
Exploring Epigenetic Modifiers in Cowpea: Genomic and Transcriptomic Insights into Histone Methyltransferases and Histone Demethylases

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Genomics
2.1.1. Identification and Classification of VuSDGS and VuJMJs in the Cowpea Genome
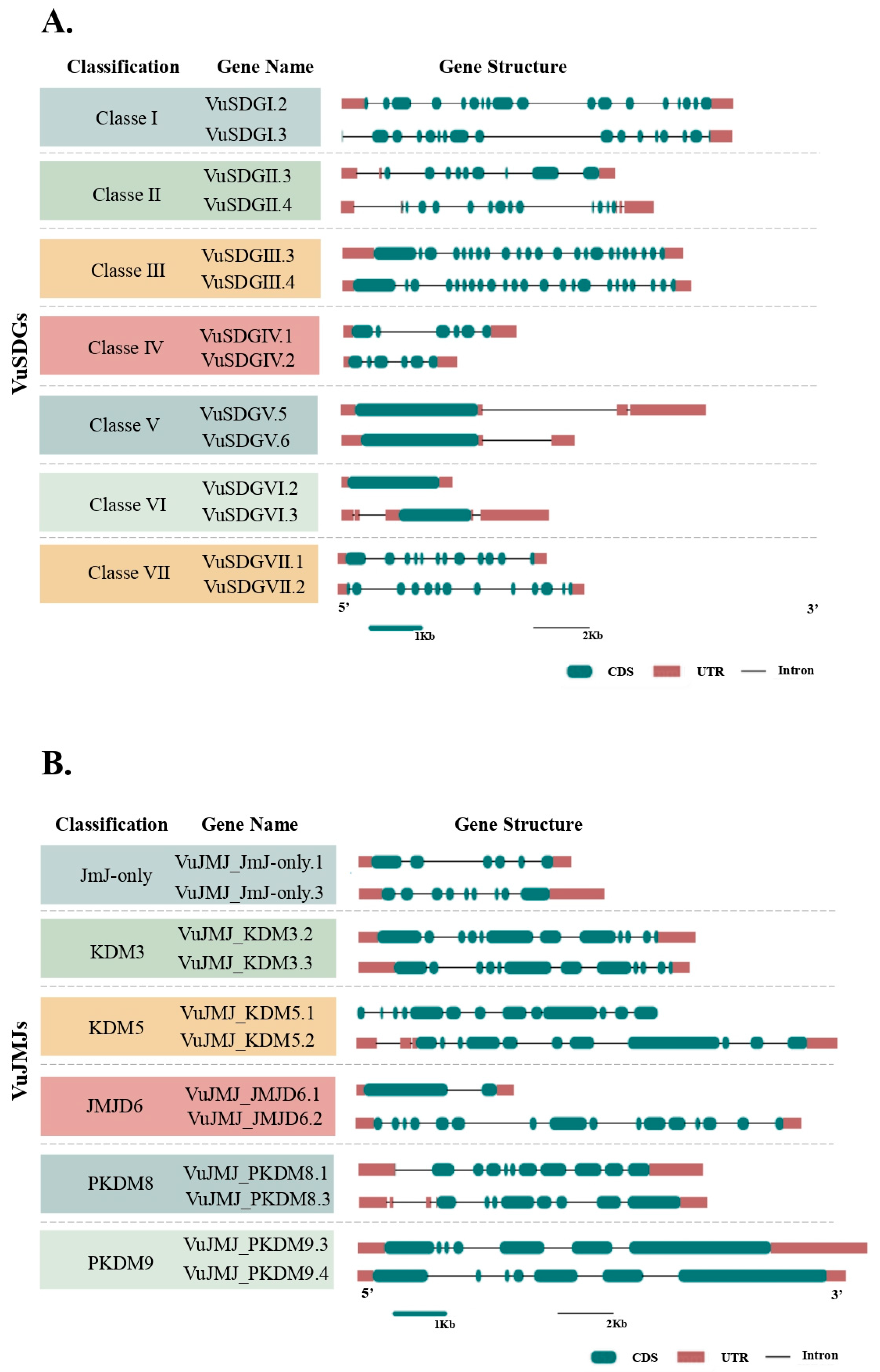
2.1.2. VuSDGs and VuJMJs Gene Structure
2.1.3. Gene Duplication Mechanisms of the VuSDGs and VuJMJs Loci
2.1.4. Selection Pressure on Duplicated VuSDGs and VuJMJs Genes
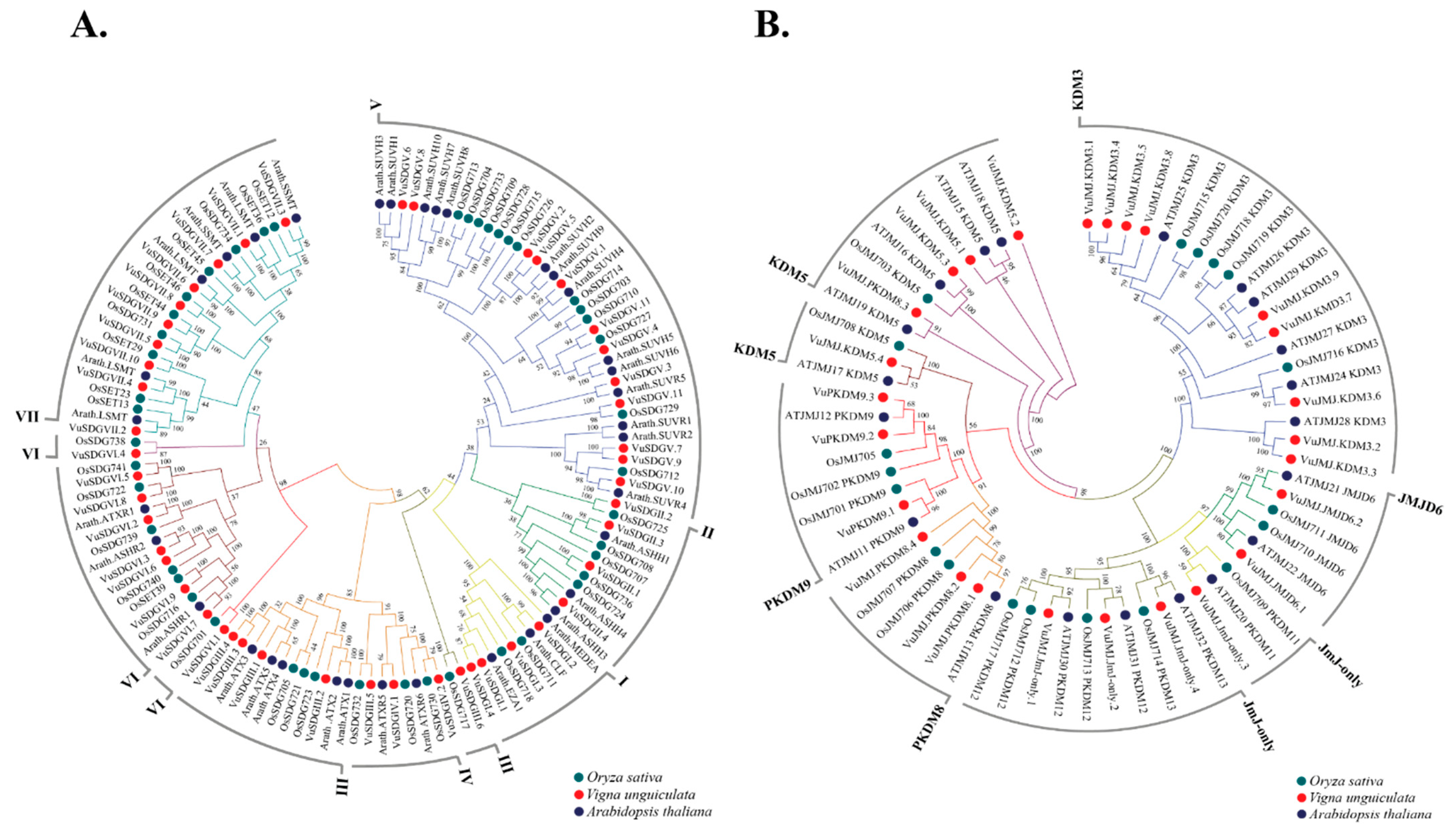
2.1.5. Orthology of VuSDGs and VuJMJs Genes in Viriplantae
2.1.6. CCREs in the Promoter Regions of the VuSDGs and VuJMJs Genes
2.2. Transcriptomics of VuSDGs and VuJMJs in Cowpea Under Different Stresses
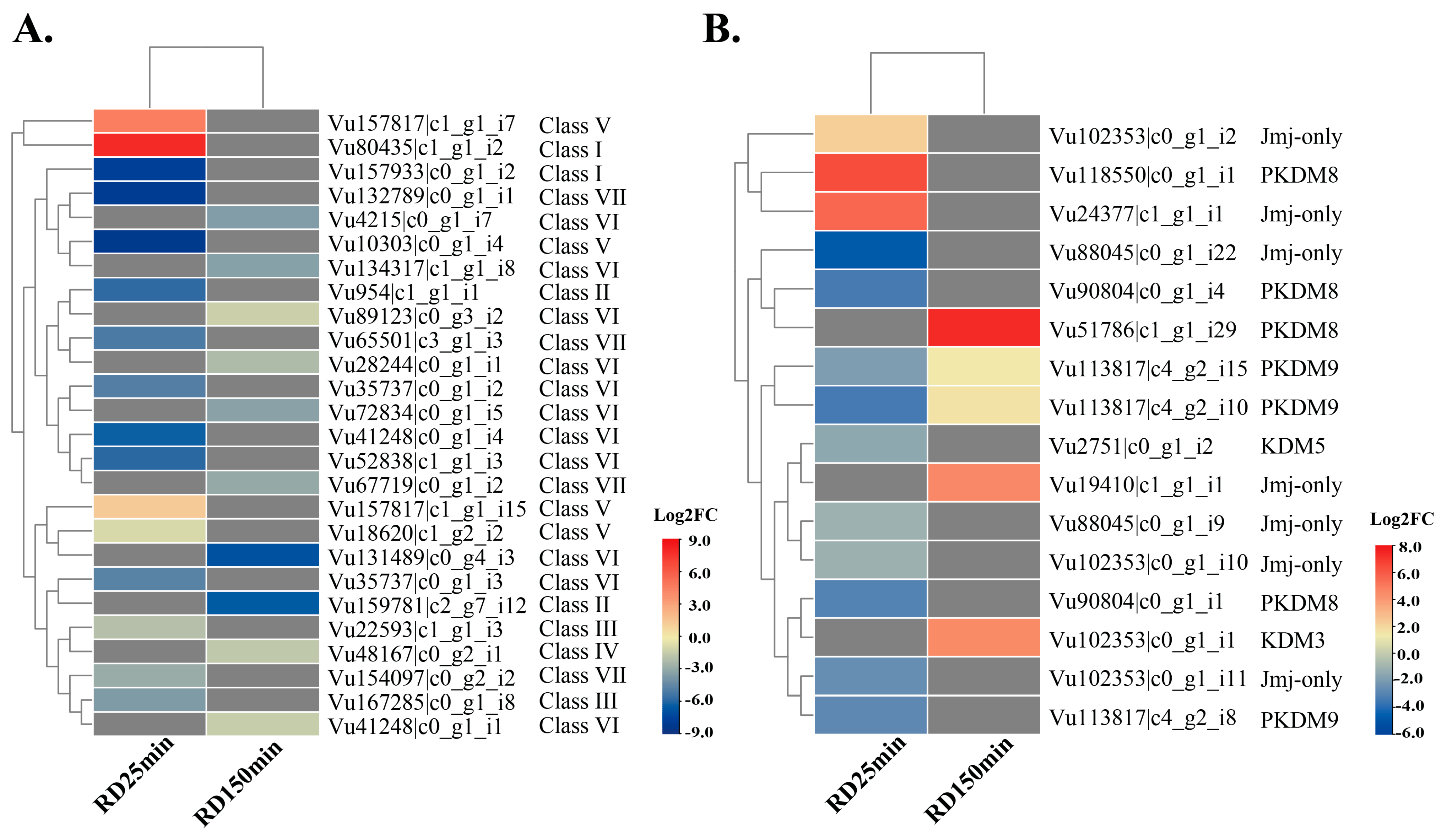
2.2.1. VuSDG and VuJMJ Expression Profiles in the Root Dehydration Assay
2.2.2. VuSDG and VuJMJ Expression Profiles in the Mechanical Injury Followed by Virus Inoculation Assays
2.2.3. VuSDG and VuJMJ Crosstalk Response Investigation
2.3. RNA-Seq Data Validation Through qPCR Assay
3. Discussion
3.1. Mining and Gene Structural Characterization of VuSDGs and VuJMJs in the Cowpea Genome
3.2. Protein Properties and Prediction of Subcellular Localization
3.3. Duplication Mechanisms of VuSDG and VuJMJ Genes in the Cowpea Genome
3.4. Comparative Genomics of VuSDGs and VuJMJs
3.5. Mining and Identification of Candidate CCREs
3.6. Transcriptomics of VuSDGs and VuJMJs in Cowpea Under Biotic and Abiotic Stress
4. Materials and Methods
4.1. Genomic Section
4.1.1. Mining and Identification of VuSDG and VuJMJ Coding Loci
4.1.2. Classification of VuSDG and VuJMJ Genes
- Profiling (presence/absence) of different domains to each VuSDG or VuJMJ class/group;
- Conducting phenetic analysis of curated protein sequences from various species, focusing on SDG and JMJ sequences. Candidate VuSDGs and VuJMJs were aligned using ClustalW [49], and a phenetic tree was constructed with the Neighbor-Joining (NJ) method [50] in MEGA 7 [51], employing 1000 bootstrap replicates. Genes were categorized based on established classifications in Arabidopsis thaliana and rice, as well as the tree topology.
4.1.3. Structural Characteristics of VuSDG and VuJMJ Genes
4.1.4. Protein Properties and Subcellular Localization Prediction
4.1.5. Expansion Mechanisms of VuSDG and VuJMJ Genes
4.1.6. Synonymous (Ks) and Non-Synonymous (Ka) Substitution Rates for WGD/Segmental Duplicated Genes
4.1.7. Orthology Analysis of VuSDG and VuJMJ Loci Across Viridiplantae Species
4.1.8. Candidate Cis-Regulatory Element (CCRE) Mining and Identification
4.2. Transcriptomics
4.2.1. Biological Material, Experimental Design, and Stress Application
Root Dehydration Assay
Mechanical Injury and Viral Inoculation Assays
4.2.2. RNA-Seq Libraries: Synthesis and Sequencing
4.2.3. RNA-Seq Library Assembly and Differential Expression Analysis
4.2.4. qPCR: Setup, Efficiency, and Relative Expression Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kornberg, R.D. Structure of Chromatin. Ann. Rev. Biochem. 1997, 46, 932–954. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Neto, J.R.C.; Silva, M.; Pandolfi, V.; Crovella, S.; Benko-Iseppon, A.; Kido, E. Epigenetic Signals on Plant Adaptation: A Biotic Stress Perspective. Curr. Protein Pept. Sci. 2017, 18, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Pfluger, J.; Wagner, D. Histone modifications and dynamic regulation of genome accessibility in plants. Curr. Opin. Plant Biol. 2007, 10, 645–652. [Google Scholar] [CrossRef]
- Ng, D.W.K.; Wang, T.; Chandrasekharan, M.B.; Aramayo, R.; Kertbundit, S.; Hall, T.C. Plant SET domain-containing proteins: Structure, function and regulation. Biochim. Biophys. Acta 2007, 1769, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Chen, X.; Hu, Y.; Zhou, D.X. Epigenetic gene regulation by plant Jumonji group of histone demethylase. Biochim. Biophys. Acta 2011, 1809, 421–426. [Google Scholar] [CrossRef]
- Elkins, J.M.; Hewitson, K.S.; McNeill, L.A.; Seibel, F.; Schlemminger, I.; Pugh, C.W.; Ratcliffe, P.J.; Schofield, C.J. Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1 alpha. J. Biol. Chem. 2003, 278, 1802–1806. [Google Scholar] [CrossRef]
- Trewick, S.C.; McLaughlin, J.; Allshire, R.C. Methylation: Lost in hydroxylation? EMBO Rep. 2005, 6, 315–320. [Google Scholar] [CrossRef]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.A.; Morohashi, K.; Grotewold, E.; Harmer, S.L. Arabidopsis JMJD5/JMJ30 acts Independently of LUX ARRHYTHMO within the plant circadian clock to enable temperature compensation. Front. Plant Sci. 2019, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhang, A.; Jin, J.B.; Zhao, B.; Wang, T.-J.; Wu, Y.; Wang, S.; Liu, Y.; Wang, J.; Guo, P.; et al. Arabidopsis histone H3K4 demethylase JMJ17 functions in dehydration stress response. New Phytol. 2019, 223, 1372–1387. [Google Scholar] [CrossRef]
- Liu, G.; Khan, N.; Ma, X.; Hou, X. Identification, evolution, and expression profiling of histone lysine methylation moderators in Brassica rapa. Plants 2019, 8, 526. [Google Scholar] [CrossRef]
- Qian, Y.; Chen, C.; Jiang, L.; Zhang, J.; Ren, Q. Genome-wide identification, classification and expression analysis of the JmjC domain-containing histone demethylase gene family in maize. BMC Genom. 2019, 20, 256. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, X.; Qiao, K.; Fan, S.; Ma, Q. Genome-wide analysis of JMJ-C histone demethylase family involved in salt-tolerance in Gossypium hirsutum L. Plant Physiol. Biochem. 2021, 158, 420–433. [Google Scholar] [CrossRef]
- Yan, L.; Fan, G.; Li, X. Genome-wide analysis of three histone marks and gene expression in Paulownia fortunei with phytoplasma infection. BMC Genom. 2019, 20, 234. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, C.; Shen, W.; Ruan, Y. Phylogenetic analysis and classification of the Brassica rapa SET domain protein family. BMC Plant Biol. 2011, 11, 175. [Google Scholar] [CrossRef]
- Lu, Z.; Huang, X.; Ouyang, Y.; Yao, J. Genome-wide identification, phylogenetic and co-expression analysis of OsSET gene family in rice. PLoS ONE 2013, 8, e65426. [Google Scholar] [CrossRef]
- Aiese, C.R.; Sanseverino, W.; Cremona, G.; Ercolano, M.R.; Conicella, C.; Consiglio, F.M. Genome-wide analysis of histone modifiers in tomato: Gaining an insight into their developmental roles. BMC Genom. 2013, 14, 57. [Google Scholar]
- Qian, Y.; Xi, Y.; Cheng, B.; Zhu, S.; Kan, X. Identification and characterization of the SET domain gene family in maize. Mol. Biol. Rep. 2014, 41, 1341–1354. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xu, H.; Liu, Y.; Wang, X.; Xu, Q.; Deng, X. Genome-wide identification of sweet orange (Citrus sinensis) histone modification gene families and their expression analysis during the fruit development and fruit-blue mold infection process. Front. Plant Sci. 2015, 6, 607. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, X.; Cheng, L.; Liu, Y.; Wang, H.; Ke, D.; Yuan, H.; Zhang, L.; Wang, L. Genome-wide analysis of soybean jmjc domain-containing proteins suggests evolutionary conservation following whole-genome duplication. Front. Plant Sci. 2016, 7, 1800. [Google Scholar] [CrossRef]
- Fan, S.; Wang, J.; Lei, C.; Gao, C.; Yang, Y.; Li, Y.; An, N.; Zhang, D.; Han, M. Identification and characterization of histone modification gene family reveal their critical responses to flower induction in apple. BMC Plant Biol. 2018, 18, 173. [Google Scholar] [CrossRef]
- Huang, Y.; Mo, Y.; Chen, P.; Yuan, X.; Meng, F.; Zhu, S.; Liu, Z. Identification of SET domain-containing proteins in Gossypium raimondii and their response to high temperature stress. Sci. Rep. 2016, 6, 32729. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, J.; Liu, W.; Ren, Z.; Zhao, J.; Pei, X.; Liu, Y.; Yang, D.; Ma, X. Characterization and stress response of the jmjc domain-containing histone demethylase gene family in the allotetraploid cotton species Gossypium hirsutum. Plants 2020, 9, 1617. [Google Scholar] [CrossRef]
- Batra, R.; Gautam, T.; Pal, S.; Chaturvedi, D.; Rakhi; Jan, I.; Balyan, H.S.; Gupta, P.K. Identification and characterization of SET domain family genes in bread wheat (Triticum aestivum L.). Sci. Rep. 2020, 10, 14624. [Google Scholar] [CrossRef]
- Freire- Filho, F.R. Feijão-Caupi no Brasil: Produção, Melhoramento Genético, Avanços e Desafios, 1st ed.; Embrapa Meio-Norte: Teresina, Brazil, 2011. [Google Scholar]
- Boukar, O.; Fatokun, C.A.; Huynh, B.L.; Roberts, A.; Close, T.J. Genomic tools in cowpea breeding programs: Status and perspectives. Front. Plant Sci. 2016, 7, 757. [Google Scholar] [CrossRef]
- Lonardi, S.; Muñoz-Amatriaín, M.; Liang, Q.; Shu, S.; Wanamaker, S.I.; Lo, S.; Tanskanen, J.; Schulman, A.H.; Zhu, T.; Luo, M.-C.; et al. The genome of cowpea (Vigna unguiculata [L.] Walp.). Plant J. 2019, 98, 767–782. [Google Scholar] [CrossRef]
- Lu, F.; Li, G.; Cui, X.; Liu, C.; Wang, X.-J.; Cao, X. Comparative Analysis of JmjC Domain-containing Proteins Reveals the Potential Histone Demethylases in Arabidopsis and Rice. J. Integr. Plant Biol. 2008, 50, 886–896. [Google Scholar] [CrossRef]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Puiu, D.; Hall, R.; Kingan, S.; Clavijo, B.J.; Salzberg, S.L. The first near-complete assembly of the hexaploid bread wheat genome, Triticum aestivum. Gigascience 2017, 6, gix097. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.H.; Qiu, H.L.; Huang, Y.; Zhang, L.; Si, J.P. Genome-wide identification and expression profiling of SET DOMAIN GROUP family in Dendrobium catenatum. BMC Plant Biol. 2020, 20, 40. [Google Scholar] [CrossRef]
- Liu, C.; Lu, F.; Cui, X.; Cao, X. Histone methylation in higher plants. Annu. Rev. Plant Biol. 2010, 61, 395–420. [Google Scholar] [CrossRef]
- Gu, T.; Han, Y.; Huang, R.; McAvoy, R.J.; Li, Y. Identification and characterization of histone lysine methylation modifiers in Fragaria vesca. Sci. Rep. 2016, 6, 23581. [Google Scholar] [CrossRef] [PubMed]
- Cerbin, S.; Jiang, N. Duplication of host genes by transposable elements. Curr. Opin. Genet. Dev. 2018, 49, 63–69. [Google Scholar] [CrossRef]
- Kang, Y.J.; Satyawan, D.; Shim, S.; Lee, T.; Lee, J.; Hwang, W.J.; Kim, S.K.; Lestari, P.; Laosatit, K.; Kim, K.H.; et al. Draft genome sequence of adzuki bean, Vigna angularis. Sci. Rep. 2015, 5, 8069. [Google Scholar] [CrossRef]
- Kang, W.H.; Kim, S.; Lee, H.A.; Choi, D.; Yeom, S.I. Genome-wide analysis of Dof transcription factors reveals functional characteristics during development and response to biotic stresses in pepper. Sci. Rep. 2016, 6, 33332. [Google Scholar] [CrossRef]
- Wen, C.L.; Cheng, Q.; Zhao, L.; Mao, A.; Yang, J.; Yu, S.; Weng, Y.; Xu, Y. Identification and characterisation of Dof transcription factors in the cucumber genome. Sci. Rep. 2016, 6, 23072. [Google Scholar] [CrossRef]
- He, Q.; Cai, H.; Bai, M.; Zhang, M.; Chen, F.; Huang, Y.; Priyadarshani, S.V.G.N.; Chai, M.; Liu, L.; Liu, Y.; et al. A soybean bZIP transcription factor GmbZIP19 confers multiple biotic and abiotic stress responses in plant. Int. J. Mol. Sci. 2020, 21, 4701. [Google Scholar] [CrossRef]
- Yang, S.; Xu, K.; Chen, S.; Li, T.; Xia, H.; Chen, L.; Liu, H.; Luo, L. A stress-responsive bZIP transcription factor OsbZIP62 improves drought and oxidative tolerance in rice. BMC Plant Biol. 2019, 19, 260. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Prado, J.S.; Latrasse, D.; Rodriguez-Granados, N.Y.; Huang, Y.; Manza-Mianza, D.; Brik-Chaouche, R.; Jaouannet, M.; Citerne, S.; Bendahmane, A.; Hirt, H.; et al. The Polycomb protein LHP1 regulates Arabidopsis thaliana stress responses through the repression of the MYC2-dependent branch of immunity. Plant J. 2019, 100, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Fu, F.; Xu, S.; Lee, S.Y.; Yun, D.J.; Mengiste, T. Global regulation of plant immunity by histone lysine methyl transferases. Plant Cell. 2016, 28, 1640–1661. [Google Scholar] [CrossRef] [PubMed]
- Ayyappan, V.; Kalavacharla, V.; Thimmapuram, J.; Bhide, K.P.; Sripathi, R.; Smolinski, T.G.; Manoharan, M.; Thurston, Y.; Todd, A.; Kingham, B. Genome Wide Profiling of Histone Modifications (H3K9me2 and H4K12ac) and Gene Expression in Rust (Uromyces appendiculatus) infected Common Bean (Phaseolus vulgaris L.). PLoS ONE 2015, 10, e0132176. [Google Scholar] [CrossRef]
- Crespo-Salvador, Ó.; Sánchez-Giménez, L.; López-Galiano, M.J.; Fernández-Crespo, E.; Schalschi, L.; García-Robles, I.; Rausell, C.; Real, M.D.; González-Bosch, C. The histone marks signature in exonic and intronic regions is relevant in early response of tomato genes to Botrytis cinerea and in miRNA regulation. Plants 2020, 9, 300. [Google Scholar] [CrossRef]
- Gan, E.S.; Xu, Y.; Ito, T. Dynamics of H3K27me3 methylation and demethylation in plant development. Plant Signal. Behav. 2015, 10, e1027851. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, P.; Jing, H.; Zhou, X.F.; Zhao, B.; Li, Y.; Jin, J.B. JMJ27-mediated histone H3K9 demethylation positively regulates drought-stress responses in Arabidopsis. New Phytol. 2021, 232, 221–236. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 21, 2947–2948. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 7, 1870–1874. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar]
- Hiller, K.; Schobert, M.; Hundertmark, C.; Jahn, D.; Münch, R. JVirGel: Calculation of virtual two-dimensional protein gels. Nucleic Acids Res. 2003, 31, 3862–3865. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of protein subcellular localization. Proteins Struct. Funct. Bioinform. 2006, 64, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.; Östlund, G. InParanoid 8: Orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 2015, 43, 234–239. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers; ISMB-94 Proceedings. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Gupta, S.; Stamatoyannopolous, J.A.; Bailey, T.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef]
- Bastos, E.A.; Nascimento, S.P.D.; Silva, E.M.D.; Freire-Filho, F.R.; Gomide, R.L. Identification of cowpea genotypes for drought tolerance. Rev. Ciênc. Agronôm. 2011, 42, 100–107. [Google Scholar] [CrossRef]
- Rodrigues, E.V.; Damasceno-Silva, K.J.; Rocha, M.M.; Bastos, E.A.; Teodoro, E. Selection of cowpea populations tolerant to water deficit by selection index. Rev. Ciênc. Agron. 2017, 48, 889–896. [Google Scholar] [CrossRef]
- Rodrigues, F.A.; Marcolino-Gomes, J.; Carvalho, J.F.; Nascimento, L.C.; Neumaier, N.; Farias, J.R.; Carazzolle, M.F.; Marcelino, F.C.; Nepomuceno, A.L. Subtractive libraries for prospecting differentially expressed genes in the soybean under water deficit. Genet. Mol. Biol. 2012, 35, 304–314. [Google Scholar] [CrossRef]
- Hoagland, D.R.; Arnon, D.I. The Water-Culture Method for Growing Plants Without soil. Circular. California Agricultural Experiment Station 347. 1950. Available online: https://www.cabdirect.org/cabdirect/abstract/19500302257 (accessed on 20 February 2021).
- Rocha, M.M.; Lima, J.A.A.; Freire-Filho, F.R.R.; Lima, C.V. Resistencia de Caupi de Tegumento Branco a Algumas Estirpes de Comovirus, Potyvirus e Cucumovirus. 1996. Available online: https://www.alice.cnptia.embrapa.br/handle/doc/53796 (accessed on 20 February 2021).
- Oliveira, C.R.R.; Filho, F.R.F.; Nogueira, M.S.; Barros, G.B.; Eiras, M.; Ribeiro, V.Q.; Lopes, Â.A. Reação de Genótipos de Feijão-caupi Revela Resistência às Coinfecções Pelo Cucumber Mosaic Virus, Cowpea Aphid-Borne Mosaic Virus e Cowpea Severe Mosaic Virus. 2012. Available online: https://www.alice.cnptia.embrapa.br/handle/doc/940785 (accessed on 20 February 2021).
- Cardoso, M.J.; Freire-Filho, F.R.; Athayde-Sobrinho, C. BR 14-mulato: Nova Cultivar de Feijão Macassar Para o Estado do Piauí; Comunicado Técnico, 48; Embrapa-UEPAE de Teresina: Teresina, Brazil, 1990; p. 4. [Google Scholar]
- Barna, B.; Király, L. Host–pathogen relations: Diseases caused by viruses, subviral organisms, and phytoplasmas. Plant Toxicol. 2004, 1, 533–568. [Google Scholar]
- Ferreira-Neto, J.R.C.; da Costa Borges, A.N.; da Silva, M.D.; de Lima Morais, D.A.; Bezerra-Neto, J.P.; Bourque, G.; Kido, E.A.; Benko-Iseppon, A.M. The Cowpea Kinome: Genomic and Transcriptomic Analysis Under Biotic and Abiotic Stresses. Front. Plant Sci. 2021, 12, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Amorim, L.L.B.; Ferreira-Neto, J.R.C.; Bezerra-Neto, J.P.; Pandolfi, V.; de Araújo, F.T.; da Silva Matos, M.K.; Santos, M.G.; Kido, E.A.; Benko-Iseppon, A.M. Cowpea and abiotic stresses: Identification of reference genes for transcriptional profiling by qPCR. Plant Methods 2018, 14, 88. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viana, J.B.V.; Ferreira-Neto, J.R.C.; Binneck, E.; Oliveira Silva, R.L.d.; Costa, A.F.d.; Benko-Iseppon, A.M. Exploring Epigenetic Modifiers in Cowpea: Genomic and Transcriptomic Insights into Histone Methyltransferases and Histone Demethylases. Stresses 2025, 5, 13. https://doi.org/10.3390/stresses5010013
Viana JBV, Ferreira-Neto JRC, Binneck E, Oliveira Silva RLd, Costa AFd, Benko-Iseppon AM. Exploring Epigenetic Modifiers in Cowpea: Genomic and Transcriptomic Insights into Histone Methyltransferases and Histone Demethylases. Stresses. 2025; 5(1):13. https://doi.org/10.3390/stresses5010013
Chicago/Turabian StyleViana, Jéssica Barbara Vieira, José Ribamar Costa Ferreira-Neto, Eliseu Binneck, Roberta Lane de Oliveira Silva, Antônio Félix da Costa, and Ana Maria Benko-Iseppon. 2025. "Exploring Epigenetic Modifiers in Cowpea: Genomic and Transcriptomic Insights into Histone Methyltransferases and Histone Demethylases" Stresses 5, no. 1: 13. https://doi.org/10.3390/stresses5010013
APA StyleViana, J. B. V., Ferreira-Neto, J. R. C., Binneck, E., Oliveira Silva, R. L. d., Costa, A. F. d., & Benko-Iseppon, A. M. (2025). Exploring Epigenetic Modifiers in Cowpea: Genomic and Transcriptomic Insights into Histone Methyltransferases and Histone Demethylases. Stresses, 5(1), 13. https://doi.org/10.3390/stresses5010013