


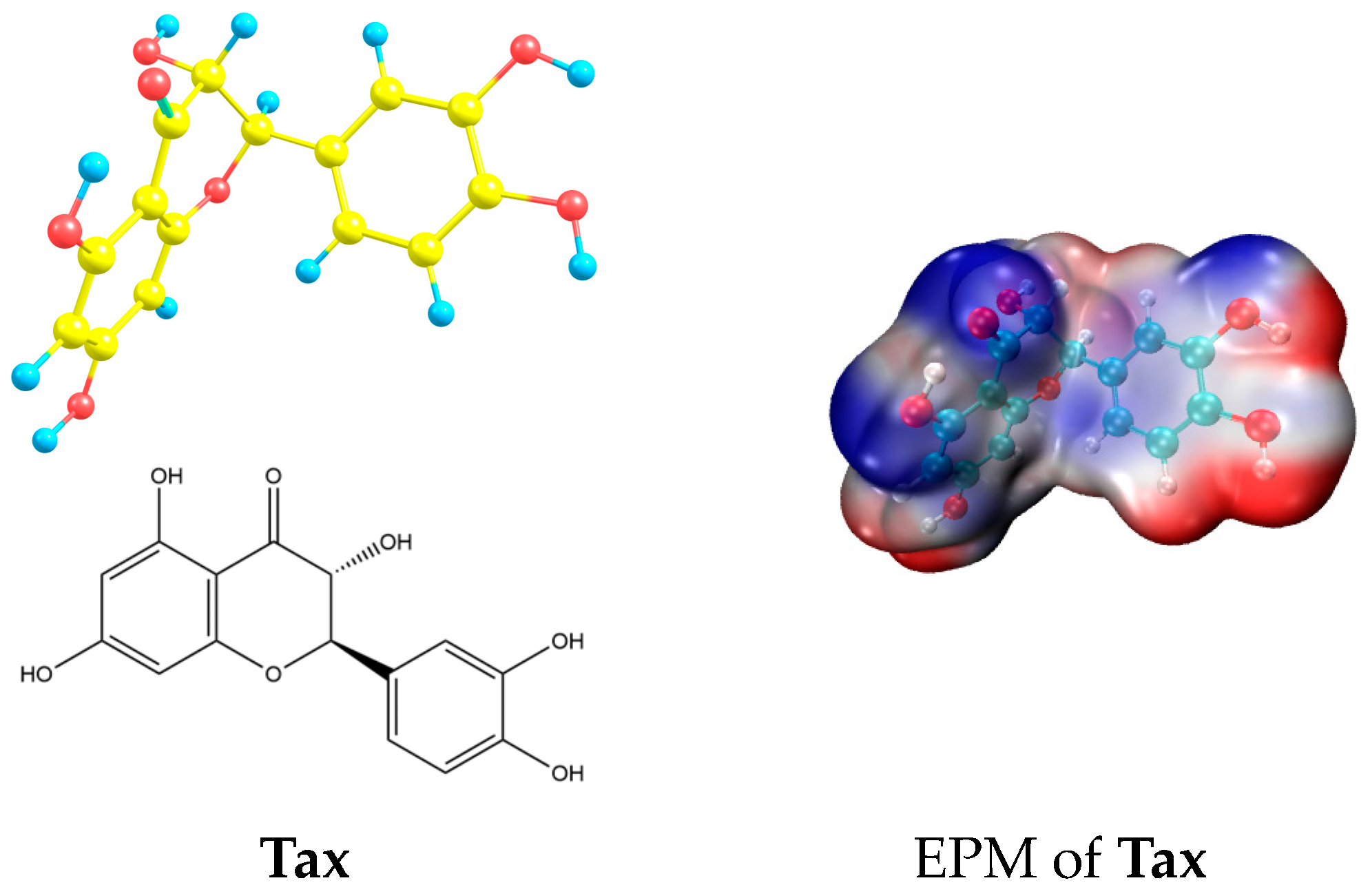
Taxifolin Adsorption on Nitrogenated Graphenes: Theoretical Insights


Abstract
:
1. Introduction
2. Computational Details
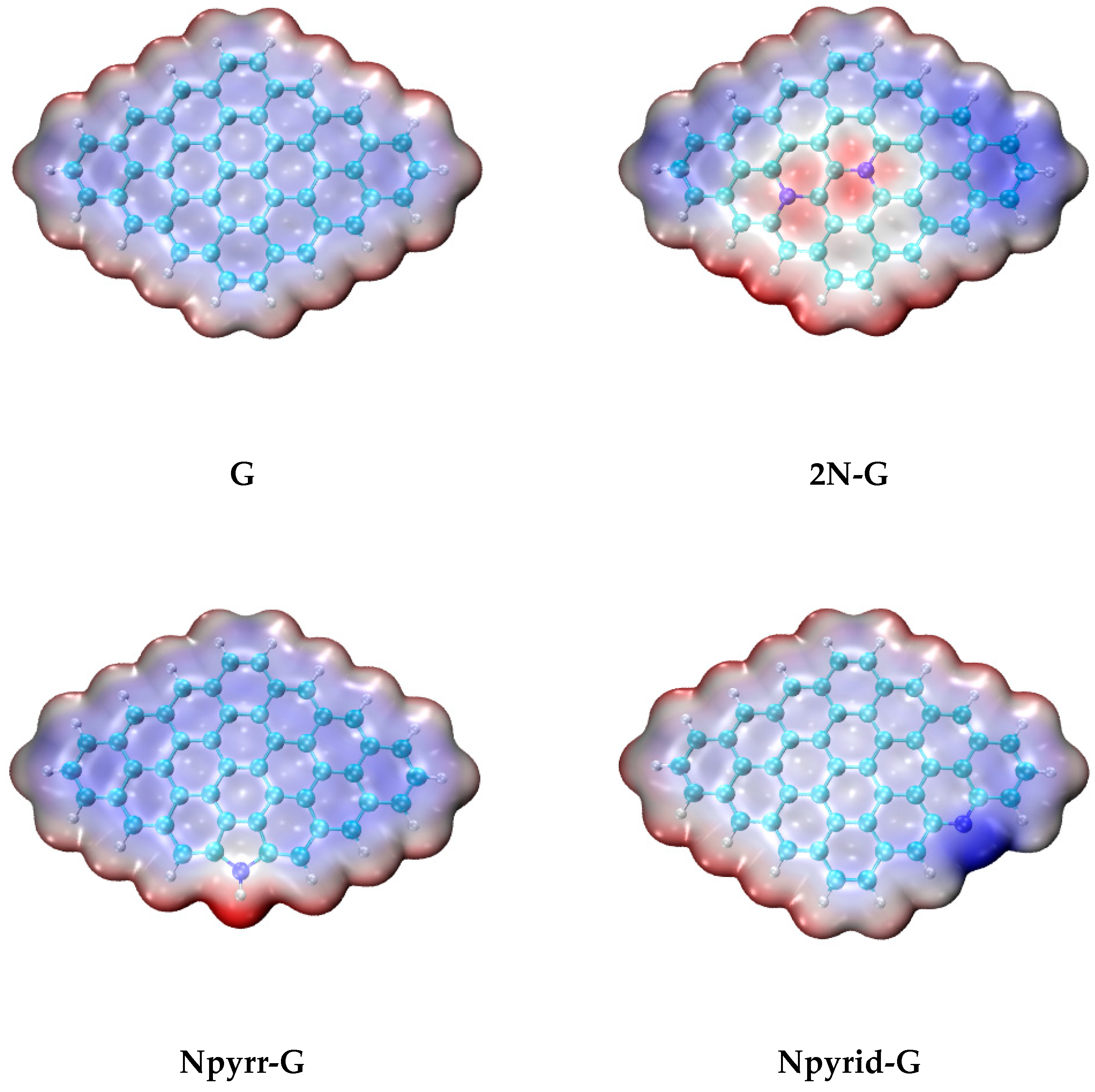
3. Results and Discussions
4. Conclusions
- (1)
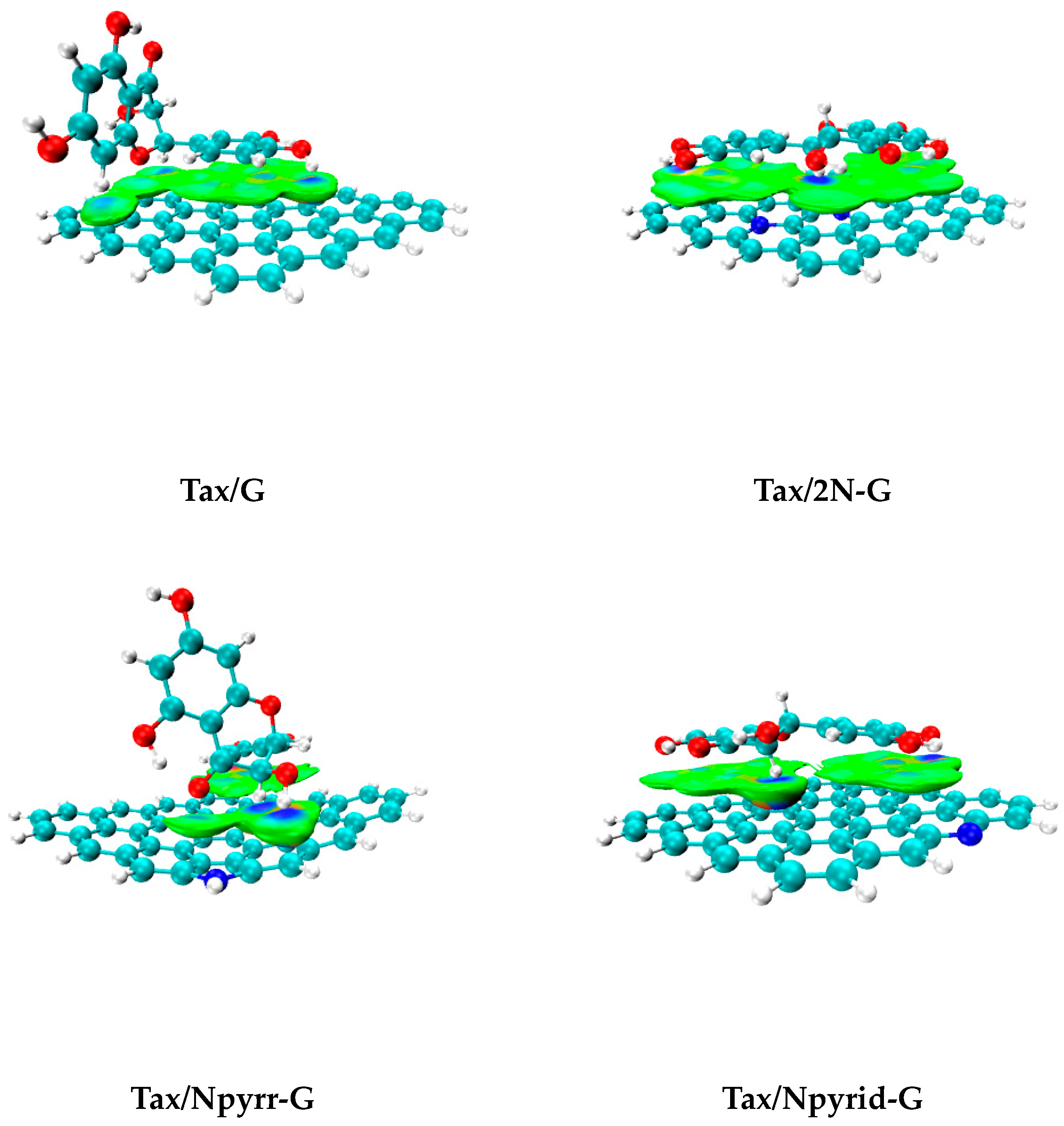
- N-doping of G leads to the substantial increase in Eint for Tax adsorption. The highest Eint value is in the case of 2N-G (−43.42 kcal/mol), and it is much larger than that for G (−30.04 kcal/mol).
- (2)
- SAPT0 calculations revealed that dispersion interactions play the main role in the stabilization of Tax (64–68%). Induction interactions contribute little to total attractions (7–8%), and the effects of electrostatic interactions fall between them (23–26%).
- (3)
- IGM graphs visually represent the dominant character of dispersion interactions. Besides this, the existence of strong interactions (H-bonding) was revealed. The QTAIM method approves the existence of H-bonding in all the studied complexes.
- (4)
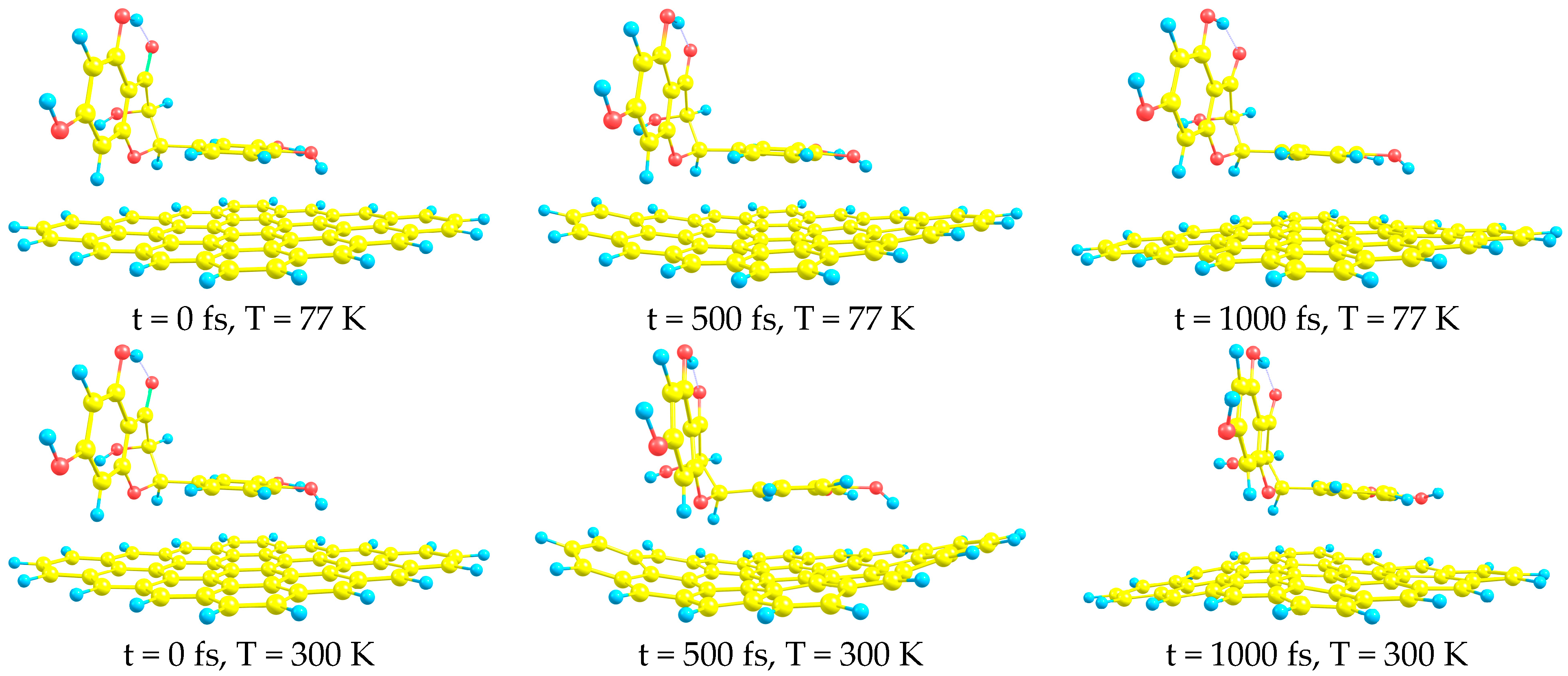
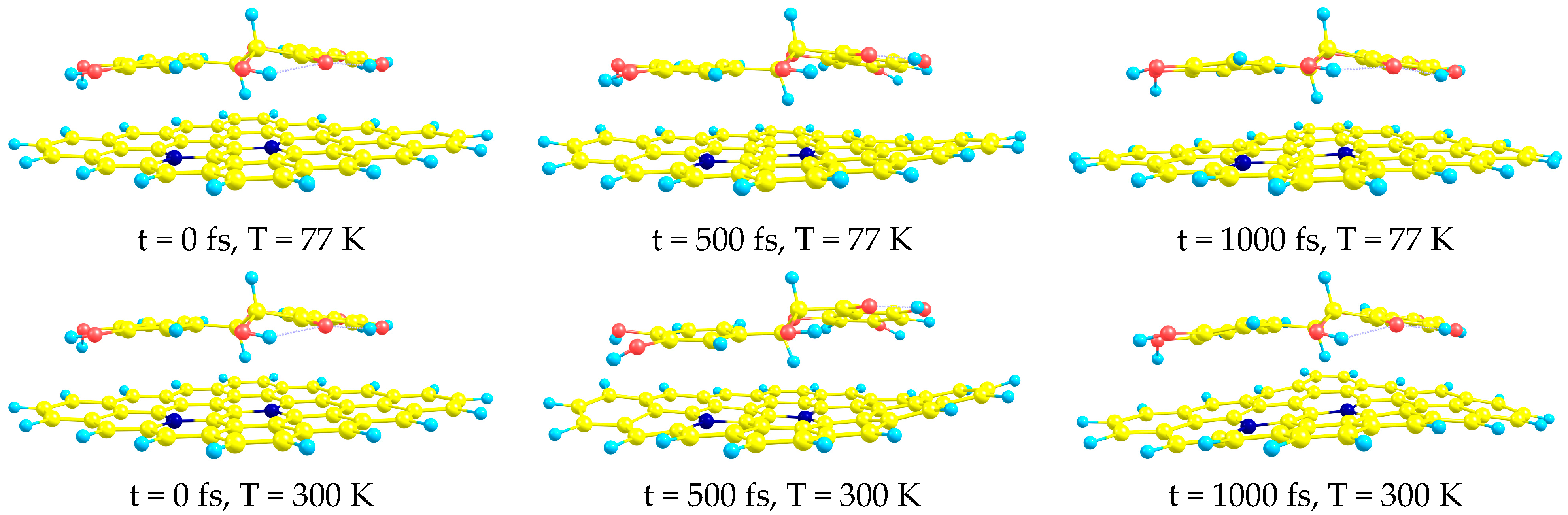
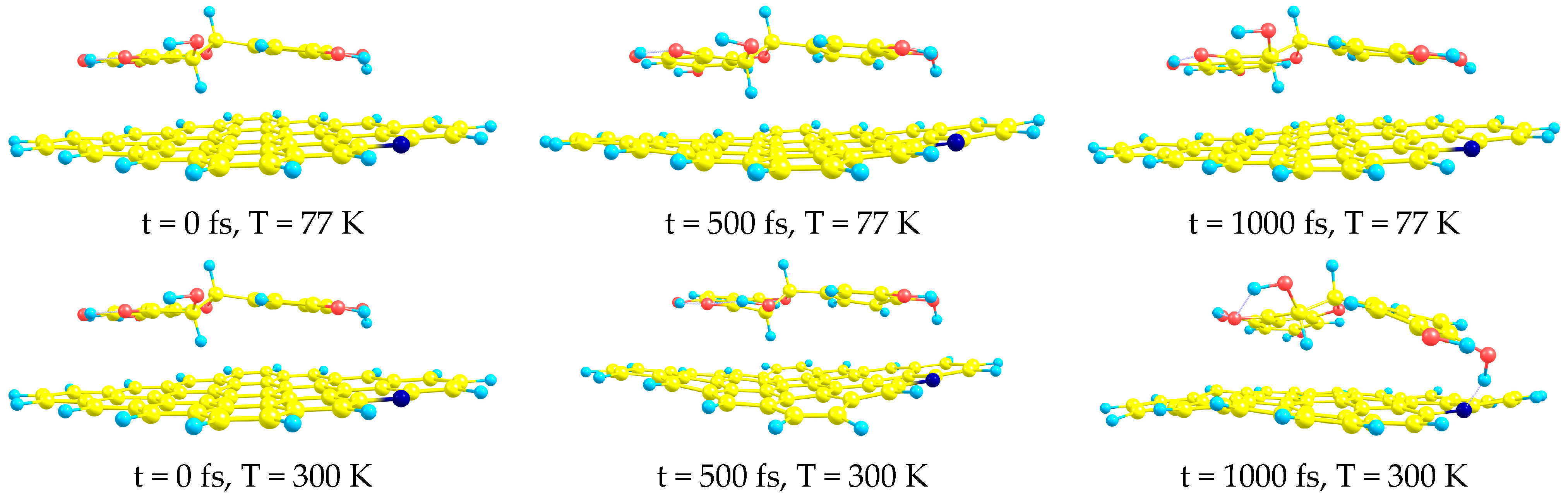
- AIMD simulations confirm the stability of all complexes at both studied temperatures (T = 77 and T = 300 K).
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laffleur, F.; Keckeis, V. Advances in drug delivery systems: Work in progress still needed? Int. J. Pharm. X 2020, 2, 100050. [Google Scholar] [CrossRef] [PubMed]
- Ezike, T.C.; Okpala, U.S.; Onoja, U.L.; Nwike, C.P.; Ezeako, E.C.; Okpara, O.J.; Okoroafor, C.C.; Eze, S.C.; Kalu, O.L.; Odoh, E.C.; et al. Advances in drug delivery systems, challenges and future directions. Heliyon 2023, 9, e17488. [Google Scholar] [CrossRef] [PubMed]
- Zhdanov, V.P. Diffusion-Limited Release of Molecules from Nanocarriers with Appreciable Energetic Heterogeneity. J. Phys. Chem. C 2024, 128, 1216–1221. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Din, F.U.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309. [Google Scholar] [CrossRef] [PubMed]
- Huda, S.; Alam, M.A.; Sharma, P.K. Smart nanocarriers-based drug delivery for cancer therapy: An innovative and developing strategy. J. Drug Deliv. Sci. Technol. 2020, 60, 102018. [Google Scholar] [CrossRef]
- Liu, J.; Cui, L.; Losic, D. Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta Biomater. 2013, 9, 9243–9257. [Google Scholar] [CrossRef]
- Oliveira, A.M.L.; Machado, M.; Silva, G.A.; Bitoque, D.B.; Tavares Ferreira, J.; Pinto, L.A.; Ferreira, Q. Graphene Oxide Thin Films with Drug Delivery Function. Nanomaterials 2022, 12, 1149. [Google Scholar] [CrossRef]
- Khodadadei, F.; Safarian, S.; Ghanbari, N. Methotrexate-loaded nitrogen-doped graphene quantum dots nanocarriers as an efficient anticancer drug delivery system. Mater. Sci. Engineering. C Mater. Biol. Appl. 2017, 79, 280–285. [Google Scholar] [CrossRef]
- Zare, H.; Ahmadi, S.; Ghasemi, A.; Ghanbari, M.; Rabiee, N.; Bagherzadeh, M.; Karimi, M.; Webster, T.J.; Hamblin, M.R.; Mostafavi, E. Carbon Nanotubes: Smart Drug/Gene Delivery Carriers. Int. J. Nanomed. 2021, 16, 1681–1706. [Google Scholar] [CrossRef]
- Bartkowski, M.; Giordani, S. Carbon nano-onions as potential nanocarriers for drug delivery. Dalton Trans. 2021, 50, 2300–2309. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kim, J.H. Synthesis, toxicity, biocompatibility, and biomedical applications of graphene and graphene-related materials. Int. J. Nanomed. 2016, 11, 1927–1945. [Google Scholar] [CrossRef] [PubMed]
- Shim, G.; Kim, M.G.; Park, J.Y.; Oh, Y.K. Graphene-based nanosheets for delivery of chemotherapeutics and biological drugs. Adv. Drug Deliv. Rev. 2016, 105, 205–227. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Baidya, R.; Chakraborty, T.; Samanta, A.K.; Roy, S. Pharmacological basis and new insights of taxifolin: A comprehensive review. Biomed. Pharmacother. Biomed. Pharmacother. 2021, 142, 112004. [Google Scholar] [CrossRef] [PubMed]
- Sundraraman, G.; Jayakumari, L.S. Meticulous Taxifolin Releasing Performance by the Zinc Oxide Nanoparticles: As a Short Road to Drug delivery System for Cancer Therapeutics. J. Clust. Sci. 2019, 31, 241–255. [Google Scholar] [CrossRef]
- Luneva, O.V.; Bikova, K.; Kashurin, A.I.; Motovilov, V.V.; Uspenskaya, M.V. Thermosensitive and pH-sensitive Drug Release from Taxifolin-Containing Collagen-Acrylic Hydrogels. In Proceedings of the 2022 IEEE-EMBS Conference on Biomedical Engineering and Sciences (IECBES), Kuala Lumpur, Malaysia, 7–9 December 2022; pp. 350–354. [Google Scholar] [CrossRef]
- Li, Y.; Su, H.; Wang, W.; Yin, Z.; Li, J.e.; Yuan, E.; Zhang, Q. Fabrication of taxifolin loaded zein-caseinate nanoparticles and its bioavailability in rat. Food Sci. Hum. Wellness 2023, 12, 2306–2313. [Google Scholar] [CrossRef]
- Kalinina, I.; Potoroko, I.; Sonawane, S.H. Sonochemical encapsulation of taxifolin into cyclodextrine for improving its bioavailability and bioactivity for food. In Encapsulation of Active Molecules and Their Delivery System; Elsevier: Amsterdam, The Netherlands, 2020; pp. 85–102. [Google Scholar] [CrossRef]
- Zhao, M.; Zhang, Y.; Qin, W.; Guo, J. First-principles study of the adsorption of flavonoids on graphene oxide. J. Phys. Org. Chem. 2022, 35, e4411. [Google Scholar] [CrossRef]
- Tiwari, H.; Karki, N.; Pal, M.; Basak, S.; Verma, R.K.; Bal, R.; Kandpal, N.D.; Bisht, G.; Sahoo, N.G. Functionalized graphene oxide as a nanocarrier for dual drug delivery applications: The synergistic effect of quercetin and gefitinib against ovarian cancer cells. Colloids Surfaces. B Biointerfaces 2019, 178, 452–459. [Google Scholar] [CrossRef]
- García, G.; Atilhan, M.; Aparicio, S. Flavonols on graphene: A DFT insight. Theor. Chem. Acc. 2015, 134, 57. [Google Scholar] [CrossRef]
- Lv, Q.; Si, W.; He, J.; Sun, L.; Zhang, C.; Wang, N.; Yang, Z.; Li, X.; Wang, X.; Deng, W.; et al. Selectively nitrogen-doped carbon materials as superior metal-free catalysts for oxygen reduction. Nat. Commun. 2018, 9, 3376. [Google Scholar] [CrossRef]
- Ganyecz, A.; Kallay, M. Oxygen Reduction Reaction on N-Doped Graphene: Effect of Positions and Scaling Relations of Adsorption Energies. J. Phys. Chemistry. C Nanomater. Interfaces 2021, 125, 8551–8561. [Google Scholar] [CrossRef] [PubMed]
- Dinadayalane, T.; Lazare, J.; Alzaaqi, N.F.; Herath, D.; Hill, B.; Campbell, A.E. Structures, properties, and applications of nitrogen-doped graphene. In Theoretical and Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2022; Volume 21, pp. 211–248. [Google Scholar] [CrossRef]
- Olteanu, D.; Filip, A.; Socaci, C.; Biris, A.R.; Filip, X.; Coros, M.; Rosu, M.C.; Pogacean, F.; Alb, C.; Baldea, I.; et al. Cytotoxicity assessment of graphene-based nanomaterials on human dental follicle stem cells. Colloids Surfaces. B Biointerfaces 2015, 136, 791–798. [Google Scholar] [CrossRef]
- Yokwana, K.; Ntsendwana, B.; Nxumalo, E.N.; Mhlanga, S.D. Recent advances in nitrogen-doped graphene oxide nanomaterials: Synthesis and applications in energy storage, sensor electrochemical applications and water treatment. J. Mater. Res. 2023, 38, 3239–3263. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Patkowski, K. Recent developments in symmetry-adapted perturbation theory. WIREs Comput. Mol. Sci. 2019, 10, e1452. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Qu, Z.; Sun, F.; Liu, X.; Gao, J.; Qie, Z.; Zhao, G. The effect of nitrogen-containing functional groups on SO2 adsorption on carbon surface: Enhanced physical adsorption interactions. Surf. Sci. 2018, 677, 78–82. [Google Scholar] [CrossRef]
- Petrushenko, I.K.; Tikhonov, N.I.; Petrushenko, K.B. Graphene-BN-organic nanoflake complexes: DFT, IGM and SAPT0 insights. Diam. Relat. Mater. 2020, 107, 107905. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Wavefunction methods for noncovalent interactions. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 304–326. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting and Cholesky decomposition approximations in symmetry-adapted perturbation theory: Implementation and application to probe the nature of π-π interactions in linear acenes. J. Chem. Phys. 2010, 132, 184111. [Google Scholar] [CrossRef]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.A.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L.; et al. Psi4: An open-source ab initio electronic structure program. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 556–565. [Google Scholar] [CrossRef]
- de la Roza, A.O.; DiLabio, G.A. Non-Covalent Interactions in Quantum Chemistry and Physics: Theory and Applications; Elsevier: Amsterdam, The Netherlands, 2017; p. 476. [Google Scholar]
- Wang, Y.; Shao, Y.; Matson, D.W.; Li, J.; Lin, Y. Nitrogen-doped graphene and its application in electrochemical biosensing. ACS Nano 2010, 4, 1790–1798. [Google Scholar] [CrossRef]
- Petrushenko, I.K.; Petrushenko, K.B. Hydrogen physisorption on nitrogen-doped graphene and graphene-like boron nitride-carbon heterostructures: A DFT study. Surf. Interfaces 2019, 17, 100355. [Google Scholar] [CrossRef]
- Petrushenko, I.K.; Petrushenko, K.B. Adsorption of diatomic molecules on nitrogenated holey graphene: Theoretical insights. Surf. Interfaces 2021, 27, 101446. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. Available online: http://www.chemcraftprog.com (accessed on 1 February 2024).
- Pykal, M.; Jurecka, P.; Karlicky, F.; Otyepka, M. Modelling of graphene functionalization. Phys. Chem. Chem. Phys. PCCP 2016, 18, 6351–6372. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Petrushenko, I.K.; Ivanov, N.A. Non-covalent interactions between dihydroquercetin and arabinogalactan molecules: Theoretical study. Comput. Theor. Chem. 2023, 1230, 114394. [Google Scholar] [CrossRef]
- Gotzias, A.; Tocci, E.; Sapalidis, A. Solvent-Assisted Graphene Exfoliation from Graphite Using Umbrella Sampling Simulations. Langmuir 2023, 39, 18437–18446. [Google Scholar] [CrossRef] [PubMed]
- Crabb, E.; France-Lanord, A.; Leverick, G.; Stephens, R.; Shao-Horn, Y.; Grossman, J.C. Importance of Equilibration Method and Sampling for Ab Initio Molecular Dynamics Simulations of Solvent-Lithium-Salt Systems in Lithium-Oxygen Batteries. J. Chem. Theory Comput. 2020, 16, 7255–7266. [Google Scholar] [CrossRef]
- Manna, K.; Huang, H.-N.; Li, W.-T.; Ho, Y.-H.; Chiang, W.-H. Toward Understanding the Efficient Exfoliation of Layered Materials by Water-Assisted Cosolvent Liquid-Phase Exfoliation. Chem. Mater. 2016, 28, 7586–7593. [Google Scholar] [CrossRef]
- Lin, H.A.; Sato, Y.; Segawa, Y.; Nishihara, T.; Sugimoto, N.; Scott, L.T.; Higashiyama, T.; Itami, K. A Water-Soluble Warped Nanographene: Synthesis and Applications for Photoinduced Cell Death. Angew. Chem. 2018, 57, 2874–2878. [Google Scholar] [CrossRef]
- Khakpour, E.; Salehi, S.; Naghib, S.M.; Ghorbanzadeh, S.; Zhang, W. Graphene-based nanomaterials for stimuli-sensitive controlled delivery of therapeutic molecules. Front. Bioeng. Biotechnol. 2023, 11, 1129768. [Google Scholar] [CrossRef] [PubMed]
- Hoseini-Ghahfarokhi, M.; Mirkiani, S.; Mozaffari, N.; Abdolahi Sadatlu, M.A.; Ghasemi, A.; Abbaspour, S.; Akbarian, M.; Farjadian, F.; Karimi, M. Applications of Graphene and Graphene Oxide in Smart Drug/Gene Delivery: Is the World Still Flat? Int. J. Nanomed. 2020, 15, 9469–9496. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.; Liu, Z.; Ma, Y.; Huang, Y.; Chen, Y. High-Efficiency Loading and Controlled Release of Doxorubicin Hydrochloride on Graphene Oxide. J. Phys. Chem. C 2008, 112, 17554–17558. [Google Scholar] [CrossRef]
- Zhan, J.; Lei, Z.; Zhang, Y. Non-covalent interactions of graphene surface: Mechanisms and applications. Chem 2022, 8, 947–979. [Google Scholar] [CrossRef]
- Mehrabani, M.; Khatibi, M.M.; Ashory, M.R.; Sadeghzadeh, S. Effect of nitrogen or boron impurities on the mechanical and vibrational properties of graphene nanosheets: A molecular dynamics approach. Micro Nano Lett. 2020, 15, 977–983. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorbent | Eint | Eel | Eex | Eind | Edisp |
|---|---|---|---|---|---|
| G | −30.04 | −17.02 (25.0) | 38.11 | −4.80 (7.0) | −46.32 (68.0) |
| Npyrid-G | −37.16 | −22.01 (23.8) | 55.33 | −7.46 (8.1) | −63.01 (68.1) |
| Npyrr-G | −32.63 | −17.64 (23.3) | 43.06 | −6.21 (8.2) | −51.84 (68.5) |
| 2N-G | −43.42 | −28.34 (26.9) | 62.12 | −9.18 (8.7) | −68.02 (64.4) |
| No. | BCPs (3,−1) | ρ(r) | ∇2ρ(r) | G(r) | V(r) | H(r) | −G(r)/V(r) |
|---|---|---|---|---|---|---|---|
| 1 | 117 | 0.009 | 0.026 | 0.006 | −0.005 | 0.0008 | 1.2 |
| 2 | 159 | 0.009 | 0.029 | 0.006 | −0.005 | 0.0010 | 1.2 |
| 3 | 188 | 0.007 | 0.018 | 0.004 | −0.003 | 0.0008 | 1.3 |
| 4 | 216 | 0.010 | 0.032 | 0.007 | −0.006 | 0.0009 | 1.2 |
| Species | BCPs (3,−1) | ρ(r) | ∇2ρ(r) | G(r) | V(r) | H(r) | −G(r)/V(r) |
|---|---|---|---|---|---|---|---|
| 1 | 203 | 0.017 | 0.050 | 0.012 | −0.011 | 0.0008 | 1.09 |
| 2 | 246 | 0.006 | 0.017 | 0.004 | −0.003 | 0.0004 | 1.33 |
| 3 | 277 | 0.008 | 0.022 | 0.005 | −0.004 | 0.0007 | 1.25 |
| 4 | 287 | 0.015 | 0.036 | 0.008 | −0.008 | 0.0006 | 1.00 |
| Species | BCPs (3,−1) | ρ(r) | ∇2ρ(r) | G(r) | V(r) | H(r) | −G(r)/V(r) |
|---|---|---|---|---|---|---|---|
| 1 | 153 | 0.013 | 0.041 | 0.009 | −0.008 | 0.0009 | 1.13 |
| 2 | 161 | 0.008 | 0.024 | 0.005 | −0.004 | 0.0009 | 1.25 |
| 3 | 199 | 0.015 | 0.039 | 0.009 | −0.009 | 0.0005 | 1.00 |
| 4 | 226 | 0.011 | 0.033 | 0.007 | −0.006 | 0.0009 | 1.17 |
| Species | BCPs (3,−1) | ρ(r) | ∇2ρ(r) | G(r) | V(r) | H(r) | −G(r)/V(r) |
|---|---|---|---|---|---|---|---|
| 1 | 138 | 0.007 | 0.018 | 0.004 | −0.003 | 0.0008 | 1.33 |
| 2 | 168 | 0.016 | 0.050 | 0.011 | −0.009 | 0.0015 | 1.22 |
| 3 | 236 | 0.005 | 0.013 | 0.003 | −0.002 | 0.0006 | 1.50 |
| Adsorbent | Eint,s No Solvent | Eint,s 3:3 w:e | Eint,s 6 w |
|---|---|---|---|
| G | −33.02 | −130.00 | −141.58 |
| Npyrid-G | −44.30 | −146.90 | −144.52 |
| Npyrr-G | −32.63 | −143.68 | −139.42 |
| 2N-G | −50.65 | −142.12 | −145.85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrushenko, I. Taxifolin Adsorption on Nitrogenated Graphenes: Theoretical Insights. Solids 2024, 5, 341-354. https://doi.org/10.3390/solids5030023
Petrushenko I. Taxifolin Adsorption on Nitrogenated Graphenes: Theoretical Insights. Solids. 2024; 5(3):341-354. https://doi.org/10.3390/solids5030023
Chicago/Turabian StylePetrushenko, Igor. 2024. "Taxifolin Adsorption on Nitrogenated Graphenes: Theoretical Insights" Solids 5, no. 3: 341-354. https://doi.org/10.3390/solids5030023
APA StylePetrushenko, I. (2024). Taxifolin Adsorption on Nitrogenated Graphenes: Theoretical Insights. Solids, 5(3), 341-354. https://doi.org/10.3390/solids5030023