
Unusual Para-Substituent Effects on the Intramolecular Hydrogen Bond in Hydrazone-Based Switches: Insights from Chemical Landscape Analysis and DFT Calculations

 , ,
, ,  and
and 
Abstract
:
1. Introduction
2. Computational Details
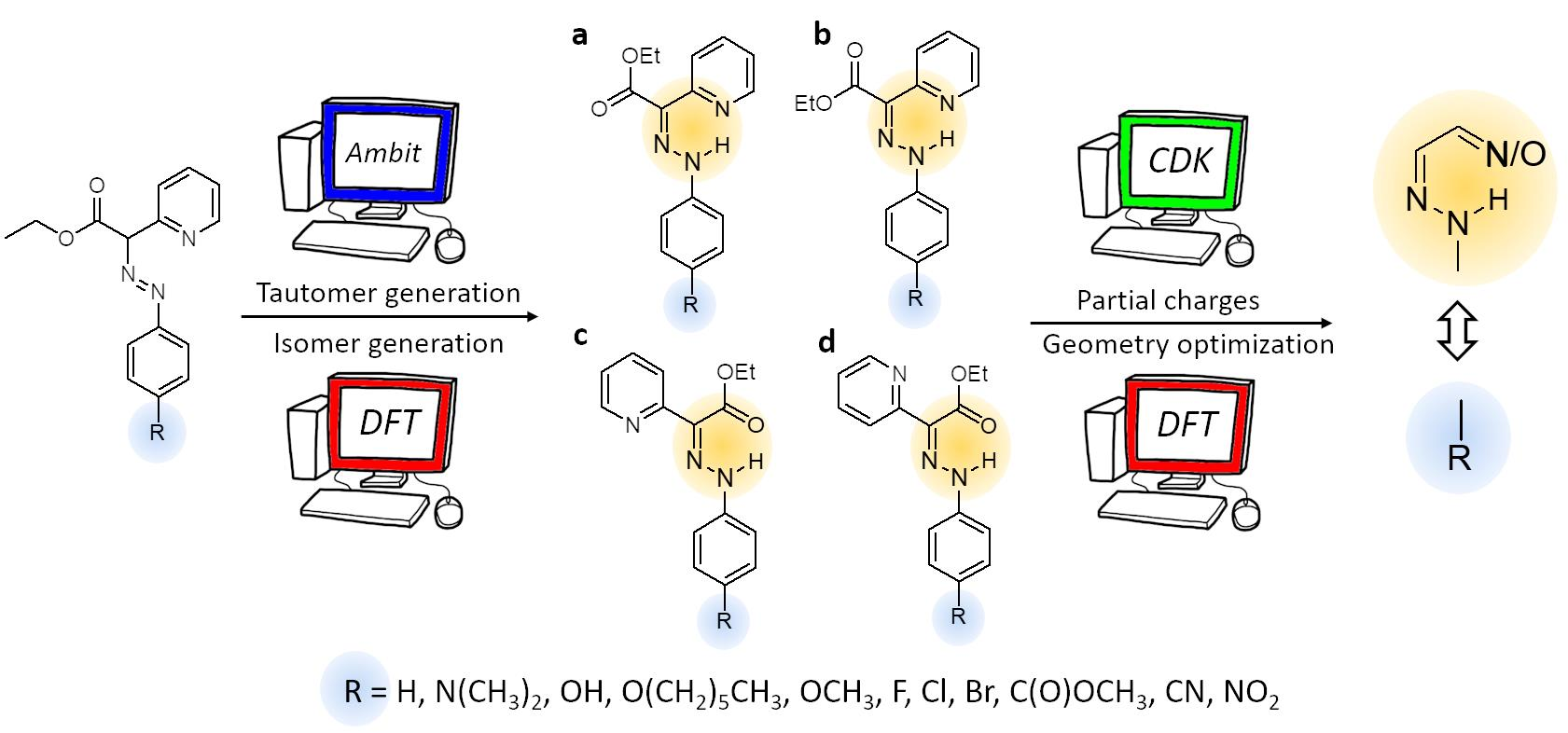
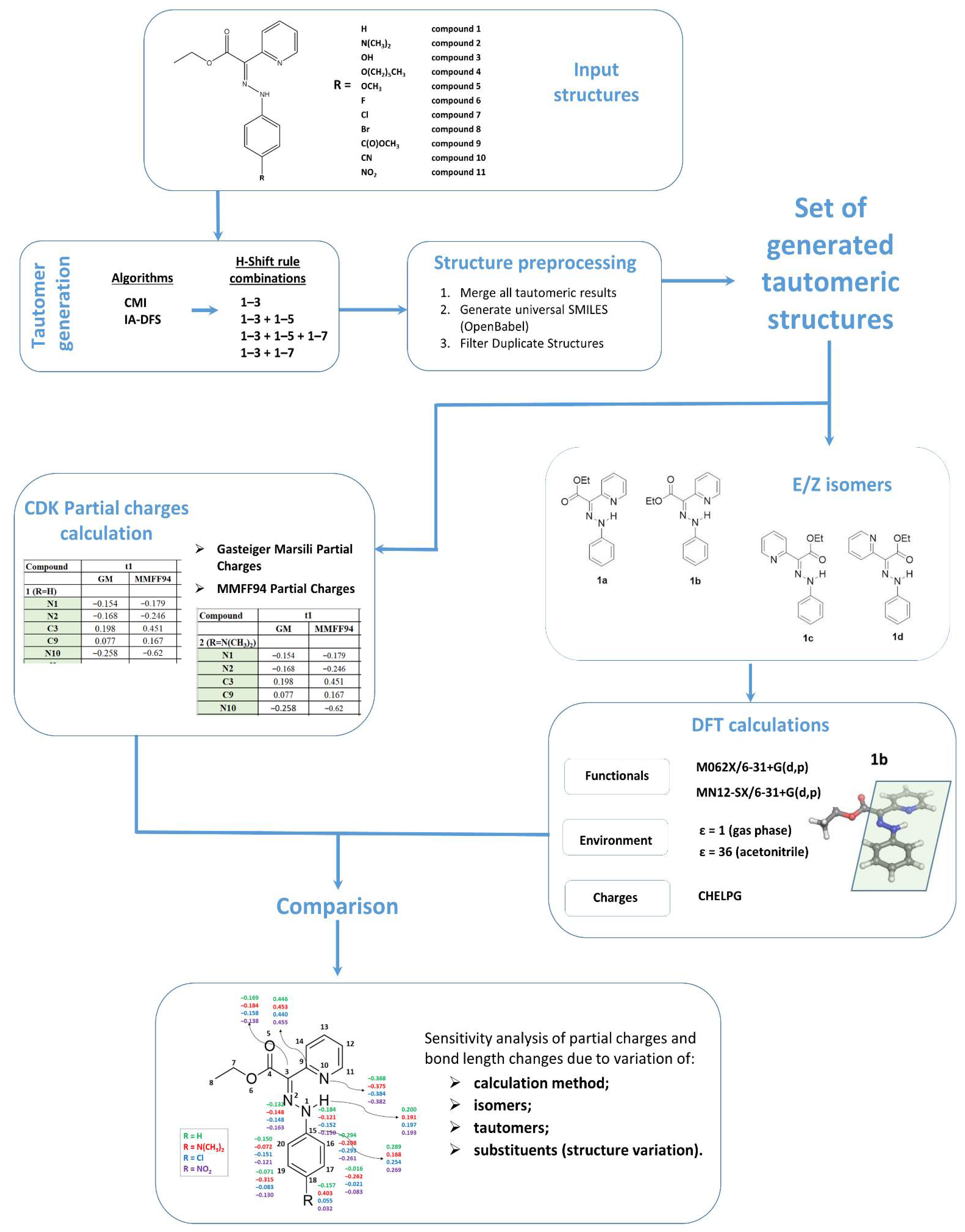
2.1. Chemical Landscape Analysis: Tautomer Generation
- Combination 1 (1–3 rules);
- Combination 2 (1–3 and 1–5 rules);
- Combination 3 (1–3, 1–5 and 1–7 rules);
- Combination 4 (1–3 and 1–7 rules).
2.2. DFT Calculations
3. Results and Discussion
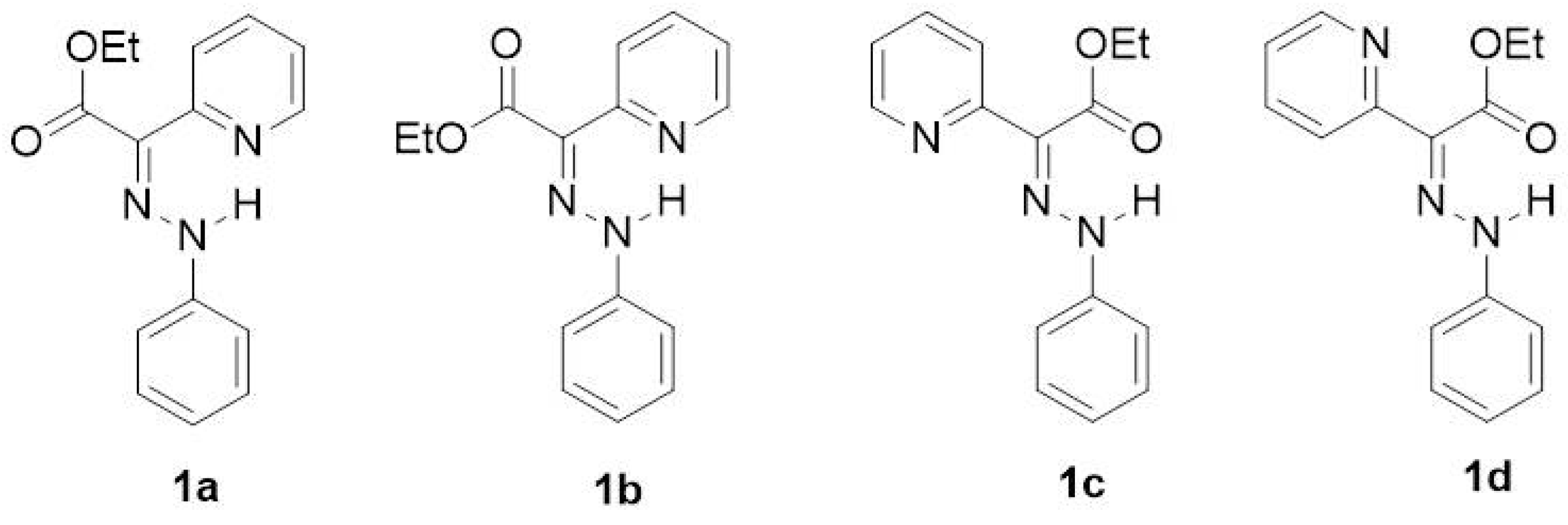
3.1. Unsubstituted System (Compound 1)
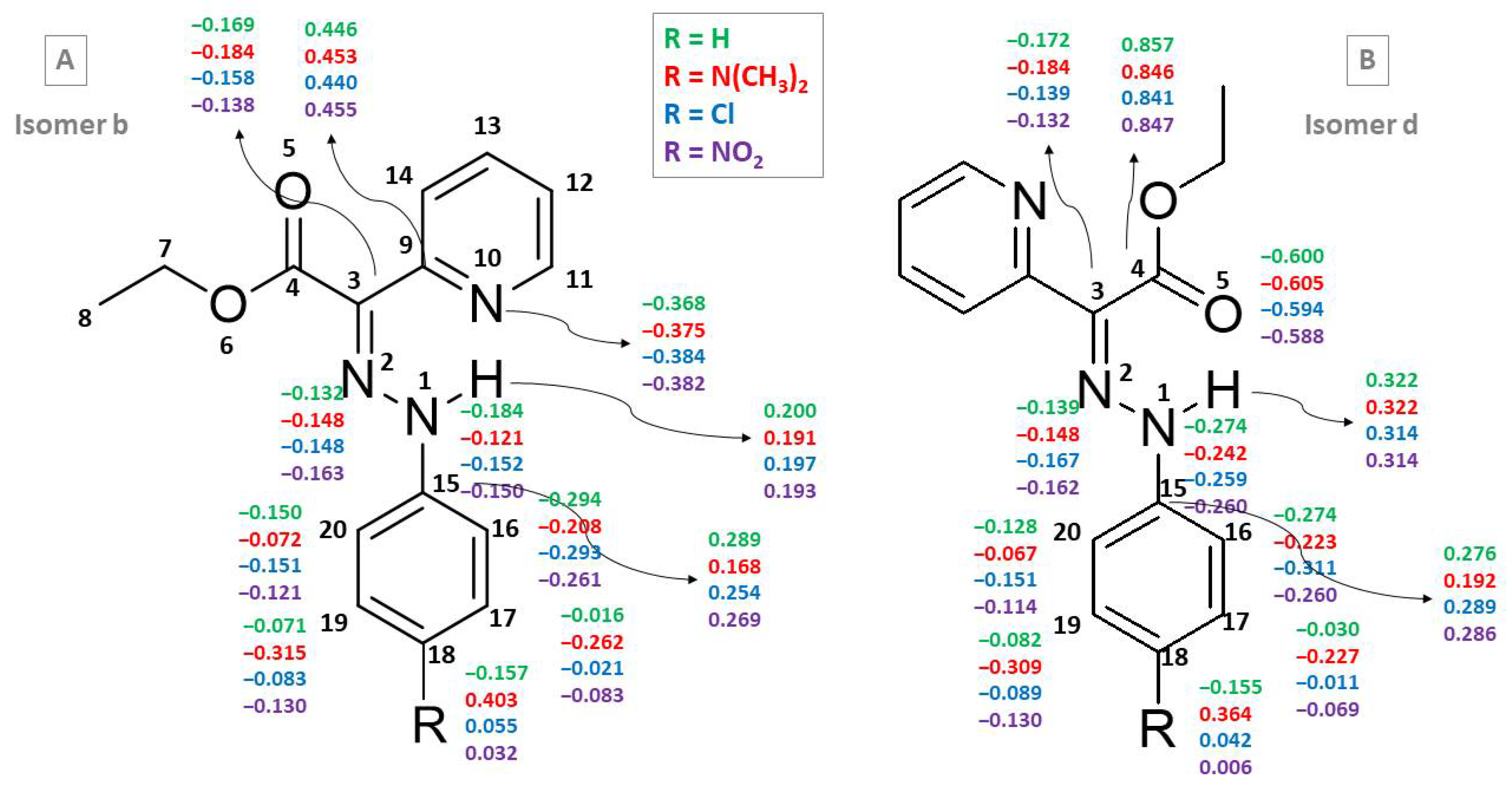
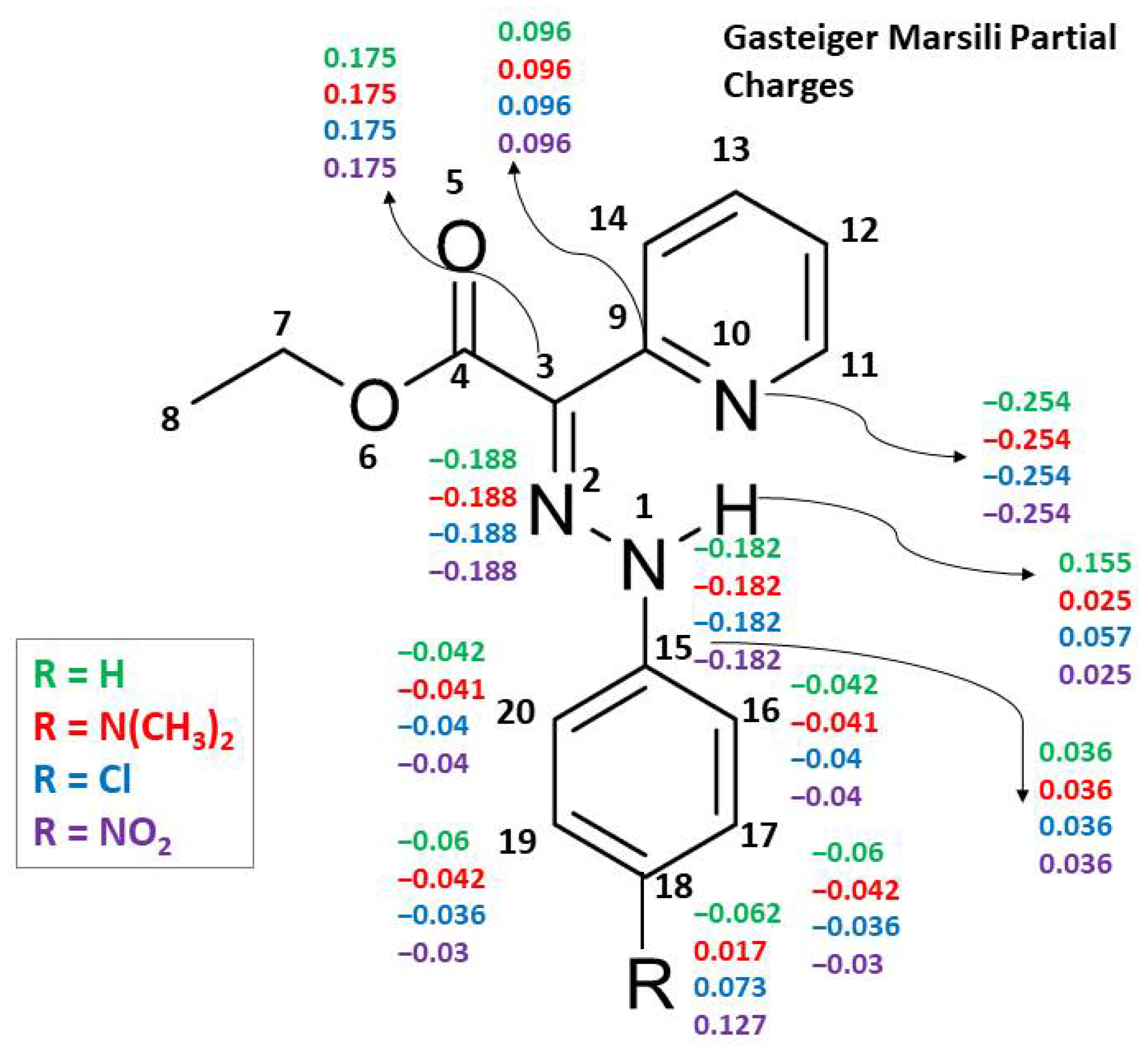
3.2. Para-Substituted Phenylhydrazones (Compounds 2–11)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Courtot, P.; Pichon, R.; Le Saint, J. Determination du site de chelation chez les arylhydrazones de tricetones et D’ α-dicetones substituees. Tetrahedron Lett. 1976, 17, 1177–1180. [Google Scholar] [CrossRef]
- Courtot, P.; Pichon, R.; Le Saint, J. Photochromisme par isomerisation syn-anti de phenylhydrazones-2- de tricetones-1,2,3 et de dicetones-1,2 substituees. Tetrahedron Lett. 1976, 17, 1181–1184. [Google Scholar] [CrossRef]
- Courtot, P.; Pichon, R.; Le Saint, J. Échanges inter et intra-molćulaires du proton entre deux atomes d’azote de cycles chélatés hydrazone-imine et azo-énamine. Tetrahedron Lett. 1979, 20, 1591–1594. [Google Scholar] [CrossRef]
- Pichon, R.; Le Saint, J.; Courtot, P. Photoisomerisation d’arylhydrazones-2 de dicetones-1,2 substituees en 2. Tetrahedron 1981, 37, 1517–1524. [Google Scholar] [CrossRef]
- Nedeltcheva-Antonova, D.; Antonov, L. Controlled Tautomerism: Is It Possible? Wiley-VCH: Weinheim, Germany, 2016; pp. 273–294. [Google Scholar] [CrossRef]
- Landge, S.M.; Aprahamian, I. A pH Activated Configurational Rotary Switch: Controlling the E/Z Isomerization in Hydrazones. J. Am. Chem. Soc. 2009, 131, 18269–18271. [Google Scholar] [CrossRef]
- Landge, S.M.; Tkatchouk, E.; Benítez, D.; Lanfranchi, D.A.; Elhabiri, M.; Goddard, W.A.; Aprahamian, I. Isomerization Mechanism in Hydrazone-Based Rotary Switches: Lateral Shift, Rotation, or Tautomerization? J. Am. Chem. Soc. 2011, 133, 9812–9823. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Lõkov, M.; Kütt, A.; Leito, I.; Aprahamian, I. Unusual para-substituent effects on the intramolecular hydrogen-bond in hydrazone-based switches. Chem. Commun. 2012, 48, 10490–10492. [Google Scholar] [CrossRef]
- Hristova, S.; Kamounah, F.S.; Crochet, A.; Vassilev, N.; Fromm, K.M.; Antonov, L. OH Group Effect in the Stator of β-Diketones Arylhydrazone Rotary Switches. Chemistry 2020, 2, 24. [Google Scholar] [CrossRef]
- Deneva, V.; Vassilev, N.; Hristova, S.; Yordanov, D.; Hayashi, Y.; Kawauchi, S.; Fennel, F.; Völzer, T.; Lochbrunner, S.; Antonov, L. Chercher de l’eau: The switching mechanism of the rotary switch ethyl-2-(2-(quinolin-8-yl)hydrazono)-2-(pyridin-2-yl)acetate. Comput. Mater. Sci. 2020, 177, 109570. [Google Scholar] [CrossRef]
- Kochev, N.T.; Paskaleva, V.; Jeliazkova, N. Ambit-Tautomer: An Open Source Tool for Tautomer Generation. Mol. Inform. 2013, 32, 481–504. [Google Scholar] [CrossRef]
- Angelova, S.; Paskaleva, V.; Kochev, N.; Antonov, L. DFT study of hydrazone-based molecular switches: The effect of different stators on the on/off state distribution. Mol. Phys. 2018, 117, 1604–1612. [Google Scholar] [CrossRef]
- Gilli, P.; Gilli, G. Hydrogen bond models and theories: The dual hydrogen bond model and its consequences. J. Mol. Struct. 2010, 972, 2–10. [Google Scholar] [CrossRef]
- Ambit Project. Available online: https://www.ambitprojects.co.uk (accessed on 17 June 2021).
- Steinbeck, C.; Hoppe, C.; Kuhn, S.; Floris, M.; Guha, R.; Willighagen, E. Recent Developments of the Chemistry Development Kit (CDK)—An Open-Source Java Library for Chemo- and Bioinformatics. Curr. Pharm. Des. 2006, 12, 2111–2120. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Peverati, R.; Truhlar, D. Screened-exchange density functionals with broad accuracy for chemistry and solid-state physics. Phys. Chem. Chem. Phys. 2012, 14, 16187–16191. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; A Pople, J. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Mennucci, B. Polarizable continuum model. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 386–404. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 1.7.6.6; Schrödinger: New York, NY, USA, 2015.


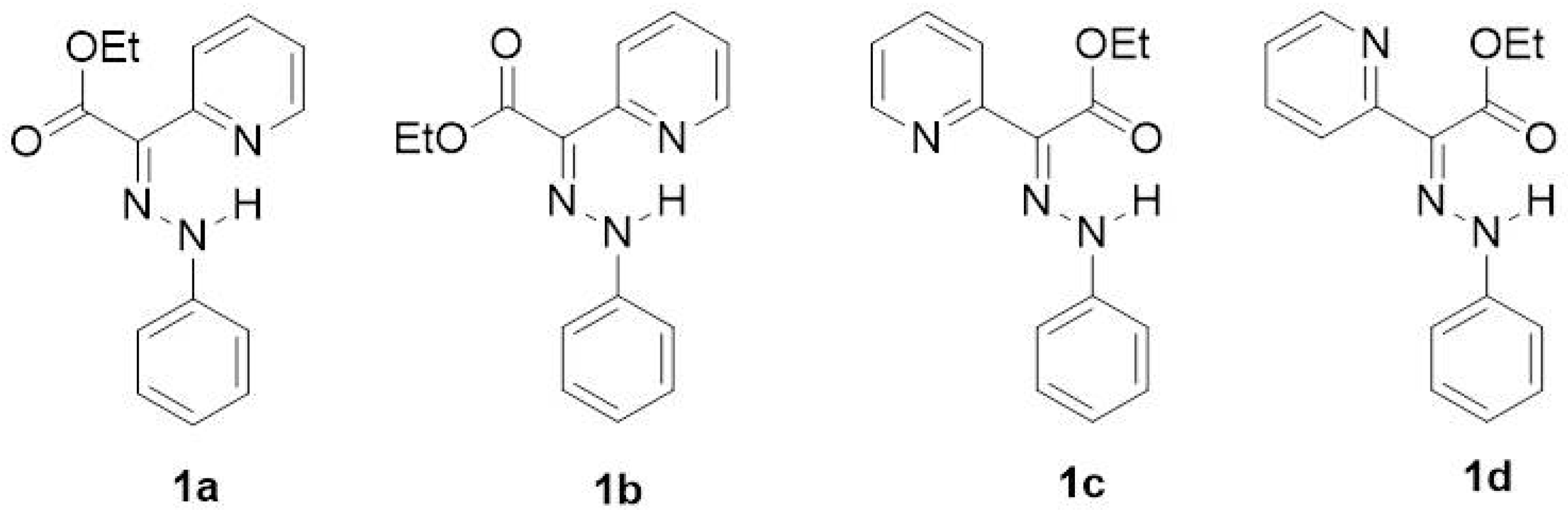



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substituent R | Compound | M062X/6-31+G(d,p) | MN12-SX/6-31+G(d,p) | ||
|---|---|---|---|---|---|
| ε = 1 | ε = 36 | ε = 1 | ε = 36 | ||
| H | 1a | 2.54 | 1.11 * | 2.51 | 1.32 * |
| 1b | 0.00 | 0.00 * | 0.00 | 0.00 * | |
| 1c | 2.93 | 2.09 * | 2.82 | 2.35 * | |
| 1d | 1.09 | 1.49 * | 1.17 | 2.10 * | |
| N(CH3)2 | 2a | 2.56 | 0.85 | 2.35 | 1.62 |
| 2b | 0.00 | 0.00 | 0.00 | 0.00 | |
| 2c | 3.43 | 1.65 | 1.49 | 1.70 | |
| 2d | 1.99 | 1.01 | 0.62 | 1.79 | |
| Cl | 7a | 2.32 | 0.83 | 2.46 | 1.06 |
| 7b | 0.00 | 0.00 | 0.00 | 0.00 | |
| 7c | 2.61 | 2.20 | 2.39 | 1.97 | |
| 7d | 1.10 | 1.14 | 1.24 | 1.30 | |
| NO2 | 11a | 1.92 | 1.05 | 2.67 | 1.93 |
| 11b | 0.00 | 0.00 | 0.00 | 0.00 | |
| 11c | 2.69 | 2.06 | 3.23 | 3.13 | |
| 11d | 0.99 | 1.34 | 1.78 | 2.18 | |
| Main Structure | Substituent R | Compound | Effect |
|---|---|---|---|
 | H | 1 | standard; no effect |
| N(CH3)2 | 2 | EDG | |
| OH | 3 | EDG | |
| O(CH2)5CH3 | 4 | EDG | |
| OCH3 | 5 | EDG | |
| F | 6 | EWG | |
| Cl | 7 | EWG | |
| Br | 8 | EWG | |
| C(O)OCH3 | 9 | EWG | |
| CN | 10 | EWG | |
| NO2 | 11 | EWG |
| Compound | a | b | c | d | |||||
|---|---|---|---|---|---|---|---|---|---|
| ε = 1 | ε = 36 | ε = 1 | ε = 36 | ε = 1 | ε = 36 | ε = 1 | ε = 36 | ||
| 1 (R=H) | |||||||||
| N1-H | 1.022 | 1.023 | 1.025 | 1.025 | N1-H | 1.019 | 1.019 | 1.019 | 1.019 |
| N1-N2 | 1.307 | 1.312 | 1.304 | 1.309 | N1-N2 | 1.307 | 1.310 | 1.313 | 1.313 |
| N2=C3 | 1.301 | 1.301 | 1.304 | 1.303 | N2=C3 | 1.301 | 1.300 | 1.298 | 1.299 |
| C3-C9 | 1.481 | 1.484 | 1.484 | 1.483 | C3-C4 | 1.479 | 1.482 | 1.486 | 1.486 |
| C9=N10 | 1.347 | 1.346 | 1.349 | 1.347 | C4=O5 | 1.223 | 1.224 | 1.223 | 1.224 |
| N10⋯H | 1.903 | 1.909 | 1.857 | 1.881 | O5⋯H | 1.885 | 1.896 | 1.902 | 1.918 |
| 2 (R=N(CH3)2) | |||||||||
| N1-H | 1.023 | 1.024 | 1.027 | 1.026 | N1-H | 1.020 | 1.020 | 1.019 | 1.019 |
| N1-N2 | 1.301 | 1.304 | 1.298 | 1.301 | N1-N2 | 1.301 | 1.303 | 1.308 | 1.306 |
| N2=C3 | 1.306 | 1.306 | 1.310 | 1.310 | N2=C3 | 1.306 | 1.306 | 1.303 | 1.304 |
| C3-C9 | 1.479 | 1.482 | 1.480 | 1.482 | C3-C4 | 1.473 | 1.476 | 1.481 | 1.480 |
| C9=N10 | 1.348 | 1.347 | 1.350 | 1.348 | C4=O5 | 1.225 | 1.226 | 1.224 | 1.226 |
| N10⋯H | 1.889 | 1.899 | 1.839 | 1.859 | O5⋯H | 1.876 | 1.889 | 1.893 | 1.913 |
| 7 (R=Cl) | |||||||||
| N1-H | 1.022 | 1.023 | 1.026 | 1.026 | N1-H | 1.019 | 1.019 | 1.019 | 1.019 |
| N1-N2 | 1.309 | 1.314 | 1.306 | 1.312 | N1-N2 | 1.309 | 1.312 | 1.316 | 1.315 |
| N2=C3 | 1.300 | 1.299 | 1.304 | 1.302 | N2=C3 | 1.300 | 1.299 | 1.297 | 1.297 |
| C3-C9 | 1.482 | 1.484 | 1.484 | 1.484 | C3-C4 | 1.480 | 1.484 | 1.488 | 1.487 |
| C9=N10 | 1.347 | 1.346 | 1.349 | 1.347 | C4=O5 | 1.223 | 1.224 | 1.223 | 1.224 |
| N10⋯H | 1.900 | 1.908 | 1.845 | 1.868 | O5⋯H | 1.880 | 1.895 | 1.897 | 1.921 |
| 11 (R=NO2) | |||||||||
| N1-H | 1.023 | 1.024 | 1.026 | 1.026 | N1-H | 1.020 | 1.019 | 1.019 | 1.019 |
| N1-N2 | 1.317 | 1.324 | 1.314 | 1.322 | N1-N2 | 1.316 | 1.321 | 1.324 | 1.325 |
| N2=C3 | 1.296 | 1.295 | 1.299 | 1.296 | N2=C3 | 1.296 | 1.294 | 1.293 | 1.292 |
| C3-C9 | 1.484 | 1.486 | 1.487 | 1.486 | C3-C4 | 1.486 | 1.491 | 1.493 | 1.494 |
| C9=N10 | 1.346 | 1.345 | 1.348 | 1.346 | C4=O5 | 1.222 | 1.222 | 1.221 | 1.222 |
| N10⋯H | 1.896 | 1.896 | 1.841 | 1.866 | O5⋯H | 1.876 | 1.890 | 1.896 | 1.919 |
| Compound | a | b | c | d | |||||
|---|---|---|---|---|---|---|---|---|---|
| ε = 1 | ε = 36 | ε = 1 | ε = 36 | ε = 1 | ε = 36 | ε = 1 | ε = 36 | ||
| 1 (R=H) | |||||||||
| N1 | −0.170 | −0.218 | −0.184 | −0.232 | N1 | −0.228 | −0.240 | −0.274 | −0.264 |
| N2 | −0.137 | −0.168 | −0.132 | −0.153 | N2 | −0.138 | −0.176 | −0.139 | −0.165 |
| C3 | −0.207 | −0.236 | −0.169 | −0.205 | C3 | −0.217 | −0.217 | −0.172 | −0.195 |
| C9 | 0.535 | 0.598 | 0.446 | 0.562 | C4 | 0.861 | 0.609 | 0.857 | 0.591 |
| N10 | −0.451 | −0.543 | −0.368 | −0.509 | О5 | −0.612 | −0.724 | −0.600 | −0.725 |
| H | 0.216 | 0.277 | 0.200 | 0.271 | H | 0.319 | 0.343 | 0.323 | 0.341 |
| 2 (R=N(CH3)2) | |||||||||
| N1 | −0.120 | −0.115 | −0.121 | −0.111 | N1 | −0.192 | −0.177 | −0.242 | −0.198 |
| N2 | −0.151 | −0.204 | −0.148 | −0.177 | N2 | −0.153 | −0.182 | −0.148 | −0.178 |
| C3 | −0.218 | −0.229 | −0.184 | −0.231 | C3 | −0.202 | −0.256 | −0.184 | −0.235 |
| C9 | 0.534 | 0.566 | 0.440 | 0.552 | C4 | 0.836 | 0.925 | 0.846 | 0.909 |
| N10 | −0.455 | −0.531 | −0.375 | −0.498 | О5 | −0.612 | −0.663 | −0.605 | −0.653 |
| H | 0.209 | 0.244 | 0.191 | 0.238 | H | 0.310 | 0.327 | 0.322 | 0.327 |
| 7 (R=Cl) | |||||||||
| N1 | −0.150 | −0.151 | −0.152 | −0.175 | N1 | −0.219 | −0.230 | −0.259 | −0.202 |
| N2 | −0.167 | −0.213 | −0.148 | −0.181 | N2 | −0.155 | −0.176 | −0.167 | −0.195 |
| C3 | −0.170 | −0.192 | −0.158 | −0.180 | C3 | −0.204 | −0.218 | −0.139 | −0.172 |
| C9 | 0.525 | 0.556 | 0.453 | 0.530 | C4 | 0.865 | 0.924 | 0.841 | 0.883 |
| N10 | −0.455 | −0.525 | −0.384 | −0.494 | О5 | −0.610 | −0.653 | −0.594 | −0.632 |
| H | 0.212 | 0.249 | 0.197 | 0.247 | H | 0.312 | 0.337 | 0.314 | 0.316 |
| 11 (R=NO2) | |||||||||
| N1 | −0.187 | −0.170 | −0.150 | −0.139 | N1 | −0.224 | −0.231 | −0.260 | −0.202 |
| N2 | −0.155 | −0.214 | −0.163 | −0.218 | N2 | −0.157 | −0.193 | −0.162 | −0.202 |
| C3 | −0.175 | −0.155 | −0.138 | −0.115 | C3 | −0.174 | −0.177 | −0.132 | −0.141 |
| C9 | 0.530 | 0.553 | 0.455 | 0.502 | C4 | 0.861 | 0.926 | 0.847 | 0.889 |
| N10 | −0.454 | −0.516 | −0.382 | −0.494 | О5 | −0.604 | −0.641 | −0.588 | −0.628 |
| H | 0.217 | 0.254 | 0.193 | 0.234 | H | 0.314 | 0.338 | 0.315 | 0.321 |
| ε = 1 | ε = 36 | |||
| Substituent | a/b | c/d | a/b | c/d |
| H | 0.053 | 0.023 | 0.025 | 0.016 |
| N(CH3)2 | 0.053 | 0.023 | 0.019 | 0.015 |
| Cl | 0.043 | 0.034 | 0.024 | 0.031 |
| NO2 | 0.049 | 0.024 | 0.032 | 0.026 |
| ε = 1 | ε = 36 | |||||
| Isomer | N(CH3)2/H | Cl/H | NO2/H | N(CH3)2/H | Cl/H | NO2/H |
| a | 0.022 | 0.022 | 0.017 | 0.049 | 0.043 | 0.049 |
| b | 0.028 | 0.017 | 0.024 | 0.054 | 0.033 | 0.066 |
| c | 0.020 | 0.010 | 0.020 | 0.135 | 0.132 | 0.135 |
| d | 0.015 | 0.020 | 0.021 | 0.137 | 0.129 | 0.133 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paskaleva, V.; Dobrev, S.; Kochev, N.; Angelova, S.; Antonov, L. Unusual Para-Substituent Effects on the Intramolecular Hydrogen Bond in Hydrazone-Based Switches: Insights from Chemical Landscape Analysis and DFT Calculations. Physchem 2021, 1, 189-201. https://doi.org/10.3390/physchem1020013
Paskaleva V, Dobrev S, Kochev N, Angelova S, Antonov L. Unusual Para-Substituent Effects on the Intramolecular Hydrogen Bond in Hydrazone-Based Switches: Insights from Chemical Landscape Analysis and DFT Calculations. Physchem. 2021; 1(2):189-201. https://doi.org/10.3390/physchem1020013
Chicago/Turabian StylePaskaleva, Vesselina, Stefan Dobrev, Nikolay Kochev, Silvia Angelova, and Liudmil Antonov. 2021. "Unusual Para-Substituent Effects on the Intramolecular Hydrogen Bond in Hydrazone-Based Switches: Insights from Chemical Landscape Analysis and DFT Calculations" Physchem 1, no. 2: 189-201. https://doi.org/10.3390/physchem1020013
APA StylePaskaleva, V., Dobrev, S., Kochev, N., Angelova, S., & Antonov, L. (2021). Unusual Para-Substituent Effects on the Intramolecular Hydrogen Bond in Hydrazone-Based Switches: Insights from Chemical Landscape Analysis and DFT Calculations. Physchem, 1(2), 189-201. https://doi.org/10.3390/physchem1020013