McNally, E.J.; Gable, D.L.; Xiang, Z.; Lombard, D.B.; Antonarakis, E.S.; et al. Familial clonal hematopoiesis in a long telomere syndrome. N. Engl. J. Med. 2023, 388, 2422–2433. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Zhao, Y.; Garbuzov, A.; Corces, M.R.; Neuhofer, P.; Gillespie, V.M.; Cheung, P.; Belk, J.A.; Huang, Y.H.; Wei, Y.; et al. Clonal inactivation of TERT impairs stem cells competition. Nature 2024, 632, 201–208. [Google Scholar] [CrossRef]
- Nakao, T.; Bick, A.G.; Taub, M.A.; Zekavat, S.M.; Uddin, M.M.; Niroula, A.; Carty, C.L.; Lane, J.; Honogberg, M.C.; Weinstock, J.S.; et al. Mendelian randomization supports. Bidirectional causality between telomere length and clonal hematopoiesis of indeterminate potential. Sci. Adv. 2022, 8, eabl6579. [Google Scholar] [CrossRef]
- Weinstock, J.S.; Gopakumar, J.; Burugula, B.B.; Uddin, M.M.; Jahn, N.; Belk, J.A.; Bouzid, H.; Daniel, B.; Miao, Z.; Ly, N.; et al. Aberrant activation of TCL1A promotes stem cell expansion in clonal hematopoiesis. Nature 2023, 616, 755–763. [Google Scholar] [CrossRef]
- Niroula, A.; Sekar, A.; Murakami, M.A.; Trinder, M.; Agrawal, M.; Wong, W.J.; Bick, A.G.; Uddin, M.M.; Gibson, C.J.; Griffin, G.K.; et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat. Med. 2021, 27, 1921–1927. [Google Scholar] [CrossRef]
- Selvan, M.E.; Kuan, P.F.; Yang, X.; Mascarenhas, J.; Klein, R.J.; Luft, B.J.; Boffetta, P.; Gumus, Z. Distinct characteristics of lymphoid and myeloid clonal hematopoiesis in World Trade Center first responders. medRxiv 2024. [Google Scholar] [CrossRef]
- Von Boeck, K.; von Boeck, T.; Ferrell, P.B.; Bick, A.G.; Kishtagari, A. Lymphoid clonal hematopoiesis: Implications for malignancy, immunity, and treatment. Blood Cancer 2024, 13, 5. [Google Scholar] [CrossRef]
- Sekar, A.; Griffin, R.; Parikh, S.A.; Genovese, G.; Robinson, D.P.; Norman, A.D.; Olson, J.E.; Rabe, K.G.; Hoel, M.S.; Boddicker, N.J.; et al. Mosaic chromosomal alterations (mCAs) in individuals with monoclonal B-cell lymphocytosis (MBL). Blood Cancer J. 2024, 14, 193. [Google Scholar] [CrossRef]
- Laurie, C.C.; Laurie, C.A.; Rice, K.; Doheny, K.F.; Zelnick, L.R.; McHugh, C.P.; ling, H.; Hetrick, K.N.; Pugh, E.W.; Amos, C.; et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. 2012, 44, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.B.; Yeager, M.; Zhou, W.; Wacholder, S.; Wang, Z.; Rodriguez-Santiago, B.; Hutchinson, A.; Deng, X.; Liu, C.; Horner, M.J.; et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet. 2012, 44, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Loh, P.R.; Genovese, G.; Handsaker, R.E.; Finucane, H.K.; Reshf, J.A.; Palamara, P.F.; Birmann, B.M.; Talkowski, M.E.; Bakoum, S.F.; McCarroll, S.A.; et al. Insights into clonal hematopoiesis from 8,432 mosaic chromosomal alterations. Nature 2018, 559, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Terao, C.; Suzuki, A.; Mmomozawa, Y.; Akiytama, M.; Ishigaki, K.; Yamamoto, K.; Matsuda, K.; Murakami, Y.; NcCarroll, S.A.; Kubo, M.; et al. Chromosomal alterations among age-related hematopoietic clones in Japan. Nature 2020, 584, 130–134. [Google Scholar] [CrossRef]
- Saiki, R.; Momozawa, Y.; Nannya, Y.; Nakagawa, M.M.; Ochi, Y.; Yoshizato, T.; Terao, C.; Korada, Y.; Shiraishi, Y.; Chiba, K.; et al. Combined landscape of single-nucleotide variants and copy number alterations in clonal hematopoiesis. Nat. Med. 2021, 27, 1239–1249. [Google Scholar] [CrossRef]
- Zekavat, S.M.; Lin, S.H.; Bick, A.G.; Liu, A.; Paruchuri, K.; Wang, C.; Uddin, M.; Ye, Y.; Liu, X.; Kamatani, Y.; et al. Hematopoietic mosaic chromosomal alterations increase the risk for diverse types of infection. Nat. Med. 2021, 27, 1012–1024. [Google Scholar] [CrossRef]
- Thompson, D.J.; Genovese, G.; Ulirsch, J.C.; Wright, D.J.; Tyerao, C. Genetic predisposition to mosaic Y chromosome loss in blood. Nature 2019, 575, 652–657. [Google Scholar] [CrossRef]
- Dawouid, A.; Tapper, W.; Cross, N. Age-related loss of chromosome Y is associated with levels of hormone binding globulin and clonal hematopoiesius defined by TET2, TP53 and CBL mutations. Sci. Adv. 2023, 9, eade9746. [Google Scholar] [CrossRef]
- Ljunstrom, V.; Martisson, J.; Halvardson, J.; Pandzic, T.; Davies, H.; Rychlicka-Buiniowska, E.; Lacaze, P.; Cavelier, L.; Dumanski, J.P.; Baliakis, P.; et al. Loss of Y and clonal hematopoiesis in blood-two sides of the same coin? Leukemia 2022, 36, 889–891. [Google Scholar] [CrossRef]
- Ouseph, M.M.; Hassejian, R.P.; Dal Cin, P.; Lovitch, S.B.; Steensma, D.P.; NMardi, V.; Weinberg, O.K. Genomic alterations in patients with somatic loss of the Y chromosome as the sole cytogenetic finding in the bone marrow cells. Haematologica 2021, 106, 555–564. [Google Scholar] [CrossRef]
- Lin, S.H.; Loftfiueld, E.; Sampson, J.N.; Zhou, W.; Yeager, M.; Freedman, N.D.; Chanock, S.J.; Machiela, M.J. Mosaic chromosome Y loss is associated with alterations in blood cell counts in the UK Biobank men. Scient. Rep. 2020, 10, 3655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, L.; Yang, Y.; Li, S.; Liu, Y.; Chen, C. Mosaic loss of chromosome Y promotes leukemogenesis and clonal hematopoiesis. JCI Insight. 2022, 7, e153768. [Google Scholar] [CrossRef]
- Brown, D.W.; Cato, L.D.; Zhao, Y.; Nandakumar, S.K.; Bao, E.L.; Gardner, E.J.; Hubbard, A.K.; De Paulis, A.; Rahling, T.; Song, L.; et al. Shared and distinct etiologies for different types of clonal hematopoiesis. Nat. Commun. 2023, 14, 5536. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Blandell, J.R. Mutational rates and fitness consequences of mosaic chromosomal alterations in blood. Nat. Genet. 2023, 55, 1677–1685. [Google Scholar] [CrossRef]
- Zhao, K.; Pershad, Y.; Poisner, H.M.; Ma, X.; Quade, K.; Vlasschaert, C.; Mack, T.; Khankary, N.K.; von Beck, K.; Brogan, J.; et al. Genetic drivers and clinical consequences of mosaic chromosomal alterations in 1 million individuals. medRxiv 2025, in press. [Google Scholar]
- Jakubek, J.A.; Zhou, Y.; Shep, A.; Bacon, J.; Wang, J.W.; Orcan, Z.; Arnett, D.; Barbes, K.; Bis, J.C.; Boerwinkle, E. Mosaic chromosomal aberrations on blood across ancestries using whole genome sequencing. Nat. Genet. 2023, 55, 1912–1919. [Google Scholar] [CrossRef]
- Pershad, Y.; Mack, T.; Poisner, H.; Jakubek, Y.; Stilp, A.M.; Mitchell, B.; Lewis, J.; Boerwinkle, E.; Loos, R.; Chami, N.; et al. Determinants of mosaic chromosomal alterations fitness. Nat. Commun. 2024, 15, 3800. [Google Scholar] [CrossRef] [PubMed]
- De Zern, A.E.; Malcovati, L.; Ebert, B.L. CHIP, CCUS, and other acronyms: Definition, implications, and impact on practice. ASCO Educ. Book 2019, 39, 400–410. [Google Scholar]
- Van Zeventer, I.A.; de Graaf, A.O.; van der Klauw, M.M.; Vellenga, E.; van der Reihden, B.A.; Schuringa, J.J.; Diepstra, A.; Malcovati, L.; Jansen, J.H.; Huls, G. Peripheral blood cytopenias in the aging general population and risk of incident hematological disease and mortality. Blood Adv. 2021, 5, 3266–3278. [Google Scholar] [CrossRef]
- Kwok, B.; Hall, J.M.; Witte, J.S.; Xu, Y.; Reddy, P.; Lin, K.; Flamholz, R.; Dabbas, B.; Yung, A.; Al-Hafidh, J.; et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood 2015, 126, 2355–2361. [Google Scholar] [CrossRef]
- Cargo, C.A.; Rowbotham, N.; Evans, P.A.; Barrans, S.L.; Bowen, D.T.; Crouch, S.; Jack, A.S. Targeted sequencing identifies patients with preclinical MDS at high risk of disease progression. Blood 2015, 126, 2362–2365. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Galli, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catricalà, S.; Bono, E.; et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017, 129, 3371–3378. [Google Scholar] [CrossRef]
- Choi, E.J.; Cho, Y.U.; Hur, E.H.; Park, H.S.; Choi, Y.; Lee, J.H.; Lee, K.H.; Kim, M.; Hwang, H.S.; Jong, S.; et al. Clinical implications and genetic features of clonal cytopenia of undetermined significance compared to lower-risk myelodysplastic syndromes. Brit. J. Haematol. 2022, 198, 703–712. [Google Scholar] [CrossRef]
- Gallì, A.; Todisco, G.; Catamo, E.; Sala, C.; Elena, C.; Pozzi, S.; Bono, E.; Ferretti, V.V.; Rizzo, E.; Molteni, E.; et al. Relationship between clone metrics and clinical outcome in clonal cytopenia. Blood 2021, 138, 965–978. [Google Scholar] [CrossRef]
- Ferrone, C.K.; McNaughton, A.; Rashedi, I.; Ring, B.; Buckstein, R.; Tsui, H.; Rauh, M.J. A lower frequency of spliceosome mutations distinguishes cytopenias of undetermined significance from low-risk myelodysplastic syndromes, despite inherent similarities in genomic, laboratory, and clinical features. Mod. Pathol. 2023, 36, 100068. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, S.U.; Safavi, S.; Dimopoulos, K.; O’Rourke, C.J.; Andersen, M.K.; Holm, M.S.; Marcher, C.W.; Andersen, J.B.; Hansen, J.W.; Grinboeck, K. Structural aberrations are associated with poor survival in patients with clonal cytopenia of undetermined significance. Haematologica 2021, 106, 1762–1766. [Google Scholar] [CrossRef] [PubMed]
- Cargo, C.; Bernard, E.; Beinertas, T.; Bolton, K.L.; Glover, P.; Wanen, H.; Payne, D.; Ali, R.; Khan, A.; Short, M.; et al. Predicting cytopenias, progression, and survival in patients with clonal cytopenia of undetermined significance: A prospective cohort study. Lancet Hematol. 2024, 11, e51–e60. [Google Scholar] [CrossRef]
- Huber, S.; Baer, C.; Hutter, S.; Wossidilo, N.; Hormann, G.; Pohlkamp, C.; Walter, W.; Meggendorfer, M.; Kan, W.; Haferlach, T.; et al. Genomic landscape of CCUS compared to MDS and its implications on risk prediction. Leukemia 2024, 38, 1634–1637. [Google Scholar] [CrossRef]
- Jajosky, A.N.; Sadri, N.; Meyerson, H.J.; Oduro, K.A.; Kelkar, A.; Fitzgerald, B.; Tomlinson, B.; Moore, E.M.; Beck, R.C. Clonal cytopenia of undetermined significance (CCUS) with dysplasia in enriched for MDS-type molecular finding compared to CCUS without dysplasia. Eur. J. Haematol. 2021, 106, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Baer, C.; Meggendorfer, M.; Kern, W.; Haferlach, C.; Haferlach, T.; Hoermann, G. Molecular evolution of CCUS already follows the same rules as MDS progression. Blood 2022, 140 (Suppl. 1), 8598–8599. [Google Scholar] [CrossRef]
- Xie, Z.; Komrokji, R.; Al Ali, N.; Regelson, A.; Geter, S.; Patel, A.; Sasygin, C.; Zeidan, A.M.; Zeidan, A.M.; Bewersdorf, J.P.; et al. Risk prediction for clonal cytopenia: Multicenter real-world evidence. Blood 2024, 144, 2033–2044. [Google Scholar] [CrossRef] [PubMed]
- Traeden, D.; Tulsrtup, M.; Johansen, M.M.; Orskov, A.D.; Marcher, C.; Raauschu-Jensen, K.; Roug, A.S.; Severnsin, M.T.; Porse, B.; Theigaard-Monch, K.; et al. Overall survival in patients with CCUS depends on presence of anemia. Blood 2023, 142 (Suppl. 1), 1864–1865. [Google Scholar] [CrossRef]
- Broyan, J.; Kishtagari, A.; Carty, R.W.; Pershad, Y.; Vlaschaert, C.; Sharber, B.; Heimlich, B.; Luo, L.; Xu, y.; Bick, A. Risk of incident cytopenia in clonal hematopoiesis. medRxiv 2024. [Google Scholar]
- Yacout, M.; Katamesh, B.; Jabban, Y.; He, R.; Greipp, P.; Jevremovic, D.; Foran, J.M.; Arana Yi, C.; Saliba, A.N.; Mangaonkar, A.A.; et al. A clinical overview of clonal cytopenia of undetermined significance in the presence of mutations. Blood 2024, 144, 2667–2668. [Google Scholar] [CrossRef]
- Shah, M.V.; Mangaonkar, A.A.; Begna, K.H.; Alkhaqteeb, H.B.; Greipp, P.; Nanaa, A.; Elliott, M.A.; Hogan, W.G.; Litzow, M.R.; McCollough, K.; et al. Therapy-related clonal cytopenia as a precursor to therapy-related myeloid neoplasms. Blood Cancer J. 2022, 12, 106. [Google Scholar] [CrossRef]
- Li, M.; Baranwal, A.; Gurney, M.; Shah, S.N.; Al_kali, A.; Alkhateeb, H.; Foran, J.; Arana Yi, C.; Ongie, L.; Chen, D.; et al. The impact of cytotoxic therapy on the risk of progression and death in clonal cytopenia(s) of undetermined significance. Blood Adv. 2024, 8, 3130–3138. [Google Scholar] [CrossRef] [PubMed]
- Kaastrup, K.; Tulstrup, M.; Hansen, J.W.; Schollopf, C.; Raaschou-Jensen, K.; Orskov, A.D.; Pose, B.; Jones, P.A.; Weinschenfeldt, J.; Gillberg, L.; et al. Overlapping DNA methylation changes in enhancers in clonal cytopenia of undetermined significance and myelodysplastic neoplasm patients with TET2, IDH2 or DNAMT3A mutations. Haematologica 2025, in press. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Hasserjian, R.P.; Greenberg, P.L.; Arango Ossa, J.E.; Creignou, M.; Tuechler, H.; Gutierrez-Abril, J.; Domenico, D.; Medina-Martinez, J.S.; Lavine, M.; et al. Molecular taxonomy of myelodysplastic syndromes and its clinical implications. Blood 2024, 144, 1617–1630. [Google Scholar] [CrossRef]
- Cargo, C.; Cullen, M.; Taylor, J.; Short, M.; Glover, P.; Van Happe, S.; Smith, A.; Evans, P.; Crouch, S. The use of targeted sequencing and flow cytometry to identify patients with a clinically significant monocytosis. Blood 2019, 133, 1325–1334. [Google Scholar] [CrossRef]
- Van Zeventer, I.; de Graaf, A.; Koorenhof-Schoele, T.; van der Reijden, B.; van der Klouw, M.; Dinmohamed, A.; Diepstra, A.; Schuringa, J.J.; Malcovati, L.; Hals, G. Monocytosis and its association with clonal hematopoiesis in community-dwelling individuals. Blood Adv. 2022, 6, 4174–4184. [Google Scholar] [CrossRef]
- Dunn, W.G.; Gu, M.; Quiros, P.; Mohorianu, I.; Wiseman, D.H.; Vassiliou, G.S. Prevalence and significance of clonal monocytosis of undetermined significance (CMUS) amongst 431,531 United Kingdom biobank participants. Blood 2024, 144, 4048–4049. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.; et al. The 5th edition of the World Health Organization classification of hematolymphoid tumors: Myeloid and histiocytic/dendritic neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, A.M.; Wang, S.A.; Bugga, H.; Barbui, T.; Barnford, S.; et al. International Consensus Classification of myeloid neoplasms and acute leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, F.; Baer, C.; Bamopoulos, S.; Ayoub, E.; Truger, M.; Meggendorfer, M.; Lenk, M.; Hoermann, G.; Hutter, S.; Muller, H.; et al. Comparing malignant monocytosis across the updated WHO and ICC classifications of 2022. Blood 2024, 143, 1139–1156. [Google Scholar] [CrossRef] [PubMed]
- Sousos, N.; Murphy, L.; Wen, S.; Hamblin, A.; Clark, S.A.; Karali, C.S.; Whittle, J.; Ren, Z.; Rahman, H.; Hayder, N.; et al. Platelet-restricted clonal hematopoiesis. Blood 2023, 142 (Suppl. 1), 814. [Google Scholar] [CrossRef]
- Camacho, V.; Craminita, E.; Barrachina, M.; Becker, I.C.; Italiano, J.E.; Nachius, K.R. Tet2 age-associated somatic mutations impair megakaryopoiesis and platelet function in clonal hematopoiesis. Blood 2024, 144 (Suppl. 1), 422–423. [Google Scholar] [CrossRef]
- Van den Akker, E.B.; Pitts, S.J.; Deelen, J.; Moed, M.H.; Potluri, S.; van Rooij, J.; Suchiman, H.E.; Lakenberg, N.; de Dijcekr, W.J.; Uitterlinden, A.G.; et al. Uncompromised 10-year survival of oldest old carrying somatic mutations in DNMT3A and TET2. Blood 2016, 127, 1512–1515. [Google Scholar] [CrossRef]
- Van Zeventer, I.A.; Salzbrunn, J.B.; de Graaf, A.O.; van der Reijden, B.A.; Boezen, H.M.; Vonk, H.M.; van der Harst, P.; Schuringa, J.J.; Jansen, J.H.; Huls, G. Prevalence, predictors, and outcomes of clonal hematopoiesis in individuals aged >80 years. Blood Adv. 2021, 5, 2115–2122. [Google Scholar] [CrossRef]
- Van Zeventer, I.A.; de Graaf, A.; Wouters, H.; van der Reijden, B.; van der Klkauw, M.; de Witte, T.; Jonker, M.A.; Malcovati, L.; Jansen, J.H.; Huls, G. Mutational spectrum and dynamics of clonal hematopoiesis in anemia of older individuals. Blood 2020, 135, 1161–1170. [Google Scholar]
- Rossi, M.; Merggendorfer, M.; Zampini, M.; Tettamanti, M.; Riva, E.; Travaglino, E.; Bersanelli, M.; Mandelli, S.; Galbussera, A.A.; Tosca, E.; et al. Clinical relevance of clonal hemaatopoiesis in persons aged >80 years. Blood 2021, 138, 2093–2105. [Google Scholar] [CrossRef]
- Holstege, H.; Pfeiffer, W.; Sie, D.; Hulsman, M.; Nicholas, T.J.; Lee, C.C. Somatic mutations found in the healthy blood compartment of a 115 yr-old woman demonstrate oligoclonal hematopoiesis. Genome Res. 2014, 24, 733–742. [Google Scholar] [CrossRef]
- Van den Akker, E.; Makrodimitris, S.; Hulsman, M.; Brugman, M.H.; Nikolic, T.; Bradley, T.; Waisfisz, Q.; Baas, F.; Jakobs, M.E.; de Jong, D.; et al. Dynamic clonal hematopoiesis and functional T-cell immunity in a supercentenarian. Leukemia 2021, 35, 2125–2129. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, W.; Yi, L.; Zhao, M.; Li, P.-Y.; Fu, M.-H.; Lin, R.; Zhu, Y.-M.; Li, J.-F.; Yang, W.-P.; et al. The impact of age and number of mutations on the size of clonal hematopoiesis. Proc. Natl. Acad. Sci. USA 2024, 121, e2319364121. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, C.D.; Williams, N.; Dawson, K.J.; Watson, C.; Yousefzadeh, M.; Lo, D.; Nyamondo, K.; Kadavali, S.; Cagan, A.; Waldvogel, S.; et al. Clonal dynamics and somatic evolution of hematopoiesis in mouse. Nature 2025, in press. [Google Scholar] [CrossRef]
- Watson, C.J.; Papula, A.L.; Poon, G.V.P.; Wong, W.H.; Young, A.L.; Druley, J.E.; Fider, D.S.; Blundell, J.R. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science 2020, 367, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Fabre, M.A.; De Almeida, J.G.; Fiorillo, E.; Mitchell, E.; Damaskou, A.; Rak, J.; Orrù, V.; Marongiu, M.; Viyabaskar, M.S.; Baxter, J.; et al. The longitudinal dynamics and natural history of clinical hematopoiesis. Nature 2022, 606, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.A.; Lattore-Crespo, E.; Terrades-Fernandes, M.; Lemos-Portela, G.; Purcell, A.G.; Livosey, B.J.; Hillary, R.F.; Murphy, L.; Fawkes, A.; MacGillivroy, L. Longitudinal dynamics of clonal hematopoiesis identifies gene-specific fitness effects. Nat. Med. 2022, 28, 1439–1446. [Google Scholar] [CrossRef]
- Van Zaventer, I.; de Graaf, A.; Salzbrunn, J.; Noklte, I.; Kamphuis, P.; Dinmohamed, A.; van der Rejden, B.; Schuring, J.J.; Jansen, J.; Huls, G. Evolutionary landscape of clonal hematopoiesis in 3,359 individuals from the general population. Cancer Cell 2023, 41, 1017–1031. [Google Scholar] [CrossRef]
- Uddin, M.M.; Saadatagah, S.; Niroula, A.; Hornsby, W.E.; Geneh, S.; Lannery, K.; Schuermans, A.; Honigberg, M.C.; Bick, A.G.; Libbi, P.; et al. Long-term longitudinal analysis of 4,187 participants reveals insights into determinants of clonal hematopoiesis. Nat. Commun. 2024, 15, 7858. [Google Scholar] [CrossRef]
- Kreger, J.; Mooney, J.A.; Shibita, D.; MacLean, A. Developmental hematopoietic stem cell variation explains clonal hematopoiesis later in life. Nat. Commun. 2024, 15, 10268. [Google Scholar] [CrossRef]
- Zhou, F.J.; Wang, H.C.; Le, M.; Wang, W.; Sunshine, M.J.; Magee, J.A.; Signer, R. Age-related changes in hematopoietic stem cell proteostasis promote the emergence of clonal hematopoiesis and leukemia in older adults. Blood 2024, 144, 4047. [Google Scholar] [CrossRef]
- Fabre, M.A.; Vassiliou, G.S. The lifelong natural history of clonal hematopoiesis and its links to myeloid neoplasia. Blood 2024, 143, 573–581. [Google Scholar] [CrossRef]
- Ferrel, A. Clonal hematopoiesis and myeloid neoplasms in the context of telomere biology disorders. Curr. Hematol. Malig. Rep. 2022, 17, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Rodrigues, F.; Groarke, E.; Tongon, N.; Rodriguez-Sevilla, J.J.; Catto, L.F.; Niewisch, R.M.; Shalhoub, R.; McReynolds, L.; Clé, D.V.; Patel, B.A.; et al. Clonal landscape and clinical outcomes of telomere biology disorders: Somatic recsue and cancer mutations. Blood 2024, 144, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Burren, O.S.; Dhindsa, R.S.; Deevi, S.; Wen, S.; Nag, A.; Mitchell, J.; Hu, F.; Loesch, D.P.; Smith, K.R.; Razdan, N.; et al. Genetic architecture of telomere length in 462,666 Uk Biobank whole-genome sequences. Nat. Genet. 2024, 56, 1832–1850. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Kovilakam, S.C.; Dunn, W.; Marande, L.; Bencena, C.; Mahorianu, I.; Smith, A.; Kar, S.P.; Fabre, M.A.; Gerstung, M.; et al. Multiparameter prediction of myeloid neoplasia risk. Nat. Genet. 2023, 55, 1523–1530. [Google Scholar] [CrossRef]
- Weeks, L.D.; Niroula, A.; Neuberg, D.; Wong, W.; Lindsley, R.C.; Luskin, M.R.; Berliner, N.; Store, R.M.; DeAngelo, D.J.; Soiffer, R.J.; et al. Prediction of risk for myeloid malignancy in clonal hematopoiesis. N. Engl. J. Med. Evid. 2023, 2, 5. [Google Scholar] [CrossRef]
- Hong, Y.S.; Battle, L.; Shi, W.; Puiu, D.; Pillalamarri, V.; Xie, J.; Pankratz, N.; Lake, N.J.; Lek, M.; Rotter, J.L.; et al. Deleterious heteroplasmic mitochondrial mutations are associated with an increased risk of overall and cancer-specific mortality. Nat. Commun. 2023, 14, 6113. [Google Scholar] [CrossRef]
- Lake, N.J.; Ma, K.; Liu, W.; Battle, S.L.; Laricchia, K.M.; Tiao, G.; Puiu, D.; Ng, K.K.; Cohen, J.; Compton, A.G.; et al. Quantifying constraint in the human mitochondrial genome. Nature 2024, 635, 390–397. [Google Scholar] [CrossRef]
- Hong, Y.S.; Pasca, S.; Shi, W.; Puiu, D.; Lake, N.J.; Lek, M.; Ru, M.; Grove, M.L.; Prizment, A.; Joshu, C.E.; et al. Mitochondrial heteroplasmy improves risk prediction for myeloid neoplasms. Nat. Commun. 2024, 15, 10133. [Google Scholar] [CrossRef]
- Haferlach, C.; Huber, S.; Baer, C.R.; Hutter, S.; Meggendorfer, M.; Kern, W.; Hoermann, G.; Haferlach, T. A proposal for a classification of Ccus, MDS and AML primarily based on genetic abnormalities considering the biological continuum of these entities. Blood 2023, 142 (Suppl. 1), 6492–6494. [Google Scholar] [CrossRef]
- Baer, C.R.; Huber, S.; Hoermann, G.; Meggendorfer, M.; Hutter, S.; Kern, W.; Haferlach, T.; Haferlach, C. The frequency of clonal hematopoiesis prior to AML and MDS varies among the different molecularly defined WHO subtypes. Blood 2023, 142 (Suppl. 1), 725–727. [Google Scholar] [CrossRef]
- Marcinek, A.; Baer, C.R.; Hutter, S.; Meggendorfer, M.; Haferlach, T.; Kern, W.; Haferlach, C. Clonal hematopoiesis can precede AML and defining cytogenetic abnormalities—The frequencies vary considerably. Blood 2024, 144 (Suppl. 1), 4318–4319. [Google Scholar] [CrossRef]
- Chien, K.S.; Braish, J.S.; Li, Z.; Loghavi, S.; Bataller, A.; Montalban-Bravo, G.; Sasaki, K.; Kanagal-Shamanna, R.; Takahashi, K.; DiNardo, C.D.; et al. Clinicopathologic characteristics and outcomes of patients with clonal hematopoiesis who progress to myeloid neoplasms. Leukemia 2025, in press. [Google Scholar] [CrossRef]
- Chien, K.S.; Ong, F.; Kim, K.; Li, Z.; Kanagal-Shamanna, R.; DiNardo, C.; Takahashi, K.; Montablan-Bravo, G.; Hammond, D.; Sasaki, K.; et al. Cancer patients with clonal hematopoiesis die from primary malignancy or comorbiodities despite higher rates of transformation to myeloid neoplasms. Cancer Med. 2024, 13, e7093. [Google Scholar] [CrossRef]
- Oshima, M.U.; Higgins, J.; Jenkins, I.; Randolph, T.; Smith, T.; Valentine, C.; Salk, J.; Yeung, C.; Beppu, L.; Campbell, J.; et al. characterization and clonal dynamics using duplex sequencing in donor-recipient pairs decades after hematopoietic cell transplantation. Sci. Transl. Med. 2024, 16, eado5108. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.S.; Wilks, M.; Boettcher, S.; Mitchell, E.; Dawson, K.; Williams, N.; Muller, J.; Kovtonyuk, L.; Jung, H.; Caiado, F.; et al. Clonal dynmamics after allogeneic haematopoietic cell transplantation. Nature 2024, 635, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kazakova, V.; Weeks, L.; Greber, J.; Tai, J.; Zhang, T.; Lowsky, R.; Wu, X.; Yang, C.; Patel, S. Effects of donor -engrafted clonal hematopoiesis in allogeneic and autologous stem cell transplantation: A systematic review and meta-analysis. Bone Marrow Transpl. 2024, 59, 1585–1593. [Google Scholar] [CrossRef]
- Gillis, N.; Abied, A.; Thompson, Z.; Pidala, J. Clinical impact of clonal hematopoiesis in hematopoietic cell transplantation: A review, meta-analysis, and call to action. Haematologica 2024, 109, 3952–3964. [Google Scholar] [CrossRef]
- Gibson, C.J.; Lindsley, R.C.; Gondek, L.P. Clonal hematopoiesis in the setting of hematopoietic cell transplantation. Semin Hematol 2024, 61, 9–15. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, T.H.; Novitzky-Basso, I.; Lee, H.; Yoo, Y.; Ahn, J.S.; Pasic, I.; Law, A.; Lam, W.; Michelis, F.W.; et al. Clonal hematopoiesis in the donor does not adversely affect long-term outcomes following allogeneic hematopoietic stem cell transplantation: Result from a 13-year follow-up. Haematologica 2023, 108, 1817–1826. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, S.; Wilk, C.M.; Singer, J.; Beier, F.; Burcklen, E.; Beisel, C.; Ferreira, M.V.; Gourri, E.; Gassner, C.; Frey, B.M.; et al. Clonal hematopoiesis in donors and long-term survivors of related allogeneic hematopoietic stem cell transplantation. Blood 2020, 135, 1548–1559. [Google Scholar] [CrossRef]
- Muskens, K.; Wieringa, N.; van Bergen, M.; Bense, J.E.; Te Pas, B.; de Patger, A.; Lankester, A.C.; Bierings, M.B.; Neuberg, D.S.; Kaitjema, S.; et al. Increased clonal hematopoiesis in long-term survivors of pediatric hematopoietic cell transplantation. Blood Cancer Discov. 2025, in press. [Google Scholar] [CrossRef] [PubMed]
- Awada, H.; Gurnari, C.; Visconte, V.; Durmaz, A.; Kuzmanovic, T.; Awada, H.; Tu, Z.J.; Cook, J.R.; Bolwell, B.J.; Sobecks, R.; et al. Clonal hematopoiesis-derived therapy-related myeloid neoplasms after autologous hematopoietic cell transplant for lymphoid and non lymphoid disorders. Leukemia 2024, 38, 1266–1274. [Google Scholar] [CrossRef]
- Rhee, J.W.; Chen, S.; Pillai, R.; Boswoth, A.; Oganesyan, A.; Atenzio, L.; Estrada, C.; Kassabian, M.; Lindenfeld, L.; Goldsmith, S.R.; et al. Clonal hematopoiesis is associated with non-myeloid subsequent malignant neoplasms after autologous hematopoietic cell transplantation. Blood 2024, 144 (Suppl. 1), 947–948. [Google Scholar] [CrossRef]
- Tanaka, T.; Morita, K.; Loghavi, S.; Wang, F.; Funudate, K.; Sasaki, Y.; Little, L.; Gumbs, C.; Matthews, J.; Daver, N.; et al. Clonal dynamics and clinical implications of postremission clonal hematopoiesis in acute myeloid leukemia. Blood 2021, 138, 1733–1736. [Google Scholar] [CrossRef]
- Heuser, M.; Heida, B.; Buttner, K.; Wienecke, C.P.; Teich, K.; Funke, C.; Brandes, M.; Klement, P.; Liebich, A.; Wichmann, M.; et al. Posttransplantation MRD monitoring in patients with AML by next-generation sequencing using DTA and non-DTA mutations. Blood Adv. 2021, 5, 2294–2304. [Google Scholar] [CrossRef] [PubMed]
- Bischof, L.; Ussmann, J.; Grimm, J.; Bill, M.; Brauer, D.; Backaus, D.; Hermann, L.; Merz, M.; Herling, M.; Metzler, K.H.; et al. Prognostic impact of measurable residual clonal hematopoiesis in acute myeloid leukemia patients after allogeneic hematopoietic stem cell transplantation. Leukemia 2024, 38, 198–201. [Google Scholar] [CrossRef]
- Imus, P.H.; Pasca, S.; Tsai, H.L.; Aljawai, Y.M.; Cooke, K.R.; Waleton, J.D.; Gocke, C.D.; Varadhan, R.; Jones, R.J.; Gondek, L.P. Recipient clonal hematopoiesis in allogeneic bone marrow transplantation for lymphoid malignancies. Blood Adv. 2024, 6, 3849–3857. [Google Scholar] [CrossRef]
- Ogawa, S. Clonal hematopoiesis in acquired aplastic anemia. Blood 2016, 128, 337–347. [Google Scholar] [CrossRef]
- Yoshizato, T.; Mumitriu, B.; Hosokawa, K.; Makishima, H.; Yoshida, K.; Toownsley, D.; Sato-Otsubo, A.; Liu, D.; Suzuki, H.; Wu, C.O.; et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N. Engl. J. Med. 2015, 373, 35–47. [Google Scholar] [CrossRef]
- Patel, B.A.; Ghannam, J.; Groarke, E.; Goswami, M.; Dillon, L.; Gutierrez-Rodrigues, F.; Rios, O.; Raffo, D.Q.; Lotter, J.; Young, N.S.; et al. Detectable mutations precede late myeloid neoplasia in aplastic anemis. Haematologica 2021, 106, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Groarke, E.M.; Patel, B.A.; Shalhoub, R.; Gutierrez-Rodrigues, F.; Desai, P.; Leuva, H.; Zaimoku, Y.; Paton, C.; Spitofsky, N.; Lotter, J.; et al. Predictors of clonal evolution and myeloid neoplasia following immunosuppressive therapy in severe aplastic anemia. Leukemia 2022, 36, 2328–2337. [Google Scholar] [CrossRef] [PubMed]
- Gurnari, C.; Pagliuca, S.; Prata, P.H.; Galimard, J.E.; Catto, L.F.; Larcher, L.; Sebert, M.; Allain, V.; Patel, B.; Durmaz, A.; et al. Clinical and molecular determinants of clonal evolution in aplastic anemia and paroxysmal nocturnal hemoglobinuria. J. Clin. Oncol. 2022, 41, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Rodrigues, F.; Groarke, E.M.; Alemu, L.; Rios, O.; Raffo, Q.D.; Lotter, J.; Young, N.S.; Patel, B.A. Longitudinal contribution of distinct hematopoietic clones during marrow recovery in immune aplastic anemia. Blood 2024, 144 (Suppl. 1), 32–33. [Google Scholar] [CrossRef]
- Kaya, D.; Cook, R.; Iacobelli, S.; Kulasekararaj, A.G.; Napolitani, G.; Gervelik, S.; Seymen, N.; Sicre de Fontbrune, F.; Griffin, M.; Frieri, C.; et al. Dynamics of oligoclonal hematopoiesis in severe aplastic anemia undergoing immunosuppressive treatment: Longitudinal somatic mutation analysis from the EBMT race trial. Blood 2024, 144 (Suppl. 1), 33–35. [Google Scholar] [CrossRef]
- Luzzatto, L.; Nakao, S. Pathogenesis of paroxysmal nocturnal hemoglobinuria. Blood 2025, in press. [Google Scholar] [CrossRef]
- Shan, W.; Clemente, M.J.; Hosono, N.; Yoshida, K.; Przychodzen, B.; Yoshizato, T.; Shiraishi, Y.; Miyano, S.; Ogawa, S.; Maciejewski, J.P.; et al. Deppe sequencing reveals stepwise mutation acquisition in paroxysmal hemoglobinuria. J. Clin. Investig. 2014, 124, 4529–4538. [Google Scholar] [CrossRef]
- Li, J.; Lin, Y.; Chen, L.; Qin, L.; Tan, H.; Zou, J.; Zhang, D.; Nie, Y.; Wang, G.; Zhang, H.; et al. Identification of acquired PIGA mutations and additional variants by next-generation sequencing in paroxysmal nocturnal hemoglobinuria. Int. J. Lab. Hematol. 2020, 42, 473–481. [Google Scholar] [CrossRef]
- Sebert, M.; Gachet, S.; Leblanc, T.; Rousseau, A.; Bluteu, O.; Kim, R.; Abdelali, R.B.; Sicre de Fontbrune, F.; Maillard, L.; Fredonie, C.; et al. Clonal hematopoiesis driven by chromosome 1q/MDM4 trisomy defines a canonical route toward leukemia in Fanconi anemia. Cell Stem. Cell 2023, 30, 153–170. [Google Scholar] [CrossRef]
- Liu, Y.; Karlson, S. Perspectives of current understanding and therapeutics of Diamond-Blackfan anemia. Leukemia 2024, 38, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Voit, R.A.; Liao, X.; Antoszewski, M.; Cohen, B.; Armant, M.; Lu, H.Y.; Fleming, T.J.; Kamal, E.; Wahlster, L.; Roche, A.M.; et al. Regulated GATA1 expression as a universal gene therapy for Diamond-Blackfan anemia. Cell Stem. Cell 2025, 32, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Nash, M.; bVlachos, A.; Wlodarski, M.W.; Lipton, J.M. Clonal hematopoiesis in patients with Diamond Blackfan anemia. Blood 2020, 136 (Suppl. 1), 25–26. [Google Scholar] [CrossRef]
- Pedigones, N.; Babushok, D.V.; Tian, L.; Bradfield, J.; Kim, C.E.; Perin, J.C.; Hakonarson, H.; Bessler, M.; Mason, P.J. An acquired mutation in deuquitinating enzyme USP11 associated with clonal hematopoiesis in Diamond Blackfan anemia. Blood 2014, 124, 1596. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef]
- Xia, J.; Miller, C.A.; Baty, J.; Ramesh, A.; Jotte, M.; Fulton, R.S.; Vogel, T.P.; Cooper, M.A.; Wankovich, K.J.; Makaryan, V.; et al. Somatic mutations and clonal hematopoiesis in congenital neutropenia. Blood 2018, 131, 408–416. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Myers, K.C.; Bowman, J.; Gibson, C.J.; Camarda, N.D.; Furutani, E.; Muscato, G.M.; Klein, R.H.; Ballotti, K.; Liu, S.; et al. Distinct genetic pathways define pre-malignant versus compensatory clonal hematopoiesis in Shwachman-Diamond syndrome. Nat. Commun. 2021, 12, 1334. [Google Scholar] [CrossRef]
- Sahoo, S.S.; Erlacher, M.; Wlodarski, M.W. Genetic and clinical spectrum of SAMD9 and SAMD9L syndromes: From variant interpretation to patient management. Blood 2025, 145, 475–485. [Google Scholar] [CrossRef]
- Sahoo, S.S.; Pastor, V.B.; Goodings, C.; Voss, R.K.; Kozyra, E.J.; Svetnik, A.; Noelike, P.; Dworzak, M.; Stary, J.; Locatelli, F.; et al. Clinical evolution, genetic landscape landscape and trajectories of clonal hematopoiesis in SAMD9/SAMD9L syndromes. Nat. Med. 2021, 27, 1806–1817. [Google Scholar] [CrossRef]
- Pastor, V.B.; Sahoo, S.S.; Boklan, J.; Schwabe, G.C.; Saribeyoglu, E.; Strahm, B.; Lebrecht, D.; Voss, M.; Bryceson, Y.Y.; Erlacher, M.; et al. Constitutional SAMD9L mutations cause familial myelodysplastic syndrome and transient monosomy 7. Haematologica 2018, 103, 427–437. [Google Scholar] [CrossRef]
- Wong, J.C.; Bryant, V.; Lamprecht, T.; Ma, J.; Walsh, M.; Schwartz, J.; Alzamora, M.; Mullighan, C.G.; Loh, M.L.; Ribeiro, R.; et al. Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. JCI Insight. 2018, 3, e121086. [Google Scholar] [CrossRef] [PubMed]
- Revy, P.; Kannengiesser, C.; Bertuch, A.A. Genetics of human telomere biology disorders. Nat. Rev. Genet. 2013, 24, 86–108. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P.; Giri, N.; Savage, S.A.; Rosenberg, P.S. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 2018, 103, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Schratz, K.E.; Armanios, M. Cancer and myeloid clonal evolution in the short telomere syndromes. Curr. Opin. Genet. Dev. 2020, 60, 112–118. [Google Scholar] [CrossRef]
- Schratz, K.E.; Haley, L.; Danoff, S.K.; Blackford, A.L.; DeZern, A.E.; Gocke, C.D.; Duffield, A.S.; Armanios, M. Cancer spectrum and outcomes in the Mendelian short telomere syndromes. Blood 2020, 135, 1946–1956. [Google Scholar] [CrossRef]
- Reilly, C.R.; Myllymaki, M.; Redd, R.; Padmanaban, S.; Karunakaran, D.S.; Tesmer, V.; TsaI, F.D.; Gibson, C.J.; Rana, H.Q.; Zhanbg, L. The clinical and functional effects of TERT variants in myelodysplastic syndrome. Blood 2021, 138, 898–911. [Google Scholar] [CrossRef]
- Gutierrez-Rodrigues, F.; Groarke, E.M.; Clé, D.V.; Patel, B.A.; Donaires, F.S.; Spitofsky, N.; Alemu, L.; Santana, B.A.; Kajigaya, S.; Colla, S.; et al. Clonal hematopoiesis in telomere biology disorders associated with the underlying germline defect and somatic mutations in POT1, PPM1D and TERT promoter. Blood 2021, 138, 111–1112. [Google Scholar] [CrossRef]
- Groarke, A.M.; Gutierrez-Ropdrigues, F.; Ma, X.; Patel, B.A.; Spitofsky, N.; Browne, P.E.; Chien, K.S.; Wu, C.O.; DiNardo, C.D.; Colla, S.; et al. U2AF1 and the splicing factor gene mutations in telomere biology disorders are associated with hematologic neoplasis and worse overall survival. Blood 2021, 138 (Suppl. 1), 862. [Google Scholar] [CrossRef]
- Sande, C.M.; Chen, S.; Mitchell, D.V.; Liu, P.; SAbraham, D.M.; Cheng, J.; Gebhard, T.; Deolikan, R.J.; Freeman, C.; Zhou, M. ATM-dependent DNA damage response constrains cell growth and drives clonal hematopoiesis in telomere biology disorders. J. Clin. Investig. 2025, 135, e181659. [Google Scholar] [CrossRef]
- Ferrer, A.; Lasho, T.; Fernandez, J.A.; Steinauer, N.P.; Simon, R.A.; Finke, C.M.; Carmona, E.M.; Wylam, M.E.; Ongle, L.J.; Burnap, B.N.; et al. Patients with telomere biology disorders show context specific somatic mosaic states with high frequency of U2AF1 variants. Am. J. Hematol. 2023, 98, E357–E359. [Google Scholar] [CrossRef]
- Tummala, H.; Walne, A.J.; Badat, M.; Patel, M.; Walne, A.M.; Ainajar, J.; Chow, C.C.; Alburson, I.; Frost, J.M.; Ballard, D.; et al. The evolving genetic landscape of telomere biology disorder dyskeratosis congenita. EMBO Mol. Med. 2024, 16, 2560–2582. [Google Scholar] [CrossRef] [PubMed]
- Perdigones, N.; Perin, J.C.; Schiano, I.; Nicholas, P.; Biegel, J.A.; Mason, P.J.; Babushock, D.V.; Bessler, M. Clonal hematopoiesis in patients with dyskeratosis congenita. Am. J. Hematol. 2016, 91, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Krischner, M.; Maurer, A.; Wodarski, M.W.; Ventura Ferreira, M.; Bouillon, A.S.; Halfmeyer, I.; Blau, W.; Kreuter, M.; Rosewitch, M.; Carbacioglu, S.; et al. Recurrent somatic mutations are rare in patients with cryptic dyskeratosis congenita. Leukemia 2018, 32, 1762–1767. [Google Scholar] [CrossRef]
- Liu, Y.C.; Eldomery, M.K.; Maciaszek, J.L.; Klco, J.M. Inherited predisposition to myeloid neoplasms: Pathogenesis and clinical implications. Ann. Rev. Pathol. Mech. Dis. 2025, 20, 87–114. [Google Scholar] [CrossRef]
- Yu, K.; Deuitch, N.; Merguerian, M.; Cunningham, L.; Davis, J.; Bresciani, E.; Diemer, J.; Andrews, E.; Young, A.; Donovan, F.; et al. Genomic landscape of patients with germline RUNX1 variants and familial platelet disorder with myeloid malignancy. Blood Adv. 2024, 6, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Homan, C.C.; Drazer, M.; Yu, K.; Lawrence, D.M.; Feng, J.; Arriola-Martinez, L.; Pozsgai, M.J.; McNeely, K.E.; Ha, T.; Venugopal, P.; et al. Somatic mutational landscape of hereditary hematopoietic malignancies caused by germline variants in RUNX1, GATA2, and DDX1. Blood Adv. 2023, 7, 6092–6102. [Google Scholar] [CrossRef]
- Simo, L.; Spinella, J.F.; Yao, C.Y.; Lavallée, V.P.; Boivin, I.; Boucher, G.; Audemard, E.; Bordeleau, M.E.; Lemieux, S.; Hébert, J.; et al. High frequency of germline RUNX1 mutations in patients with RUNX1-mutated AML. Blood 2020, 135, 1882–1886. [Google Scholar] [CrossRef]
- Haddad, C.; Guarnera, L.; Kubota, Y.; Bravo-Perez, C.; Gurnari, C.; Ogbue, O.; Durmaz, A.; Unlu, S.; Mandala, A.; Orland, M.; et al. Genetic landscape of germline RUNX1 variants in adult neoplasia according to functional impact. Blood 2024, 144 (Suppl. 1), 1328–1329. [Google Scholar] [CrossRef]
- Drazer, M.W.; Homan, C.C.; Yu, K.; Silva, M.C.; McNeely, K.E.; Poszgai, M.J.; Acevedo-Mendez, M.G.; Segal, J.P.; Wang, P.; Feng, J.; et al. Clonal hematopoiesis in patients with ANKRD26 or ETV6 germline mutations. Blood Adv. 2022, 6, 4357–4366. [Google Scholar] [CrossRef]
- Torres-Esquius, S.; Ramil Lopez, G.; Sabira, M.G.; Acha, P.; Jauregui, S.N.; Pratcorona, M.; Saumell, S.; Valcarcel, D.; Jerez, A.; Montoro, M.J. Association of clonal hematopoiesis and dysgranulopoiesis with germline ETV6 carriers without hematological malignancies. Blood 2024, 144 (Suppl. 1), 2709–2710. [Google Scholar] [CrossRef]
- Kusne, Y.; Bactor, T.; Lasho, T.; Marando, L.; Mangaonkar, A.A.; Finke, C.; Foran, J.M.; Al-Kali, A.; Palmer, J.; Yi, C.A.; et al. Prevalence of cytopenia(s) and somatic variants in patients with DDX41 mutant germline predisposition syndrome. Brit. J. Haematol. 2025, 206, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Calvo, K.R.; Hickstein, D.D. The spectrum of GATA2 deficiency syndrome. Blood 2023, 141, 1524–1532. [Google Scholar] [CrossRef]
- Largeud, L.; Collin, M.; Mouselet, N.; Vergez, F.; Fregona, V.; Larcher, L.; Hircsh, P.; Duployez, N.; Bidet, A.; Luquet, I.; et al. Somatic genetic alterations predict hematological progression in GATA2 deficiency. Haematologica 2023, 108, 1515–1529. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Rofls, C.; Trumpp, M.; Winter, S.; Fischer, L.; Richter, M.; Menger, V.; Nenoff, K.; Grieb, N.; Kubasch, A.S.; et al. Activation of distinct inflammatory pathways in LR-MDS is determined by genetics. Blood 2022, 140 (Suppl. 1), 4011–4012. [Google Scholar] [CrossRef]
- Nielsen, A.B.; Hansen, J.W.; Orskov, A.D.; Dimopoulos, K.; Salem, M.; Grigorian, M.; Bruunsgard, H.; Gronbaek, K. Inflammatory cytokine profiles do not differ between patients with idiopathic cytopenias of undetermined significance and myelodysplastic syndromes. Hemasphere 2022, 6, e713. [Google Scholar] [CrossRef]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanithale, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem. Cell 2018, 23, 833–849. [Google Scholar] [CrossRef]
- Burns, S.S.; Kumar, R.R.; Pasupuleti, S.K.; So, K.; Zhang, C.; Kapur, T. ILK1r1 drives leukemogenesis induced by Tet2 loss. Leukemia 2022, 36, 2531–2534. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballatntyne, C.; Fonseca, F.; CANTOS trial group. Anti-inflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.C.; Madar, A.; Campbell, C.D.; He, Y.; Sultan, M.; Healey, M.L.; Xu, H.; D’Aco, K.; Fernandez, A.; Wache-Mainier, C.; et al. TET2-driven clonal hematopoiesis and response to canakinumab: An exploratory analysis of the CANTOS randomized clinical trial. JAMA Cardiol. 2022, 7, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Vallurupalli, M.; MacFadyen, J.G.; Glynn, R.J.; Thuren, T.; Libby, P.; Berliner, N.; Ridker, P.M. Effects of interleukin-1β inhibition on incident anemia: Exploratory analyses from a randomized trial. Ann. Intern. Med. 2020, 172, 523–532. [Google Scholar] [CrossRef]
- Woo, J.; Lu, D.; Lewandoski, A.; Xu, H.; Serrano, P.; Healey, M.; Yata, D.P.; Baste, M.T.; Libby, P.; Ridker, P.M.; et al. Effects of IL-1β inhibition on anemia and clonal hematopoiesis in the randomized CANTOS study. Blood Adv. 2023, 7, 7471–7484. [Google Scholar] [CrossRef]
- Woo, J.; Zhai, T.; Yang, F.; Xu, H.; Healey, M.L.; Yates, D.P.; Beste, M.T.; Steensma, D.T. Effect of clonal hematopoiesis mutations and canakinumab treatment on incidence of solid tumors in the CANTOS randomized clinical trial. Cancer Prev. Res. 2024, 17, 429–436. [Google Scholar] [CrossRef]
- Rodriguez-Seville, J.J.; Adema, V.; Chien, K.S.; Loghavi, S.; Ma, F.; Yang, H.; Montalban-Bravo, G.; Huang, X.; Calvo, X.; Joseph, J.; et al. The IL-1β inhibitor canakinumab in previously treated lower-risk myelodysplastic syndromes: A phase 2 clinical trial. Nat. Commun. 2024, 15, 9840. [Google Scholar] [CrossRef]
- Borate, U.M.; Dvorak-Kornaus, K.M.; Zhao, Q.; Eisfeld, A.K.; Mundy-Bosse, B.L. A randomized double-blind placebo-controlled phase II multi-center study of inflammation modification of canakinumab to prevent leukemic progression of clonal cytopenias of unknown significance (CCUS): Impact study. Blood 2023, 142 (Suppl. 1), 1327–1329. [Google Scholar] [CrossRef]
- Cimino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 2017, 170, 1079–1095. [Google Scholar] [CrossRef] [PubMed]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates hematopoietic stem cell function and leukemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Comazzetto, S.; Cassidy, D.; Davis, B.; Reyes, A.; Morrison, S. Vitamin C limits multipotent progenitor self-renewal and clonal expansion. Blood 2023, 142 (Suppl. 1), 4048. [Google Scholar] [CrossRef]
- Mikkelsen, S.U.; Vallentin, A.; Nielsen, A.; Hansen, J.; Mortensen, B.; Severinsen, M.; O’Connell, C.; Andersen, M.; Likkesfeldt, I.; Jones, P.; et al. Supplementation in patients with clonal cytopenia of undetermined significance or low-risk myeloid malignancies: Results from EVI-2, a randomized, placebo-controlled phase 2 study. In Proceedings of the he 29th Congress of the European Hematology Association, Brussels, Belgium, 13–16 June 2024. [Google Scholar]
- Al-Mousawi, A.; Mikkelsen, S.U.; Nielsen, A.B.; Vallentin, A.P.; Mortensen, A.; Madaj, Z.; Jespersen, J.; Gillberg, L.; Hansen, J.W.; Andersen, M.K.; et al. Oral vitamin C supplementation modulates inflammatory cytokines in clonal cytopenia of undetermined significance and low-risk myeloid malignancies: Results from Evi-2 trial. Blood 2024, 144 (Suppl. 1), 3201. [Google Scholar] [CrossRef]
- Xie, Z.; Fernandez, J.; Lasho, T.; Finke, C.; Amundson, M.; McCullogh, K.B.; LaPlant, B.R.; Mangaonkar, A.; Gangat, N.; Reichard, K.K.; et al. High-dose ascorbic acid therapy for patients with CCUS with TET2 mutations. Blood 2024, 144, 2456–2461. [Google Scholar] [CrossRef]
- Mouchel, P.L.; Bérard, E.; Tavitian, S.; Gadaud, N.; Vergez, F.; Rieu, J.B.; Luquet, I.; Sarry, A.; Huguet, F.; Largeaud, L.; et al. Vitamin C and D supplementation in acute myeloid leukemia. Blood Adv. 2023, 7, 6886–6894. [Google Scholar] [CrossRef]
- McCullogh, K.B.; Xie, Z.; Lasho, T.L.; Finke, C.; Fernandez, J.A.; Amundson, R.N.; LaPlant, B.; Witzing, T.E.; Paatnaik, M.M. Phase II trial assessing safety and preliminary efficacy of high-dose intravenous ascorbic acid and decitabine in TET2-mutant chronic myelomonocytic leukemia. Blood 2024, 144 (Suppl. 1), 6699–6700. [Google Scholar] [CrossRef]
- Kamel, M.M.; Robinson, N.N.; Kutyna, M.; Lim, K.; Thompson-Peach, C.; Lane, S.W.; Yeung, D.T.; Yong, A.; Ross, D.M.; Hiwase, D.; et al. High dose ascorbate reduces interleukin-1 beta secretion in TET2 mutant monocytes and demonstrates excellent safety and tolerability in AMML patients in combination with azacitidine. Blood 2024, 144 (Suppl. 1), 1817. [Google Scholar] [CrossRef]
- Petrone, G.; Stein, E.M. Ivosidenib for patients with clonal cytopenia of undetermined significance and mutations in IDH1. Blood 2023, 142 (Suppl. 1), 3253–3254. [Google Scholar] [CrossRef]
- Della Porta, M.G.; Garcia-Manero, G.; Santini, V.; Zeidan, A.M.; Komrokji, R.S.; Shortt, J.; Valcarel, D.; Jonasova, A.; Dimicoli-Salazar, S.; Tiong, I.S.; et al. Luspatercept versus epoietin alfa in erythropoiesis-stimulating agent-naïve, transfusion-dependent, lower-risk myelodysplastic syndromes (COMMANDS): Primary analysis of a phase 3, open-label, randomized, trial. Lancet Hematol. 2024, 11, e646–e658. [Google Scholar] [CrossRef]
- Hasan, M.; Vodala, S.; Hayati, S.; Garcia-Manero, G.; Gandhi, A.K.; Suragani, R. Clinical hematopoiesis-related mutations are associated with favorable clinical benefit following luspatercept treatment in patients with lower-risk myelodysplastic syndromes: A subgroup analysis from the phase 3 COMMANDS trial. Blood 2013, 142 (Suppl. 1), 3214–3216. [Google Scholar] [CrossRef]
- Hasan, M.; Tannu, S.; Saini, S.; Ugidos, M.; Garcia-Manero, G.; Platzbecker, U.; Chang, C.P.; Gandhi, A.K.; Garcia, R.; Suragani, R. Luspatercept reduces clonal hematopoiesis-associated cardiac stress via modulation of mTORC1 and inflammatory signaling pathways. Blood 2024, 144 (Suppl. 1), 4046–4047. [Google Scholar] [CrossRef]
- Xu, J.; Yan, Y.; Zonfg, S.; Ye, W.; Zheng, J.; Min, C.; Wang, Q.; Li, Z. Rapid and sustained response to luspaterceprt and eltrombopag combined treatment in one case of clonal cytopenias of undetermined significance with prior failure to cyclosporin and androgen therapy: A case report. Ther. Adv. Hematol. 2024, 15, 20406207241260353. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; McCloskey, J.; Griffiths, E.A.; Yee, K.; Zeidan, A.M.; Al-Kali, A.; Deeg, H.J.; Patel, P.A.; Sabloff, M.; Kaeting, M.M.; et al. Oral decitabine-cedazuridine versus intravenous decitabine for myelodysplastic syndromes and chronic myelomonocytic leukemia (ASCERTAIN): A registrational, randomized, crossover, pharmacokinetics, phase 3 study. Lancet Hematol. 2024, 11, e15–e26. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Jabbour, E.; Montalban-Bravo, G.; Darbaniyan, F.; Do, K.A.; Class, C.; Short, N.J.; Kanagal-Shamana, R.; Kadia, T.; Borthakur, G.; et al. Low-dose decitabine versus low-dose azacitidine in lower-risk MDS. NEJM Evid. 2022, 1, EVIDoa220034. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Bachiashvili, K.; Griffiths, E.A.; Zeidan, A.M.; Traer, E.; Saini, L.; Amin, H.; Mohan, S.; Lubbert, M.; Maness-Harris, L.; et al. MDS-641 randomized phase 1-2 study to assess safety and efficacy of low-dose (LD) oral decitabine/cedazuridine (ASTX727) in lower-risk myelodysplastic syndromes (LR-MDS) patients: Interim safety analysis. Clin. Lymph. Myel. Leuk. 2023, 23 (Suppl. 1), S376–S377. [Google Scholar] [CrossRef]
- Szlest, M.; Giannopoulos, K. Targeting splicing for hematological malignancies therapy. BMC Genomics. 2024, 25, 1067. [Google Scholar] [CrossRef]
- Biswas, J.; Boussi, L.; Abdel-Wahab, O. Aberrant pre-mRNA processing in cancer. J. Exp. Med. 2024, 221, e20230891. [Google Scholar] [CrossRef]
- Zhang, Q.; Abdel-Wahab, O. Molecular impact of mutations in RNA splicing factors in cancer. Mol. Cell 2024, 84, 3667–3680. [Google Scholar] [CrossRef] [PubMed]
- Bewersdorf, J.P.; Mi, X.; Lu, B.; Kuykendall, A.T.; Sallamn, D.; Patel, M.; Stevens, D.; Philipovskiy, A.; Sutamtewagul, G.; Masarova, L.; et al. Phase Ib study of PRT543, an oral protein arginine methytransferase 5 (PRMT5) inhibitor, in patients with relapsed or refractory, splicing factor-mutant myeloid malignancies. Blood 2024, 144, 3215. [Google Scholar] [CrossRef]
- Boddu, P.; Roy, R.; Baumgartner, F.; Hutter, S.; Haferlach, T.; Pillai, M. A unified post-transcriptional mechanism regulates intron retention in splicing factor-mutant MDS. Blood 2024, 144 (Suppl. 1), 2732–2733. [Google Scholar] [CrossRef]
- Biancon, G.; Busanello, E.; Cheng, M.; Halene, S.; Tebaldi, T. Dissecting the stress granule RNA world: Dynamics, strategies and data. RNA 2025, in press. [Google Scholar] [CrossRef]
- Biancon, G.; Joshl, P.; Zimmer, J.T.; Hunck, T.; Gao, Y.; Lessard, M.D.; Corchaine, E.; Barentine, A.; Machyna, M.; Botti, V.; et al. Precision analysis of mutant U2AF1 activity reveals deployment of stress granules in myeloid malignancies. Mol. Cell 2022, 82, 1107–1122. [Google Scholar] [CrossRef] [PubMed]
- Biancon, G.; Busanello, E.; Chdeng, M.; Sidoli, S.; VanOudenhove, J.; Bucciarelli, G.; Tebaldi, T.; Halene, S. Unravelling the drivers of the stress granule signature in splicing factor-mutant myeloid malignancies. Blood 2024, 144, 4117–4118. [Google Scholar] [CrossRef]
- Shannon, M.L.; Heimlich, J.B.; Olson, S.; Debevec, A.; Copeland, Z.; Kishtagari, A.; Vlasschaert, C.; Snider, C.; Silver, A.J.; Brown, D.; et al. Clonal Hematopoiesis and Inflammation in the VasculaturE (CHIVE): A prospective, longitudinal cohort and biorepository. Blood Adv. 2024, 8, 3453–3463. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Healthy Helthy Controls | CHIP CHIP | ICUS ICUS | CCUS CCUS | MDS MDS | |
|---|---|---|---|---|---|
| Cytopenia | - | - | + | + | + |
| Dysplasia | - | - | - | - | + |
| Frequently mutated genes | - | DNMT3A TET2 ASXL1 PPM1D JAK2 TP53 | - | TET2 DNMT3A ASXL1 SRSF2 ZRSR2 TP53 U2AF1 | SF3B1 TET2 ASXL1 SRSF2 TP53 RUNX1 STAG2 U2AF1 ZRSR2 |
| Average number of mutated genes | - | ≈1 | - | ≈2 | ≈3 |
| Typical allele Frequency | - | 0.1–0.15 | - | 0.2–0.4 | 0.3–0.5 |
| Risk of MN | Very low | Low | Low | Low/Moderate | High |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, U.; Castelli, G.; Pelosi, E. Clonal Hematopoiesis, a Risk Condition for Developing Myeloid Neoplasia. Hemato 2025, 6, 10. https://doi.org/10.3390/hemato6020010
Testa U, Castelli G, Pelosi E. Clonal Hematopoiesis, a Risk Condition for Developing Myeloid Neoplasia. Hemato. 2025; 6(2):10. https://doi.org/10.3390/hemato6020010
Chicago/Turabian StyleTesta, Ugo, Germana Castelli, and Elvira Pelosi. 2025. "Clonal Hematopoiesis, a Risk Condition for Developing Myeloid Neoplasia" Hemato 6, no. 2: 10. https://doi.org/10.3390/hemato6020010
APA StyleTesta, U., Castelli, G., & Pelosi, E. (2025). Clonal Hematopoiesis, a Risk Condition for Developing Myeloid Neoplasia. Hemato, 6(2), 10. https://doi.org/10.3390/hemato6020010