
Chemical and Structural Comparison of Different Commercial Food Supplements for Silicon Uptake



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
2.3. Characterization
3. Results and Discussion
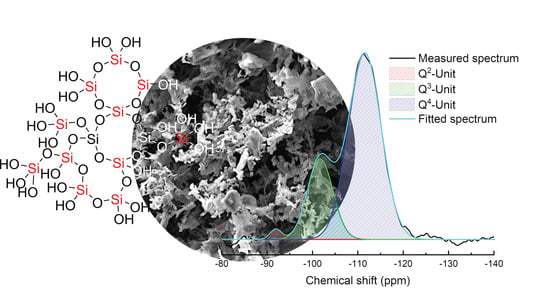
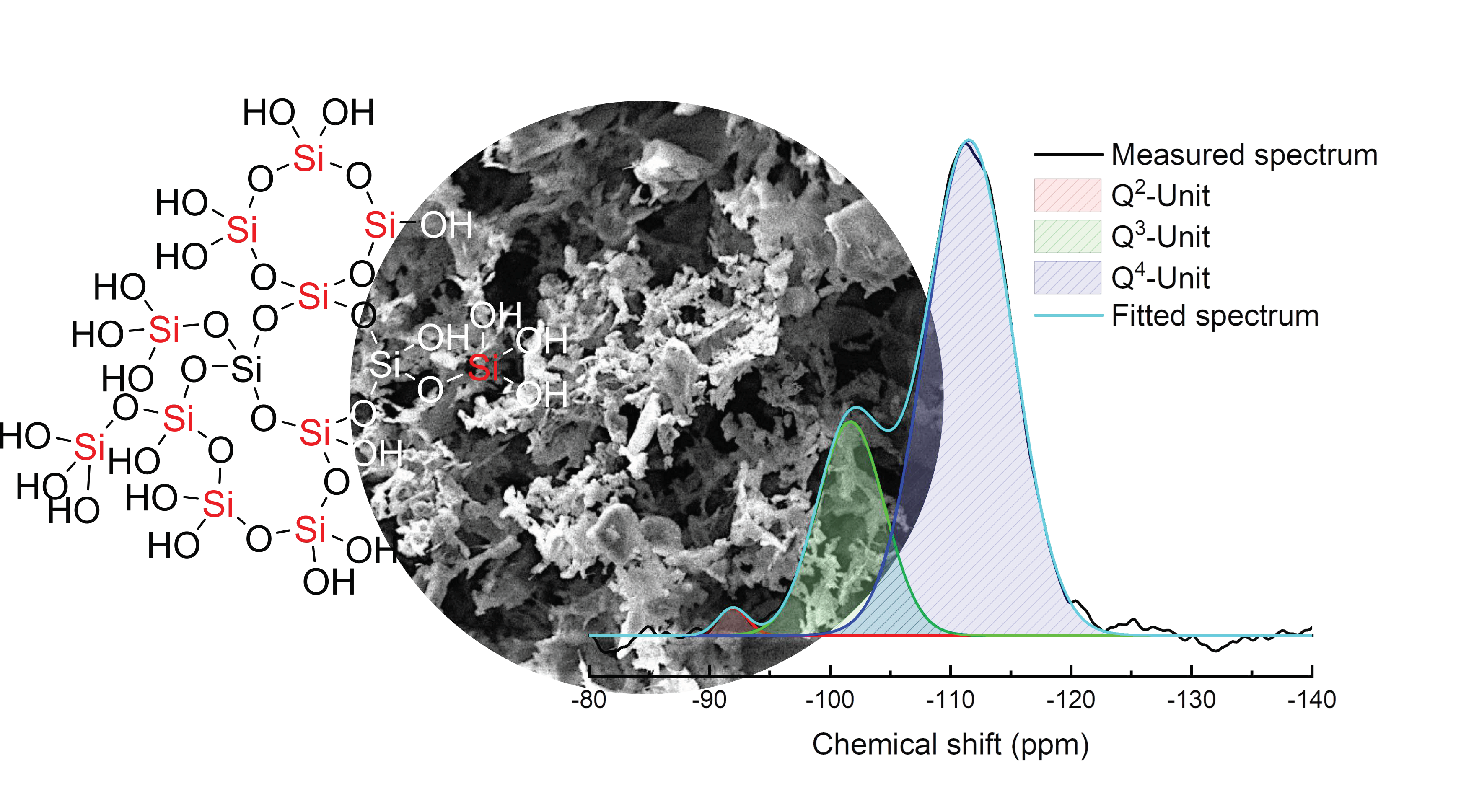
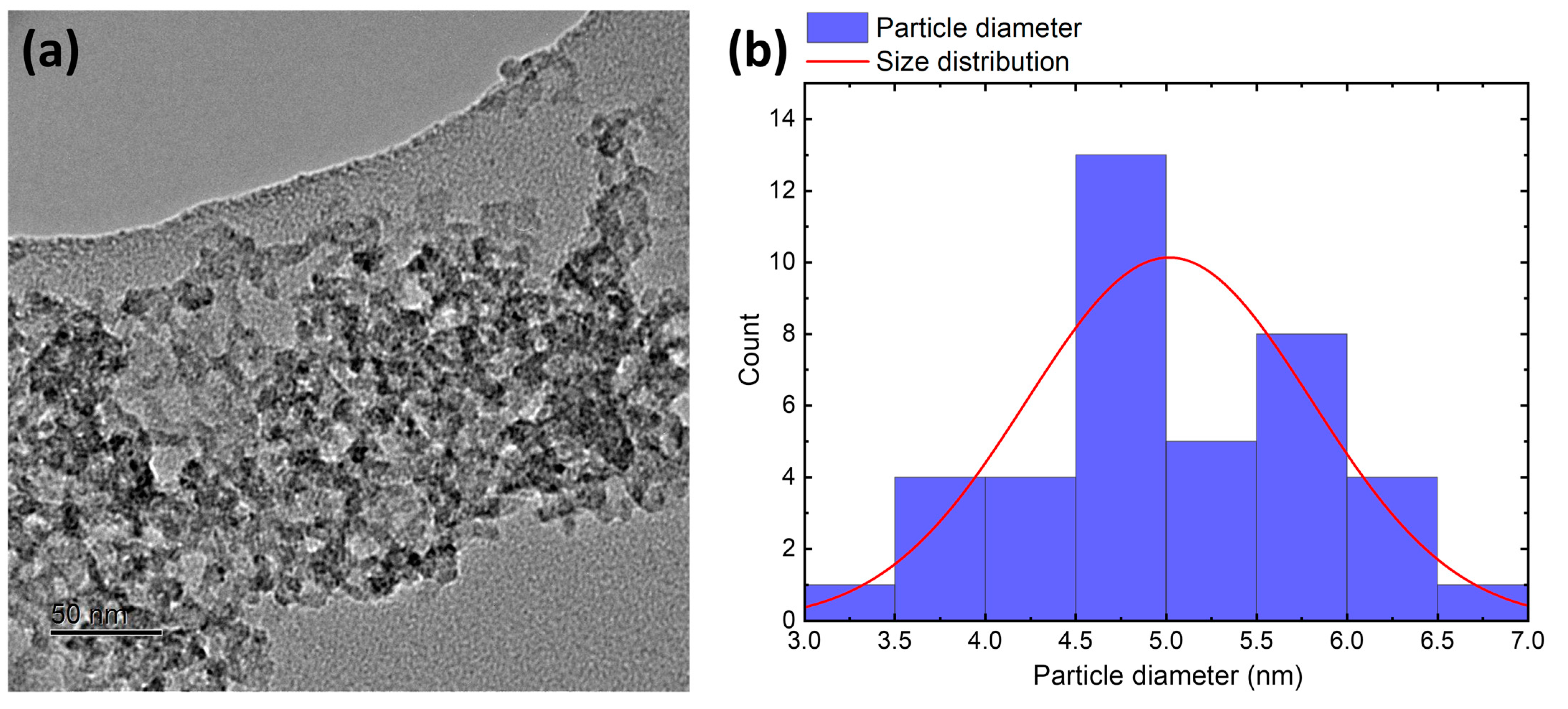
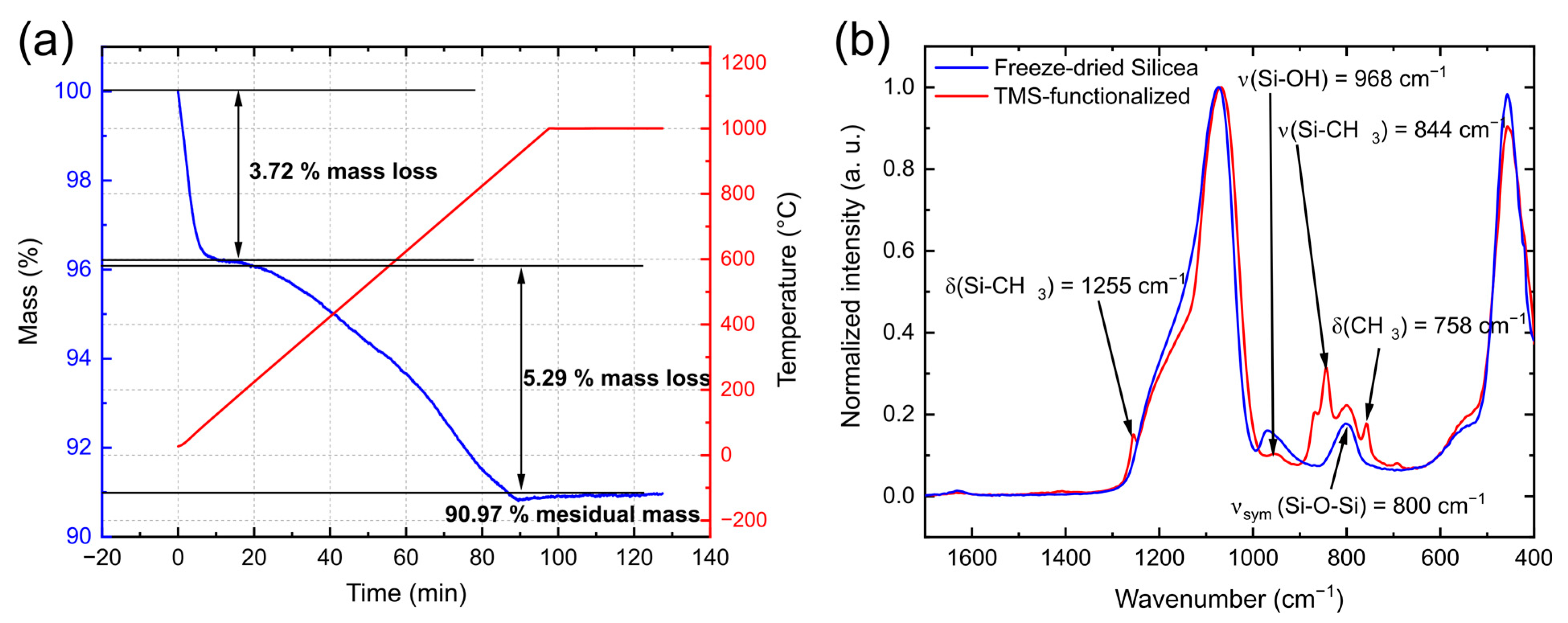
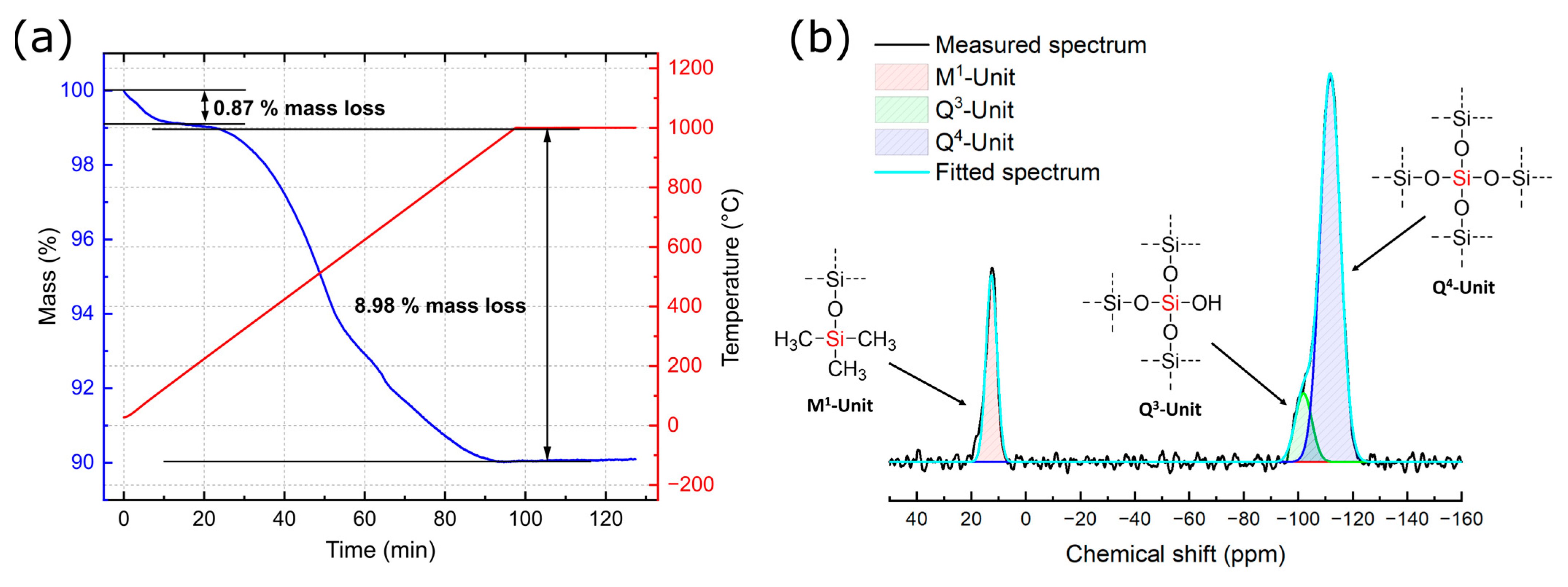
3.1. Characterization of Silicea
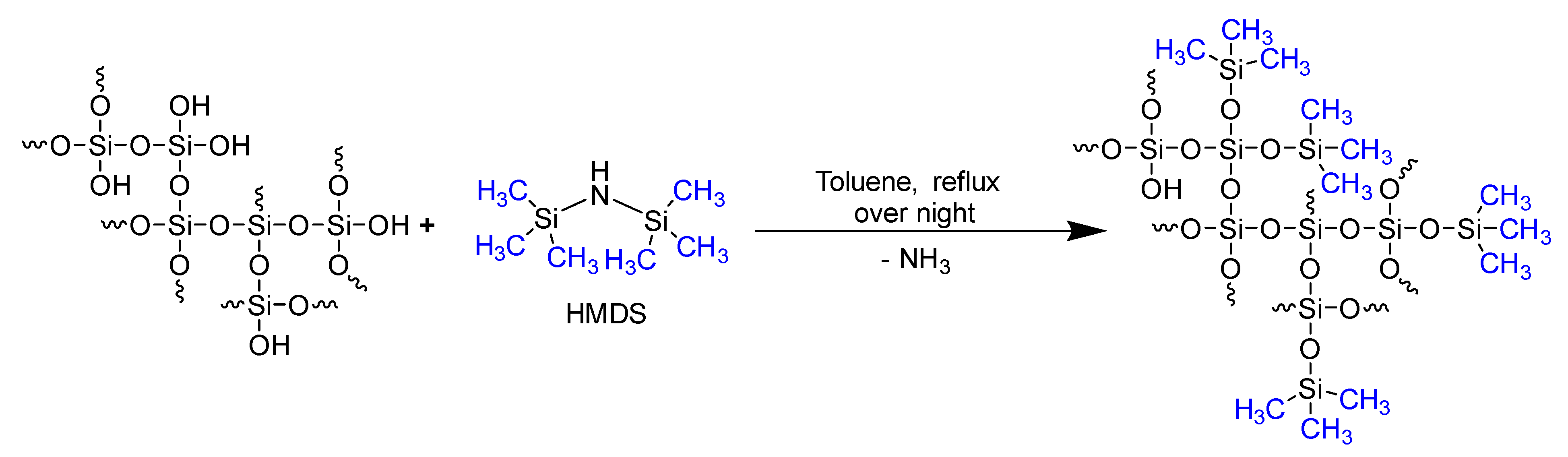
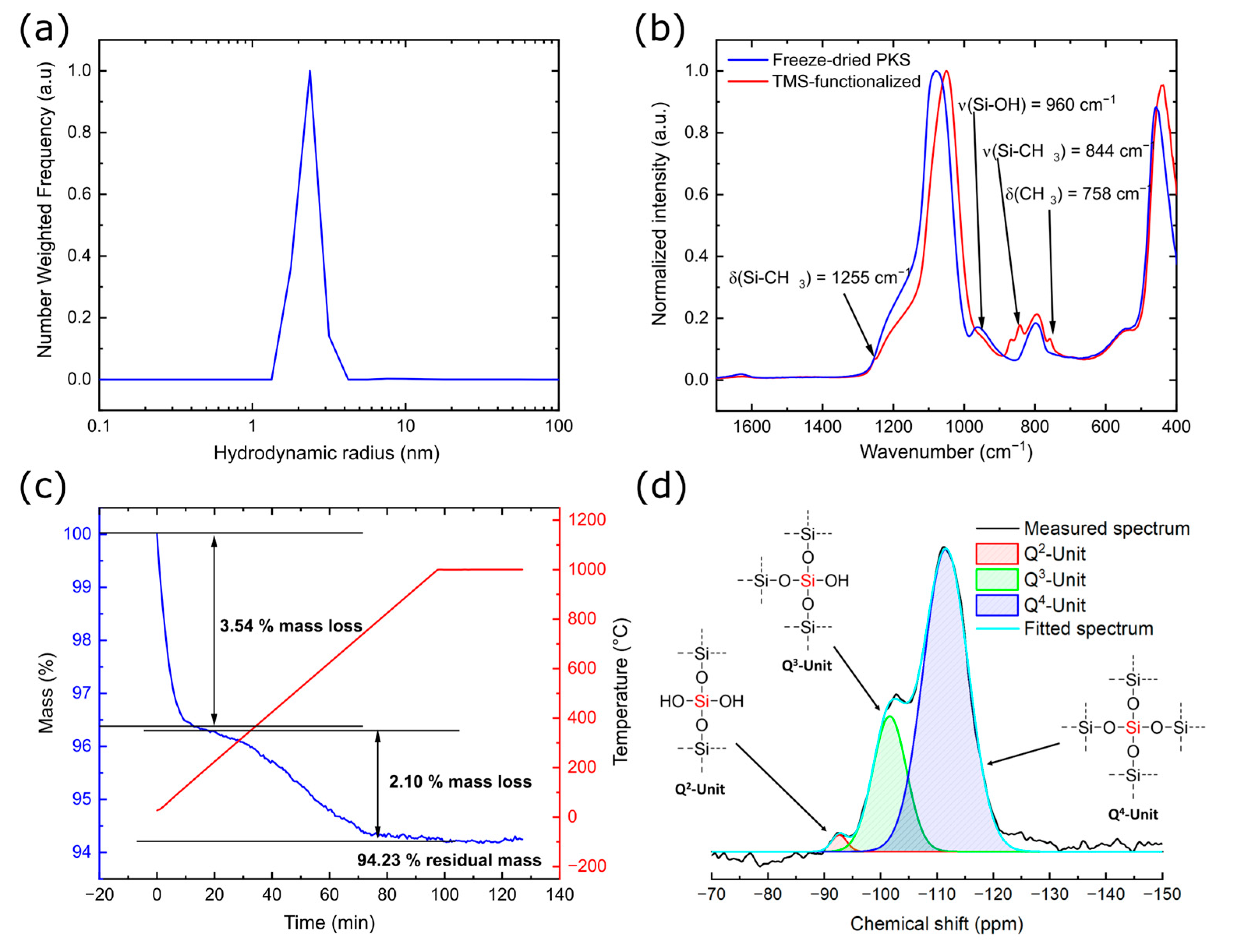
3.2. Characterization of the PKS Product
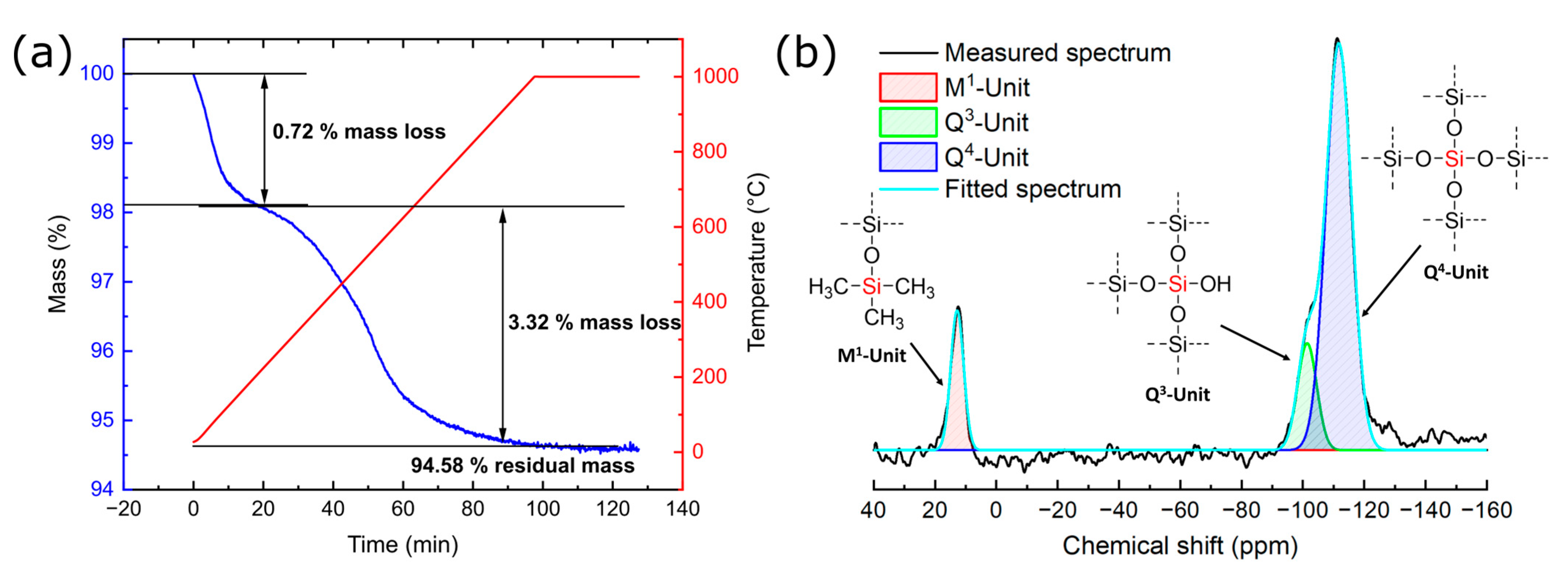
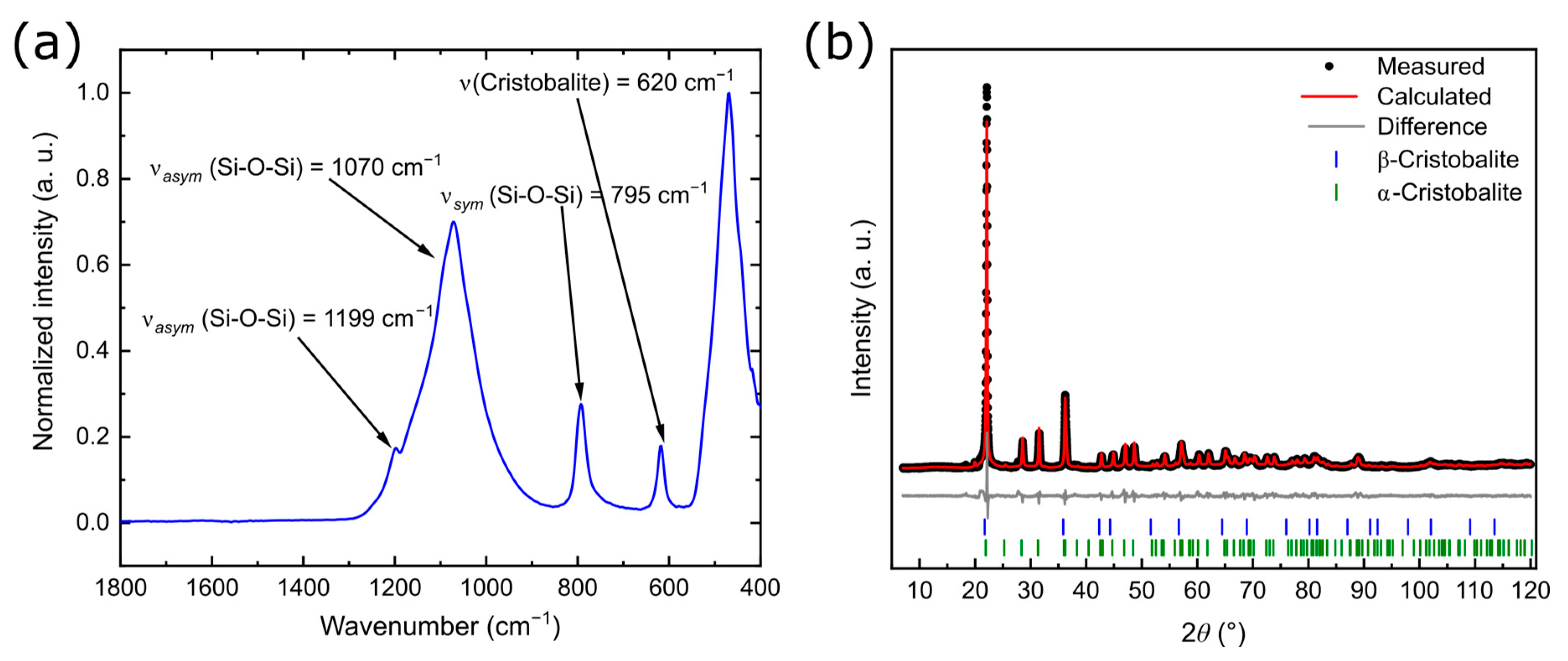
3.3. Characterization of the Product FlKe
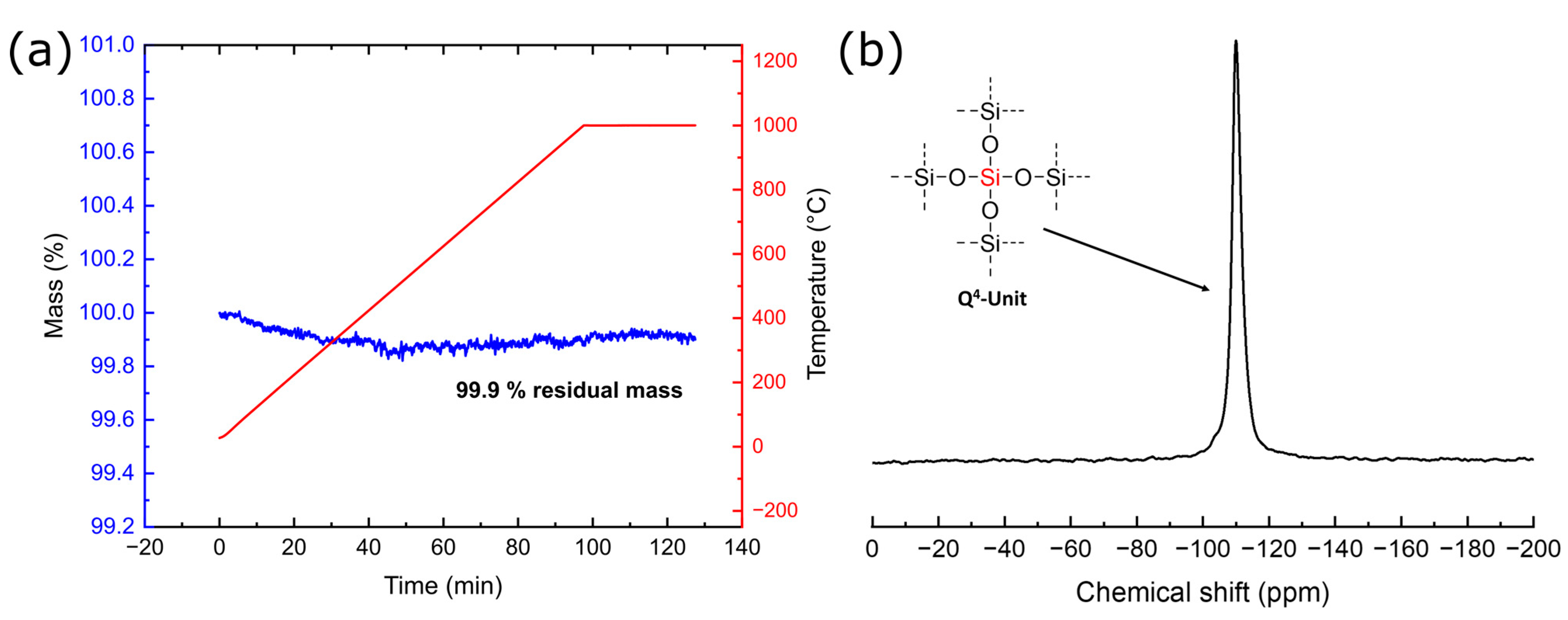
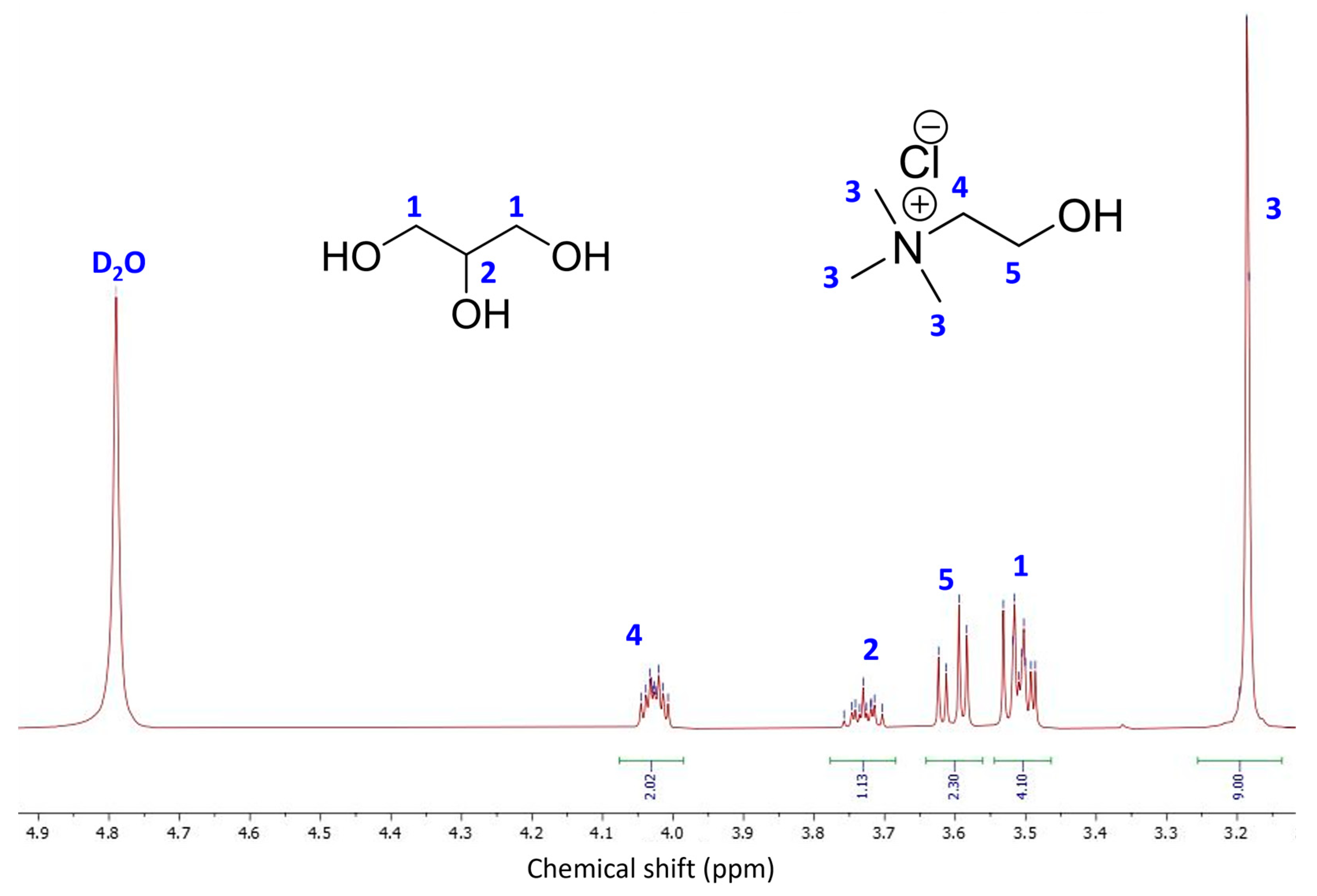
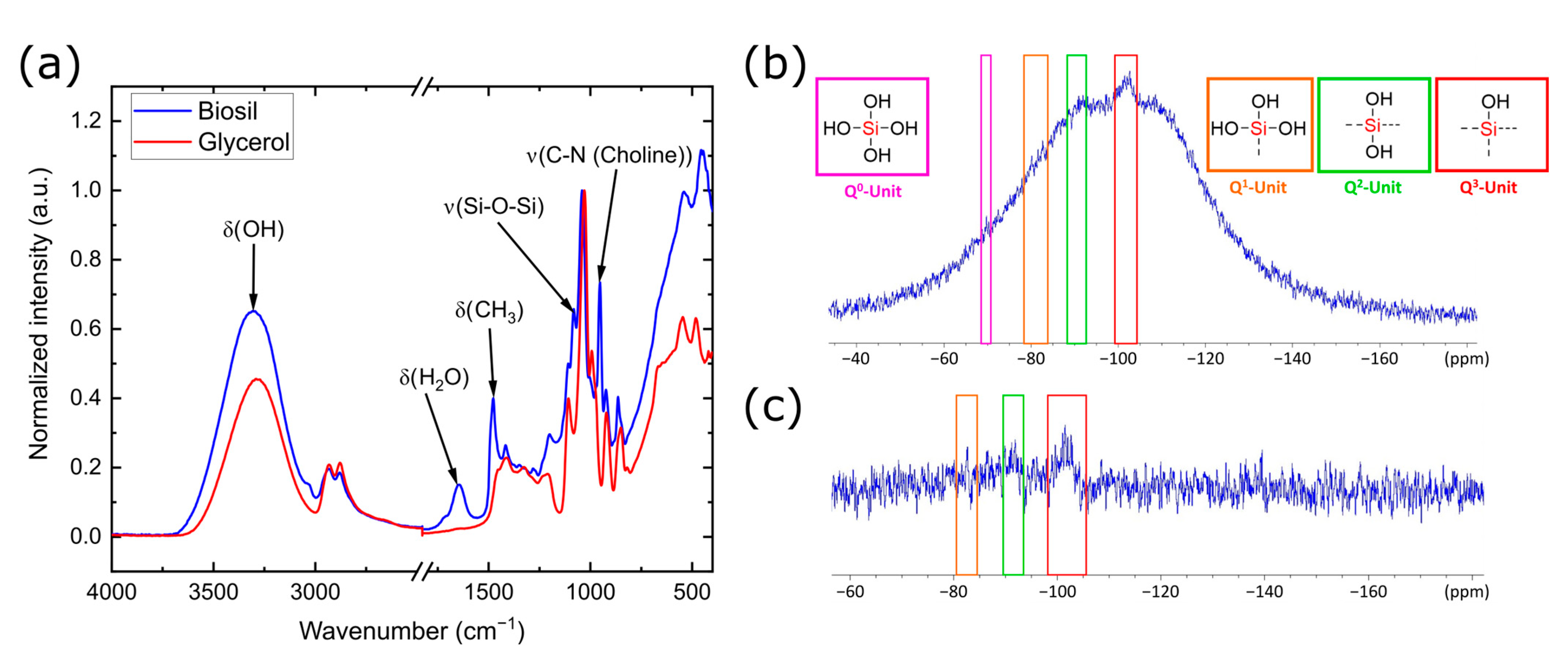
3.4. Characterization of BioSil®
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jurkić, L.M.; Cepanec, I.; Pavelić, S.K.; Pavelić, K. Biological and therapeutic effects of ortho-silicic acid and some ortho-silicic acid-releasing compounds: New perspectives for therapy. Nutr. Metab. 2013, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Reffitt, D.M.; Jugdaohsingh, R.; Thompson, R.P.H.; Powell, J.J. Silicic acid: Its gastrointestinal uptake and urinary excretion in man and effects on aluminium excretion. J. Inorg. Biochem. 1999, 76, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Sadowska, A.; Świderski, F. Sources, Bioavailability, and Safety of Silicon Derived from Foods and Other Sources Added for Nutritional Purposes in Food Supplements and Functional Foods. Appl. Sci. 2020, 10, 6255. [Google Scholar] [CrossRef]
- Ferreira, A.O.; Freire, É.S.; Polonini, H.C.; Da Silva, P.J.L.C.; Brandão, M.A.F.; Raposo, N.R.B. Anti-Aging Effects of Monomethylsilanetriol and Maltodextrin-Stabilized Orthosilicic Acid on Nails, Skin and Hair. Cosmetics 2018, 5, 41. [Google Scholar] [CrossRef]
- Carlisle, E.M. Silicon as a trace nutrient. Sci. Total Environ. 1988, 73, 95–106. [Google Scholar] [CrossRef]
- Reffitt, D.M.; Ogston, N.; Jugdaohsingh, R.; Cheung, H.F.J.; Evans, B.A.J.; Thompson, R.P.H.; Powell, J.J.; Hampson, G.N. Orthosilicic acid stimulates collagen type 1 synthesis and osteoblastic differentiation in human osteoblast-like cells in vitro. Bone 2003, 32, 127–135. [Google Scholar] [CrossRef]
- Authority, E.F.S. Opinion of the Scientific Panel on Dietetic products, nutrition and allergies [NDA] related to the Tolerable Upper Intake Level of Silicon. EFSA J. 2004, 2, 60. [Google Scholar] [CrossRef]
- Carlisle, E.M. Biochemical and Morphological Changes Associated with Long Bone Abnormalities in Silicon Deficiency. J. Nutr. 1980, 110, 1046–1056. [Google Scholar] [CrossRef]
- Sripanyakorn, S.; Jugdaohsingh, R.; Thompson, R.P.H.; Powell, J.J. Dietary silicon and bone health. Nutr. Bull. 2005, 30, 222–230. [Google Scholar] [CrossRef]
- Jugdaohsingh, R. Silicon and bone health. J. Nutr. Health Aging 2007, 11, 99–110. [Google Scholar]
- Sripanyakorn, S.; Jugdaohsingh, R.; Dissayabutr, W.; Anderson, S.H.C.; Thompson, R.P.H.; Powell, J.J. The comparative absorption of silicon from different foods and food supplements. Br. J. Nutr. 2009, 102, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Scholey, D.V.; Belton, D.J.; Burton, E.J.; Perry, C.C. Bioavailability of a novel form of silicon supplement. Sci. Rep. 2018, 8, 17022. [Google Scholar] [CrossRef]
- Kästele, X.; Klüfers, P.; Kopp, F.; Schuhmacher, J.; Vogt, M. Silicon Chelation in Aqueous and Nonaqueous Media: The Furanoidic Diol Approach. Chem. Eur. J. 2005, 11, 6326–6346. [Google Scholar] [CrossRef]
- Voronkov, M.G.; Baryshok, V.P. Atranes as a new generation of biologically active substances. Her. Russ. Acad. Sci. 2010, 80, 514–521. [Google Scholar] [CrossRef]
- Puri, J.K.; Singh, R.; Chahal, V.K. Silatranes: A review on their synthesis, structure, reactivity and applications. Chem. Soc. Rev. 2011, 40, 1791–1840. [Google Scholar] [CrossRef] [PubMed]
- Frye, C.L.; Vogel, G.E.; Hall, J.A. Triptych-siloxazolidines: Pentacoordinate bridgehead silanes resulting from transannular interaction of nitrogen and silicon. J. Am. Chem. Soc. 1961, 83, 996–997. [Google Scholar]
- Sieburth, S.M.; Chen, C.-A. Silanediol Protease Inhibitors: From Conception to Validation. Eur. J. Org. Chem. 2006, 2006, 311–322. [Google Scholar] [CrossRef]
- Chen, C.-A.; Sieburth, S.M.; Glekas, A.; Hewitt, G.W.; Trainor, G.L.; Erickson-Viitanen, S.; Garber, S.S.; Cordova, B.; Jeffry, S.; Klabe, R.M. Drug design with a new transition state analog of the hydrated carbonyl: Silicon-based inhibitors of the HIV protease. Chem. Biol. 2001, 8, 1161–1166. [Google Scholar] [CrossRef]
- Kim, J.; Glekas, A.; Sieburth, S.M. Silanediol-Based inhibitor of thermolysin. Bioorg. Med. Chem. Lett. 2002, 12, 3625–3627. [Google Scholar] [CrossRef]
- Mutahi, M.W.; Nittoli, T.; Guo, L.; Sieburth, S.M. Silicon-Based Metalloprotease Inhibitors: Synthesis and Evaluation of Silanol and Silanediol Peptide Analogues as Inhibitors of Angiotensin-Converting Enzyme1. J. Am. Chem. Soc. 2002, 124, 7363–7375. [Google Scholar] [CrossRef]
- Sieburth, S.M.; Nittoli, T.; Mutahi, A.M.; Guo, L. Silanediols: A New Class of Potent Protease Inhibitors. Angew. Chem. Int. Ed. 1998, 37, 812–814. [Google Scholar] [CrossRef]
- Blunder, M.; Hurkes, N.; Spirk, S.; List, M.; Pietschnig, R. Silanetriols as in vitro inhibitors for AChE. Bioorg. Med. Chem. Lett. 2011, 21, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Pietschnig, R.; Spirk, S. The chemistry of organo silanetriols. Coord. Chem. Rev. 2016, 323, 87–106. [Google Scholar] [CrossRef]
- Tacke, R. Milestones in the Biochemistry of Silicon: From Basic Research to Biotechnological Applications. Angew. Chem. Int. Ed. 1999, 38, 3015–3018. [Google Scholar] [CrossRef]
- Mojet, B.L.; Ebbesen, S.D.; Lefferts, L. Light at the interface: The potential of attenuated total reflection infrared spectroscopy for understanding heterogeneous catalysis in water. Chem. Soc. Rev. 2010, 39, 4643–4655. [Google Scholar] [CrossRef]
- Artemov, V.G.; Uykur, E.; Roh, S.; Pronin, A.V.; Ouerdane, H.; Dressel, M. Revealing excess protons in the infrared spectrum of liquid water. Sci. Rep. 2020, 10, 11320–11329. [Google Scholar] [CrossRef]
- Rahman, I.A.; Vejayakumaran, P.; Sipaut, C.S.; Ismail, J.; Chee, C.K. Effect of the drying techniques on the morphology of silica nanoparticles synthesized via sol–gel process. Ceram. Int. 2008, 34, 2059–2066. [Google Scholar] [CrossRef]
- Jafarzadeh, M.; Rahman, I.A.; Sipaut, C.S. Synthesis of silica nanoparticles by modified sol–gel process: The effect of mixing modes of the reactants and drying techniques. J. Sol.-Gel. Sci. Technol. 2009, 50, 328–336. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, Y.; Luo, G.; Bai, S. Effects of Precipitation and Drying Processes on the Synthesis of Silica Materials with a Large-Pore-Volume and Narrow-Pore-Diameter Distribution. Ind. Eng. Chem. Res. 2016, 55, 3579–3587. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Sarawade, P.B.; Kim, J.-K.; Hilonga, A.; Quang, D.V.; Kim, H.T. Effect of drying technique on the physicochemical properties of sodium silicate-based mesoporous precipitated silica. Appl. Surf. Sci. 2011, 258, 955–961. [Google Scholar] [CrossRef]
- Ek, S.; Root, A.; Peussa, M.; Niinistö, L. Determination of the hydroxyl group content in silica by thermogravimetry and a comparison with 1H MAS NMR results. Thermochim. Acta 2001, 379, 201–212. [Google Scholar] [CrossRef]
- de Farias, R.F.; Airoldi, C. Thermogravimetry as a Reliable tool to Estimate the Density of Silanols on a Silica Gel Surface. J. Therm. Anal. Calorim. 1998, 53, 751–756. [Google Scholar] [CrossRef]
- Peng, L.; Qisui, W.; Xi, L.; Chaocan, Z. Investigation of the states of water and OH groups on the surface of silica. Colloids Surf. A 2009, 334, 112–115. [Google Scholar] [CrossRef]
- Cheng, Z.; Shan, H.; Sun, Y.; Zhang, L.; Jiang, H.; Li, C. Evolution mechanism of surface hydroxyl groups of silica during heat treatment. Appl. Surf. Sci. 2020, 513, 145766. [Google Scholar] [CrossRef]
- Metin, C.O.; Lake, L.W.; Miranda, C.R.; Nguyen, Q.P. Stability of aqueous silica nanoparticle dispersions. J. Nanoparticle Res. 2011, 13, 839–850. [Google Scholar] [CrossRef]
- Liu, Q.X.; Xu, W.C. Study on Preparation and Properties of Precipitated Silica. Adv. Mater. Res. 2012, 468–471, 1353–1358. [Google Scholar] [CrossRef]
- Uchino, T.; Sakka, T.; Iwasaki, M. Interpretation of Hydrated States of Sodium Silicate Glasses by Infrared and Raman Analysis. J. Am. Ceram. Soc. 1991, 74, 306–313. [Google Scholar] [CrossRef]
- Jitianu, A.; Gonzalez, G.; Klein, L.C. Hybrid Sol–Gel Glasses with Glass-Transition Temperatures Below Room Temperature. J. Am. Ceram. Soc. 2015, 98, 3673–3679. [Google Scholar] [CrossRef]
- Husung, R.D.; Doremus, R.H. The infrared transmission spectra of four silicate glasses before and after exposure to water. J. Mater. Res. 1990, 5, 2209–2217. [Google Scholar] [CrossRef]
- Al-Oweini, R.; El-Rassy, H. Synthesis and characterization by FTIR spectroscopy of silica aerogels prepared using several Si(OR)4 and R′′Si(OR′)3 precursors. J. Mol. Struct. 2009, 919, 140–145. [Google Scholar] [CrossRef]
- Medvedev, E.F.; Komarevskaya, A.S. IR spectroscopic study of the phase composition for sodium silicate synthesized in aqueous medium. Glass Ceram. 2007, 64, 7–11. [Google Scholar] [CrossRef]
- Kapusuz, D. Sol–gel derived silica/polyethylene glycol hybrids as potential oligonucleotide vectors. J. Mater. Res. 2019, 34, 3787–3797. [Google Scholar] [CrossRef]
- Li, J.; Hayakawa, S.; Shirosaki, Y.; Osaka, A. Revisiting structure of silica gels from water glass: An 1H and 29Si MAS and CP-MAS NMR study. J. Sol.-Gel. Sci. Technol. 2013, 65, 135–142. [Google Scholar] [CrossRef]
- Ide, M.; El-Roz, M.; De Canck, E.; Vicente, A.; Planckaert, T.; Bogaerts, T.; Van Driessche, I.; Lynen, F.; Van Speybroeck, V.; Thybault-Starzyk, F.; et al. Quantification of silanol sites for the most common mesoporous ordered silicas and organosilicas: Total versus accessible silanols. Phys. Chem. Chem. Phys. 2013, 15, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Halasz, I.; Kierys, A.; Goworek, J.; Liu, H.; Patterson, R.E. 29Si NMR and Raman Glimpses into the Molecular Structures of Acid and Base Set Silica Gels Obtained from TEOS and Na-Silicate. J. Phys. Chem. C 2011, 115, 24788–24799. [Google Scholar] [CrossRef]
- Bahlmann, E.K.F.; Harris, R.K.; Say, B.J. Method for the quantification of silicon-29 NMR spectra, developed for viscous silicate solutions. Magn. Reson. Chem. 1993, 31, 266–267. [Google Scholar] [CrossRef]
- Bahlmann, E.F.; Harris, R.; Rockliffe, J.; Smith, E. Silicon-29 NMR self-diffusion and chemical-exchange studies of concentrated sodium silicate solutions. J. Chem. Soc. Faraday Trans. 1997, 93, 93–98. [Google Scholar] [CrossRef]
- Cabrera, Y.; Cabrera, A.; Larsen, F.H.; Felby, C. Solid-state 29Si NMR and FTIR analyses of lignin-silica coprecipitates. Holzforschung 2016, 70, 709–718. [Google Scholar] [CrossRef]
- Webb, J.D.; Seki, T.; Goldston, J.F.; Pruski, M.; Crudden, C.M. Selective functionalization of the mesopores of SBA-15. Microporous Mesoporous Mater. 2015, 203, 123–131. [Google Scholar] [CrossRef]
- Protsak, I.S.; Morozov, Y.M.; Dong, W.; Le, Z.; Zhang, D.; Henderson, I.M. A 29Si, 1H, and 13C Solid-State NMR Study on the Surface Species of Various Depolymerized Organosiloxanes at Silica Surface. Nanoscale Res. Lett. 2019, 14, 160. [Google Scholar] [CrossRef] [PubMed]
- Serra, J.; González, P.; Liste, S.; Chiussi, S.; León, B.; Pérez-Amor, M.; Ylänen, H.O.; Hupa, M. Influence of the non-bridging oxygen groups on the bioactivity of silicate glasses. J. Mater. Sci. Mater. Med. 2002, 13, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Plumeré, N.; Ruff, A.; Speiser, B.; Feldmann, V.; Mayer, H.A. Stöber silica particles as basis for redox modifications: Particle shape, size, polydispersity, and porosity. J. Colloid Interface Sci. 2012, 368, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Jae Chul, R.; In, J.C. Structures and properties of silica gels prepared by the sol—Gel method. J. Non-Cryst. Solids 1991, 130, 8–17. [Google Scholar] [CrossRef]
- ALOthman, Z.A. A Review: Fundamental Aspects of Silicate Mesoporous Materials. Materials 2012, 5, 2874–2902. [Google Scholar] [CrossRef]
- Fripiat, J.; Uytterhoeven, J. Hydroxyl content in silica gel “Aerosil”. J. Phys. Chem. 1962, 66, 800–805. [Google Scholar] [CrossRef]
- Capel-Sanchez, M.C.; Barrio, L.; Campos-Martin, J.M.; Fierro, J.L.G. Silylation and surface properties of chemically grafted hydrophobic silica. J. Colloid Interface Sci. 2004, 277, 146–153. [Google Scholar] [CrossRef]
- Khedkar, M.V.; Somvanshi, S.B.; Humbe, A.V.; Jadhav, K.M. Surface modified sodium silicate based superhydrophobic silica aerogels prepared via ambient pressure drying process. J. Non-Cryst. Solids 2019, 511, 140–146. [Google Scholar] [CrossRef]
- Bu, J.; Rhee, H.K. Silylation of Ti-MCM-41 by trimethylsilyl-imidazole and its effect on the olefin epoxidation with aqueous H2O2. Catal. Lett. 2000, 66, 245–249. [Google Scholar] [CrossRef]
- Chen, Y.; Sepahvand, S.; Gauvin, F.; Schollbach, K.; Brouwers, H.; Yu, Q. One-pot synthesis of monolithic silica-cellulose aerogel applying a sustainable sodium silicate precursor. Constr. Build. Mater. 2021, 293, 123289. [Google Scholar] [CrossRef]
- Gao, G.-M.; Liu, D.-R.; Zou, H.-F.; Zou, L.-C.; Gan, S.-C. Preparation of silica aerogel from oil shale ash by fluidized bed drying. Powder Technol. 2010, 197, 283–287. [Google Scholar] [CrossRef]
- Venkateswara Rao, A.; Kulkarni, M.M.; Amalnerkar, D.P.; Seth, T. Superhydrophobic silica aerogels based on methyltrimethoxysilane precursor. J. Non-Cryst. Solids 2003, 330, 187–195. [Google Scholar] [CrossRef]
- Haukka, S.; Root, A. The reaction of hexamethyldisilazane and subsequent oxidation of trimethylsilyl groups on silica studied by solid-state NMR and FTIR. J. Phys. Chem. 1994, 98, 1695–1703. [Google Scholar] [CrossRef]
- Koyano, K.A.; Tatsumi, T.; Tanaka, Y.; Nakata, S. Stabilization of Mesoporous Molecular Sieves by Trimethylsilylation. J. Phys. Chem. B 1997, 101, 9436–9440. [Google Scholar] [CrossRef]
- Odenwald, C.; Kickelbick, G. Additive-free continuous synthesis of silica and ORMOSIL micro- and nanoparticles applying a microjet reactor. J. Sol.-Gel. Sci. Technol. 2018, 89, 343–353. [Google Scholar] [CrossRef]
- Xue, S.-H.; Xie, H.; Ping, H.; Li, Q.-C.; Su, B.-L.; Fu, Z.-Y. Induced transformation of amorphous silica to cristobalite on bacterial surfaces. RSC Adv. 2015, 5, 71844–71848. [Google Scholar] [CrossRef]
- Sumper, M.; Kröger, N. Silica formation in diatoms: The function of long-chain polyamines and silaffins. J. Mater. Chem. 2004, 14, 2059–2065. [Google Scholar] [CrossRef]
- Spearing, D.R.; Farnan, I.; Stebbins, J.F. Dynamics of the α-β phase transitions in quartz and cristobalite as observed by in-situ high temperature 29Si and 17O NMR. Phys. Chem. Miner. 1992, 19, 307–321. [Google Scholar] [CrossRef]
- Trujillo, S.A.; Peña-Solórzano, D.; Bejarano, O.R.; Ochoa-Puentes, C. Tin(ii) chloride dihydrate/choline chloride deep eutectic solvent: Redox properties in the fast synthesis of N-arylacetamides and indolo(pyrrolo)[1,2-a] quinoxalines. RSC Adv. 2020, 10, 40552–40561. [Google Scholar] [CrossRef]
- Jansson, H.; Bernin, D.; Ramser, K. Silicate species of water glass and insights for alkali-activated green cement. AIP Adv. 2015, 5, 067167. [Google Scholar] [CrossRef]
- Bass, J.L.; Turner, G.L. Anion Distributions in Sodium Silicate Solutions. Characterization by 29SI NMR and Infrared Spectroscopies, and Vapor Phase Osmometry. J. Phys. Chem. B 1997, 101, 10638–10644. [Google Scholar] [CrossRef]
- Harris, R.K.; Bahlmann, E.K.F.; Metcalfe, K.; Smith, E.G. Quantitative silicon-29 NMR investigations of highly concentrated high-ratio sodium silicate solutions. Magn. Reson. Chem. 1993, 31, 743–747. [Google Scholar] [CrossRef]
- Schraml, J.; Sandor, P.; Korec, S.; Krump, M.; Foller, B. Improved baseline in 29Si NMR spectra of water glasses. Magn. Reson. Chem. 2013, 51, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, K.; Van Cauwenbergh, R.; Robberecht, H.; Deelstra, H. Bioavailability of silicon from food and food supplements. Fresenius J. Anal. Chem. 1999, 363, 541–544. [Google Scholar] [CrossRef]
- Horcajada, P.; Rámila, A.; Boulahya, K.; González-Calbet, J.; Vallet-Regí, M. Bioactivity in ordered mesoporous materials. Solid State Sci. 2004, 6, 1295–1300. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Q2 Unit (%) | Q3 Unit (%) | Q4 Unit (%) | Total Si-OH Content (%) |
|---|---|---|---|---|
| Silicea (FD) | 2 | 25 | 73 | 29 |
| Silicea (TMS) | 0 | 13 | 87 | 13 |
| PKS (FD) | 2 | 24 | 74 | 28 |
| PKS (TMS) | 0 | 17 | 83 | 17 |
| FlKe | 0 | 0 | 100 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curto, Y.; Koch, M.; Kickelbick, G. Chemical and Structural Comparison of Different Commercial Food Supplements for Silicon Uptake. Solids 2023, 4, 1-21. https://doi.org/10.3390/solids4010001
Curto Y, Koch M, Kickelbick G. Chemical and Structural Comparison of Different Commercial Food Supplements for Silicon Uptake. Solids. 2023; 4(1):1-21. https://doi.org/10.3390/solids4010001
Chicago/Turabian StyleCurto, Yannic, Marcus Koch, and Guido Kickelbick. 2023. "Chemical and Structural Comparison of Different Commercial Food Supplements for Silicon Uptake" Solids 4, no. 1: 1-21. https://doi.org/10.3390/solids4010001
APA StyleCurto, Y., Koch, M., & Kickelbick, G. (2023). Chemical and Structural Comparison of Different Commercial Food Supplements for Silicon Uptake. Solids, 4(1), 1-21. https://doi.org/10.3390/solids4010001