

Modification of Sympathetic and Hypothalamic Responses to Prevent Complications of COVID-19: “Dam and Wall Concept”


Abstract
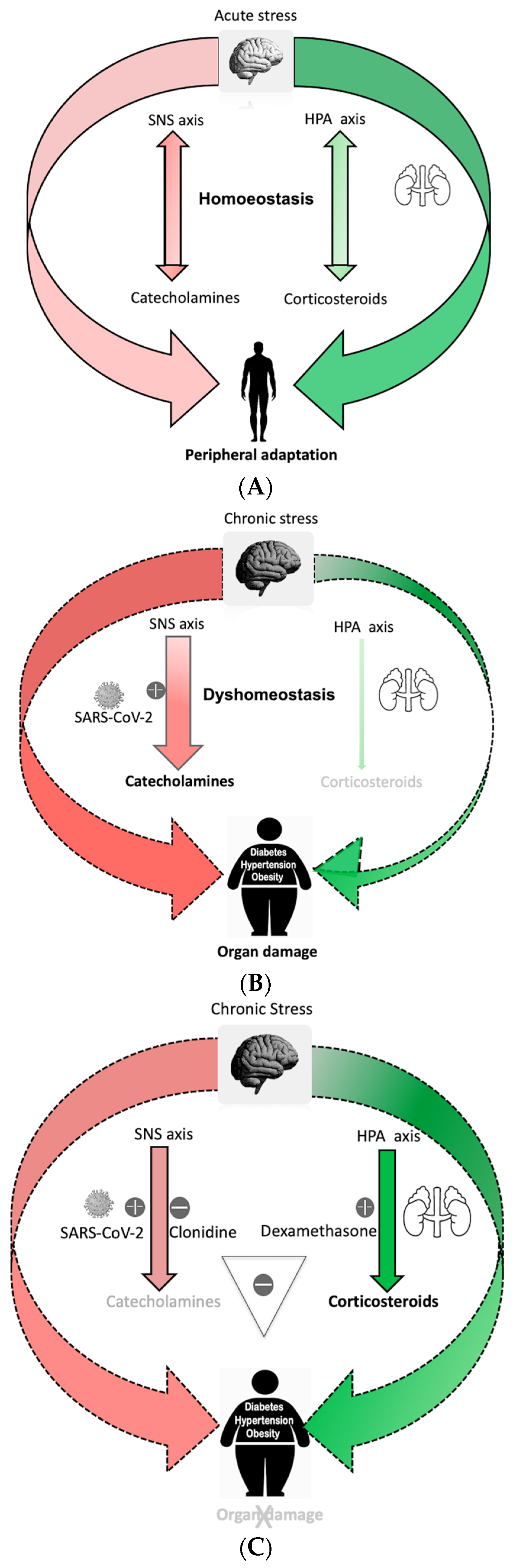
1. The Role of Sympathetic Nervous System and Hypothalamic–Pituitary–Adrenal (HPA) Axes That Drive Homeostasis in the Stress Response System
2. Chronic Stress Disrupts the Stress Response, Resulting in Maladaptation and Impaired Recovery
3. Body Dyshomeostasis and Role of Sympathetic Hyperactivity
4. SARS-CoV2 Imbalances Stress Response System in Vulnerable Population
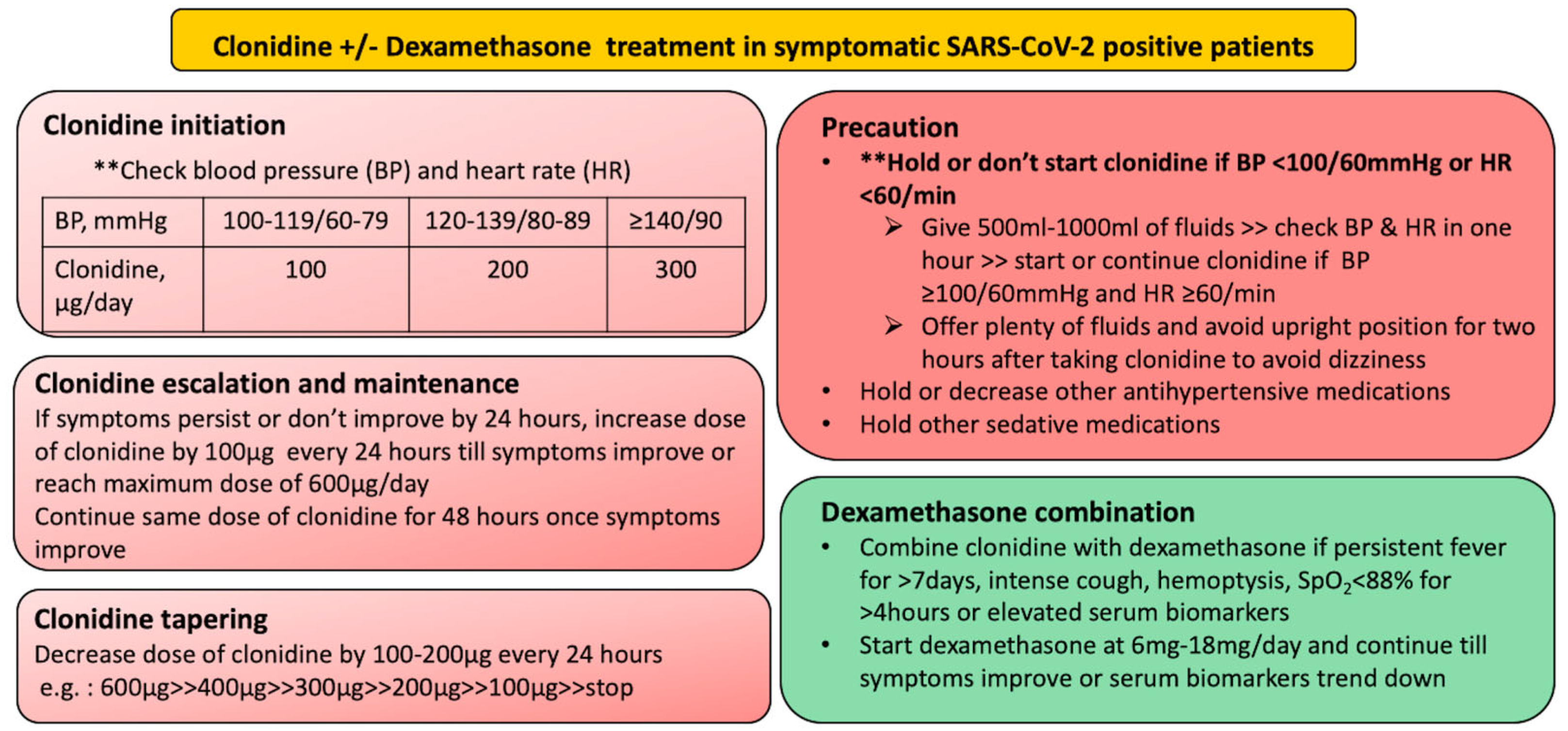
5. Clonidine and Dexamethasone Act Synergistically to Prevent Complication during SARS-CoV-2 Infection
6. Concluding Remark
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, T.M.; Fee, E. Walter Bradford Cannon: Pioneer Physiologist of Human Emotions. Am. J. Public Health 2002, 92, 1594–1595. [Google Scholar] [CrossRef]
- Chrousos, G.P.; Gold, P.W. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA 1992, 267, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S. The extended autonomic system, dyshomeostasis, and COVID-19. Clin. Auton. Res. 2020, 30, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, D.L.; Lorton, D.; Felten, S.Y.; Felten, D.L. Innervation of lymphoid organs and implications in development, aging, and autoimmunity. Int. J. Immunopharmacol. 1992, 14, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Pongratz, G.; Straub, R.H. The sympathetic nervous response in inflammation. Arthritis Res. Ther. 2014, 16, 504. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, L.A.; Woster, A.P.; Dahlman, J.; Sauter, E.R.; Combs, C.K.; Porter, J.E. Alpha1-adrenergic receptors positively regulate Toll-like receptor cytokine production from human monocytes and macrophages. J. Pharmacol. Exp. Ther. 2011, 338, 648–657. [Google Scholar] [CrossRef]
- Flierl, M.A.; Rittirsch, D.; Nadeau, B.A.; Chen, A.J.; Sarma, J.V.; Zetoune, F.S.; McGuire, S.R.; List, R.P.; Day, D.E.; Hoesel, L.M.; et al. Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature 2007, 449, 721–725. [Google Scholar] [CrossRef]
- Swanson, M.A.; Lee, W.T.; Sanders, V.M. IFN-gamma production by Th1 cells generated from naive CD4+ T cells exposed to norepinephrine. J. Immunol. 2001, 166, 232–240. [Google Scholar] [CrossRef]
- Hermoso, M.A.; Cidlowski, J.A. Putting the brake on inflammatory responses: The role of glucocorticoids. IUBMB Life 2003, 55, 497–504. [Google Scholar] [CrossRef]
- Dimitrov, S.; Benedict, C.; Heutling, D.; Westermann, J.; Born, J.; Lange, T. Cortisol and epinephrine control opposing circadian rhythms in T cell subsets. Blood 2009, 113, 5134–5143. [Google Scholar] [CrossRef]
- Elenkov, I.J.; Wilder, R.L.; Chrousos, G.P.; Vizi, E.S. The sympathetic nerve--an integrative interface between two supersystems: The brain and the immune system. Pharmacol. Rev. 2000, 52, 595–638. [Google Scholar]
- Selye, H. Stress and the general adaptation syndrome. Br. Med. J. 1950, 1, 1383–1392. [Google Scholar] [CrossRef]
- Sterling, P. Allostasis: A model of predictive regulation. Physiol. Behav. 2012, 106, 5–15. [Google Scholar] [CrossRef]
- McEwen, B.S. Protective and damaging effects of stress mediators: Central role of the brain. Dialogues Clin. Neurosci. 2006, 8, 367–381. [Google Scholar] [CrossRef]
- McEwen, B.S. Stress, adaptation, and disease. Allostasis and allostatic load. Ann. N. Y. Acad. Sci. 1998, 840, 33–44. [Google Scholar] [CrossRef]
- Harris, R.B. Role of set-point theory in regulation of body weight. FASEB J. 1990, 4, 3310–3318. [Google Scholar] [CrossRef]
- Spengler, R.N.; Allen, R.M.; Remick, D.G.; Strieter, R.M.; Kunkel, S.L. Stimulation of alpha-adrenergic receptor augments the production of macrophage-derived tumor necrosis factor. J. Immunol. 1990, 145, 1430–1434. [Google Scholar] [CrossRef]
- Walsh, C.P.; Ewing, L.J.; Cleary, J.L.; Vaisleib, A.D.; Farrell, C.H.; Wright, A.G.C.; Gray, K.; Marsland, A.L. Development of glucocorticoid resistance over one year among mothers of children newly diagnosed with cancer. Brain Behav. Immun. 2018, 69, 364–373. [Google Scholar] [CrossRef]
- Cole, S.W.; Mendoza, S.P.; Capitanio, J.P. Social stress desensitizes lymphocytes to regulation by endogenous glucocorticoids: Insights from in vivo cell trafficking dynamics in rhesus macaques. Psychosom. Med. 2009, 71, 591–597. [Google Scholar] [CrossRef]
- Walsh, C.P.; Bovbjerg, D.H.; Marsland, A.L. Glucocorticoid resistance and beta2-adrenergic receptor signaling pathways promote peripheral pro-inflammatory conditions associated with chronic psychological stress: A systematic review across species. Neurosci. Biobehav. Rev. 2021, 128, 117–135. [Google Scholar] [CrossRef]
- Grebe, K.M.; Takeda, K.; Hickman, H.D.; Bailey, A.M.; Embry, A.C.; Bennink, J.R.; Yewdell, J.W. Cutting edge: Sympathetic nervous system increases proinflammatory cytokines and exacerbates influenza A virus pathogenesis. J. Immunol. 2010, 184, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Hyoju, S.K.; Zaborina, O.; van Goor, H. SARS-CoV-2 and the sympathetic immune response: Dampening inflammation with antihypertensive drugs (Clonidine and Propranolol). Med. Hypotheses 2020, 144, 110039. [Google Scholar] [CrossRef] [PubMed]
- Kopp, W. Chronically increased activity of the sympathetic nervous system: Our diet-related “evolutionary” inheritance. J. Nutr. Health Aging 2009, 13, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.A.; Velazquez, K.T.; Herbert, K.M. Influence of high-fat diet on gut microbiota: A driving force for chronic disease risk. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Carnagarin, R.; Lambert, G.W.; Kiuchi, M.G.; Nolde, J.M.; Matthews, V.B.; Eikelis, N.; Lambert, E.A.; Schlaich, M.P. Effects of sympathetic modulation in metabolic disease. Ann. N. Y. Acad. Sci. 2019, 1454, 80–89. [Google Scholar] [CrossRef]
- Fisher, J.P.; Young, C.N.; Fadel, P.J. Central sympathetic overactivity: Maladies and mechanisms. Auton. Neurosci. 2009, 148, 5–15. [Google Scholar] [CrossRef]
- Snitker, S.; Macdonald, I.; Ravussin, E.; Astrup, A. The sympathetic nervous system and obesity: Role in aetiology and treatment. Obes. Rev. 2000, 1, 5–15. [Google Scholar] [CrossRef]
- Hyoju, S.K.; Zaborin, A.; Keskey, R.; Sharma, A.; Arnold, W.; van den Berg, F.; Kim, S.M.; Gottel, N.; Bethel, C.; Charnot-Katsikas, A.; et al. Mice Fed an Obesogenic Western Diet, Administered Antibiotics, and Subjected to a Sterile Surgical Procedure Develop Lethal Septicemia with Multidrug-Resistant Pathobionts. mBio 2019, 10, e00903-19. [Google Scholar] [CrossRef]
- Dani, M.; Dirksen, A.; Taraborrelli, P.; Torocastro, M.; Panagopoulos, D.; Sutton, R.; Lim, P.B. Autonomic dysfunction in ‘long COVID’: Rationale, physiology and management strategies. Clin. Med. 2021, 21, e63–e67. [Google Scholar] [CrossRef]
- Stute, N.L.; Stickford, J.L.; Province, V.M.; Augenreich, M.A.; Ratchford, S.M.; Stickford, A.S.L. COVID-19 is getting on our nerves: Sympathetic neural activity and haemodynamics in young adults recovering from SARS-CoV-2. J. Physiol. 2021, 599, 4269–4285. [Google Scholar] [CrossRef]
- Porzionato, A.; Emmi, A.; Barbon, S.; Boscolo-Berto, R.; Stecco, C.; Stocco, E.; Macchi, V.; De Caro, R. Sympathetic activation: A potential link between comorbidities and COVID-19. FEBS J. 2020, 287, 3681–3688. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Qusti, S.; Alshammari, E.M.; Gyebi, G.A.; Batiha, G.E. Covid-19-Induced Dysautonomia: A Menace of Sympathetic Storm. ASN Neuro 2021, 13, 17590914211057635. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Konig, M.F.; Powell, M.; Staedtke, V.; Bai, R.Y.; Thomas, D.L.; Fischer, N.; Huq, S.; Khalafallah, A.M.; Koenecke, A.; Xiong, R.; et al. Preventing cytokine storm syndrome in COVID-19 using alpha-1 adrenergic receptor antagonists. J. Clin. Investig. 2020, 130, 3345–3347. [Google Scholar] [CrossRef]
- Gubbi, S.; Nazari, M.A.; Taieb, D.; Klubo-Gwiezdzinska, J.; Pacak, K. Catecholamine physiology and its implications in patients with COVID-19. Lancet Diabetes Endocrinol. 2020, 8, 978–986. [Google Scholar] [CrossRef]
- Baker, J.; Incognito, A.V.; Wilson, R.J.A.; Raj, S.R. Syncope and silent hypoxemia in COVID-19: Implications for the autonomic field. Auton. Neurosci. 2021, 235, 102842. [Google Scholar] [CrossRef]
- Fischer, L.; Barop, H.; Ludin, S.M.; Schaible, H.G. Regulation of acute reflectory hyperinflammation in viral and other diseases by means of stellate ganglion block. A conceptual view with a focus on Covid-19. Auton. Neurosci. 2022, 237, 102903. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, L.; Lang, H.; Hu, X.; Jing, S.; Luo, M.; Xu, G.; Zhou, Z. Effect of a Stellate Ganglion Block on Acute Lung Injury in Septic Rats. Inflammation 2018, 41, 1601–1609. [Google Scholar] [CrossRef]
- Liu, L.D.; Duricka, D.L. Stellate ganglion block reduces symptoms of Long COVID: A case series. J. Neuroimmunol. 2022, 362, 577784. [Google Scholar] [CrossRef]
- Kearney, T.J.; Bentt, L.; Grode, M.; Lee, S.; Hiatt, J.R.; Shabot, M.M. Coagulopathy and catecholamines in severe head injury. J. Trauma 1992, 32, 608–611; discussion 611–612. [Google Scholar] [CrossRef]
- Ikarugi, H.; Taka, T.; Nakajima, S.; Noguchi, T.; Watanabe, S.; Sasaki, Y.; Haga, S.; Ueda, T.; Seki, J.; Yamamoto, J. Norepinephrine, but not epinephrine, enhances platelet reactivity and coagulation after exercise in humans. J. Appl. Physiol. 1999, 86, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.M.; Shah, M.; Shah, S.; Li, A.; Jauhar, S. Takotsubo Syndrome and COVID-19: Associations and Implications. Curr. Probl. Cardiol. 2021, 46, 100763. [Google Scholar] [CrossRef] [PubMed]
- Staedtke, V.; Bai, R.Y.; Kim, K.; Darvas, M.; Davila, M.L.; Riggins, G.J.; Rothman, P.B.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature 2018, 564, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Brunner, E.J.; Hemingway, H.; Walker, B.R.; Page, M.; Clarke, P.; Juneja, M.; Shipley, M.J.; Kumari, M.; Andrew, R.; Seckl, J.R.; et al. Adrenocortical, autonomic, and inflammatory causes of the metabolic syndrome: Nested case-control study. Circulation 2002, 106, 2659–2665. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef]
- Pal, R. COVID-19, hypothalamo-pituitary-adrenal axis and clinical implications. Endocrine 2020, 68, 251–252. [Google Scholar] [CrossRef]
- Hyoju, S.K.; Baral, B.; Jha, P.K. Central catecholaminergic blockade with clonidine prevent SARS-CoV-2 complication: A case series. IDCases 2021, 25, e01219. [Google Scholar] [CrossRef]
- Petitjeans, F.; Martinez, J.Y.; Danguy des Deserts, M.; Leroy, S.; Quintin, L.; Escarment, J. A Centrally Acting Antihypertensive, Clonidine, Sedates Patients Presenting With Acute Respiratory Distress Syndrome Evoked by Severe Acute Respiratory Syndrome-Coronavirus 2. Crit. Care Med. 2020, 48, e991–e993. [Google Scholar] [CrossRef]
- Hamilton, J.L.; Vashi, M.; Kishen, E.B.; Fogg, L.F.; Wimmer, M.A.; Balk, R.A. The Association of an Alpha-2 Adrenergic Receptor Agonist and Mortality in Patients With COVID-19. Front. Med. 2021, 8, 797647. [Google Scholar] [CrossRef]
- Baller, E.B.; Hogan, C.S.; Fusunyan, M.A.; Ivkovic, A.; Luccarelli, J.W.; Madva, E.; Nisavic, M.; Praschan, N.; Quijije, N.V.; Beach, S.R.; et al. Neurocovid: Pharmacological Recommendations for Delirium Associated With COVID-19. Psychosomatics 2020, 61, 585–596. [Google Scholar] [CrossRef]
- Shang, L.; Zhao, J.; Hu, Y.; Du, R.; Cao, B. On the use of corticosteroids for 2019-nCoV pneumonia. Lancet 2020, 395, 683–684. [Google Scholar] [CrossRef]
- Group, R.C.; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Russell, C.D.; Millar, J.E.; Baillie, J.K. Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet 2020, 395, 473–475. [Google Scholar] [CrossRef]
- Tomazini, B.M.; Maia, I.S.; Cavalcanti, A.B.; Berwanger, O.; Rosa, R.G.; Veiga, V.C.; Avezum, A.; Lopes, R.D.; Bueno, F.R.; Silva, M.; et al. Effect of Dexamethasone on Days Alive and Ventilator-Free in Patients With Moderate or Severe Acute Respiratory Distress Syndrome and COVID-19: The CoDEX Randomized Clinical Trial. JAMA 2020, 324, 1307–1316. [Google Scholar] [CrossRef]
- Kanczkowski, W.; Beuschlein, F.; Bornstein, S.R. Is there a role for the adrenal glands in long COVID? Nat. Rev. Endocrinol. 2022, 18, 451–452. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Parameter | Normal Range | |
|---|---|---|
| Serum ferritin | 1006.4 | 11.0–306 ng/mL |
| D-dimer | 4.6 | <0.5 mg/L |
| CRP | 138.47 | <10 mg/dL |
| PT | 20.5 | 11–16 s |
| LDH | 926 | 0–246 ng/mL |
| Age/Sex | PCR Test Result | Symptoms/Lowest Recorded SpO2/Oxygen Supplementation Received (Yes/No) | Abnormal Laboratory Reports (Normal Value) | Home/Hospital Based Treatment | Highest Doses of Clonidine and/or Dexamethasone Treatment per Day | Total Duration of Treatment with Clonidine | Outcome |
|---|---|---|---|---|---|---|---|
| 61 y/female | Unknown | High-grade fever, cough, shortness of breath/ SpO2:85/Yes | ESR:64 mm/h (0–10) CRP: ++ D dimer: 1.61 µg/mL (<0.5) Ferritin: 721 ng/mL (6.24–264) AST: 138 U/L (5–40) ALT: 153 U/L (5–45) | Home-based treatment | Clonidine 300 microgram/day | 7 days | Recovered |
| 49 y/female | Positive | Fever, cough, hemoptysis, shortness of breath/SpO2:90/Yes | D dimer 1.01 mg/L (<0.5) | Home-based treatment | Clonidine 300 microgram/day | 10 days | Recovered |
| 51 y/male | Positive | Fever, cough, loss of smell, loss of taste, weakness/ SpO2:88/No | CRP: ++ ESR:16 mm/h (0–10) LDH: 486 U/L (<460) AST:124 U/L (5–40) ALT:101 U/L (5–45) CT Chest: CORADS-6 | Home-based treatment | Clonidine 300 microgram/day | 10 days | Recovered |
| 79 y/female | Positive | Fever, cough, shortness of breath/ SpO2:85/No | TLC < 3500 (4000–11000) | Home-based treatment | Clonidine 300 microgram/day | 10 days | Recovered |
| 67 y/female | Positive | Fever, shortness of breath, weakness/ SpO2: 88/Yes | ESR:52 mm/h (0–20) CRP:48 mg/L (0–6) Ferritin: 389 ng/mL (11–307) | Home and hospital-based treatment | Clonidine 300 microgram/day and dexamethasone 6 mg/day | 10 days | Recovered |
| 32 y/male | Positive | Fever, cough, shortness of breath/ SpO2:75/Yes | CRP:31 mg/L (0–6) CT Chest: CORADS-6 LDH:400 U/L (120–246) | Hospital-based treatment | Clonidine 600 microgram/day and dexamethasone 12 mg/day | 30 days | Recovered |
| 48 y/female | Positive | High-grade fever, cough, shortness of breath, loss of smell, taste and appetite/SpO2:75/Yes | CRP = 59 mg/L (0–6) ESR: 53 mm/h (0–10) | Home-based treatment | Clonidine 600 microgram/day and dexamethasone 9 mg/day | 14 days | Recovered |
| 59 y/male | Positive | High-grade fever, cough, shortness of breath, loss of smell, taste and appetite/SpO2:75/Yes | ESR: 38 mm/h (0–10) CRP:42 mg/L (0–6) NT-Pro BNP: 20211 pg/mL (<220) | Home-based treatment | Clonidine 600 microgram/day and dexamethasone 12 mg/day | 21 days | Recovered |
| 43 y/male | Positive | High-grade fever, cough, shortness of breath, loss of smell, taste and appetite, SpO2:80/Yes | ESR: 54 mm/h (0–10) CRP: 57 mg/L (0–6) NT-Pro BNP: 8567 pg/ml(<220) | Home-based treatment | Clonidine 600 microgram/day and dexamethasone 9 mg/day | 18 days | Recovered |
| 38 y/male | Positive | High-grade fever, cough, shortness of breath/SpO2:90/Yes | CRP: 211 mg/L (0–5). ESR: 83 mm/h. (0–12) Serum ferritin > 3000 ng/mL(25–350) LDH: 535 U/L (125–220) D dimer: 600 ng/mL (<500) AST: 361 U/L (16–63) ALT: 125 U/L (16–37) | Home-based treatment | Clonidine 300 microgram/day and dexamethasone 16 mg/day | 14 days | Recovered |
| 69 y/female | Positive | Fever, cough, shortness of breath/ SpO2:85/Yes | ESR: 30 mm/h (0–9) CRP: 24 mg/L(<6) Procalcitonin 0.81 ng/mL (0–0.5) | Home-based treatment | Clonidine 300 microgram/day and dexamethasone 12 mg/day | 21 days | Recovered |
| 72 y/male | Positive | Fever, cough, shortness of breath/ SpO2:80/Yes | CRP: 38 mg/L (0–10) CT Chest: CORADS-6 | Isolation-center-based treatment | Clonidine 300 microgram/day and dexamethasone 6 mg/day | 15 days | Recovered |
| 59 y/female | Positive | Fever, cough, shortness of breath, SpO2:82,Yes | ESR: 33 mm/h (0–12) CRP: 19.8 mg/L (0–6) Glucose ®: 206 | Home-based treatment | Clonidine 300 microgram/day and dexamethasone 6 mg/day | 12 days | Recovered |
| 58 y/male | Positive | Fever, cough, whole body ache, shortness of breath, intense headache, BP:197/117 mmHg/ SpO2:82/Yes | CRP: 89 mg/L (<10) D-Dimer: 1133 ng/mL (30–400) | Home-based treatment | Clonidine 900 microgram/day and dexamethasone 12 mg/day | 18 days | Recovered |
| 45 y/male | Positive | Fever, cough, hemoptysis/SpO2:88/No | CRP: 88 mg/L (0–6) Ferritin: 544 ng/mL (17–464) AST: 48 U/L (5–40) ALT: 86 U/L (5–45) | Home treatment | Clonidine 300 microgram/day and dexamethasone 12 mg/day | 14 days | Recovered |
| 96 y/male | Unknown | Fever, shortness of breath/SpO2: 86/Yes | Not done | Home-based treatment | Clonidine 400 microgram/day and dexamethasone 6 mg/day | 10 days | Recovered |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyoju, S.K. Modification of Sympathetic and Hypothalamic Responses to Prevent Complications of COVID-19: “Dam and Wall Concept”. Stresses 2023, 3, 153-166. https://doi.org/10.3390/stresses3010012
Hyoju SK. Modification of Sympathetic and Hypothalamic Responses to Prevent Complications of COVID-19: “Dam and Wall Concept”. Stresses. 2023; 3(1):153-166. https://doi.org/10.3390/stresses3010012
Chicago/Turabian StyleHyoju, Sanjiv K. 2023. "Modification of Sympathetic and Hypothalamic Responses to Prevent Complications of COVID-19: “Dam and Wall Concept”" Stresses 3, no. 1: 153-166. https://doi.org/10.3390/stresses3010012
APA StyleHyoju, S. K. (2023). Modification of Sympathetic and Hypothalamic Responses to Prevent Complications of COVID-19: “Dam and Wall Concept”. Stresses, 3(1), 153-166. https://doi.org/10.3390/stresses3010012