Abstract
The design of molecular solar fuels is challenging because of the long list of requirements these molecules have to fulfil: storage density, solar harvesting capacity, robustness, and heat release ability. All of these features cause a paradoxical design due to the conflicting effects found when trying to improve any of these properties. In this contribution, we will review different types of compounds previously suggested for this application. Each of them present several advantages and disadvantages, and the scientific community is still struggling to find the ideal candidate suitable for practical applications. The most promising results have been found using norbornadiene-based systems, although the use of other alternatives like azobenzene or dihydroazulene cannot be discarded. In this review, we primarily focus on highlighting the optical and photochemical aspects of these three families, discussing the recently proposed systems and recent advances in the field.
1. Introduction
Energy generation and storage has become one of the major challenges in our society and are especially relevant for industry [1,2]. The current energy demand is continuously rising [3] each year by 1.3%, and this progression is expected to last at least until 2040 [4], even considering that many industries worldwide have been affected by COVID-19. According to the International Energy Agency, buildings are responsible for almost 30% of energy consumption and account for 28% of CO2 emissions [4,5]. To avoid the environmental impact from conventional energy sources, the use of renewable electricity needs to augment considerably. However, we are not yet able to avoid our dependence on fossil fuels. Consequently, significant efforts to find better alternatives to generate and store energy are under exploration. This is especially relevant for solar energy use and storage [6], which has been envisioned as an abundant, clean, and promising energy source.
Using natural photosynthesis as a working model for solar energy use, scientists are designing and preparing chemical systems capable of capturing and storing solar energy. Nowadays, different alternatives to make use of sunlight are under research, including direct use of photonic solar power and heating capacity of solar radiation. The variability in solar income is a very significant drawback to solar energy, as the power of both types of energy (photonic and heating) is not constant during the four seasons of the year [7] and strongly depends on the weather and geographical factors. This non-constant power supply unequivocally demands a storage solution, which should allow wider usability under conditions such as night or winter. Consequently, different methodologies have been developed to exploit solar power such as underground solar energy storage (USES) and molecular solar thermal (MOST) systems.
The USES system mechanism consists of the storage of sun energy underground during summer months using a pile [8,9]. There are four basic types of USES systems: hot-water-thermal storage, borehole thermal storage, aquifer thermal storage, and water gravel pit storage [10]. This mechanism requires a plant of quite large dimensions, making it quite difficult to use this technique once the building has already been built [11]. Similarly, in these approaches, the thermal insulation requirements usually imply a challenge for long-term storage.
On the other hand, MOST technology has become a promising candidate to capture and store solar energy in a sustainable and efficient manner. These systems have been expanded significantly in the last decades [7], even though the first idea dates a while back [12]. The MOST approach is based on the storage of solar energy as chemical energy using a photoactive molecule, which, after being exposed to sunlight, isomerizes into a metastable high-energy photoisomer [13]. The release of chemical energy as heat can be performed during the back conversion step using an external stimulus (Figure 1a) either through heat, through a catalyst, or electrochemically [14].
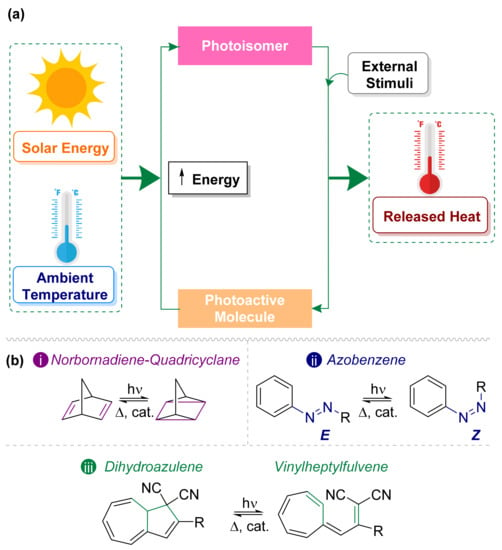
Figure 1.
(a) Concept of the MOST system [15]. (b) Photoswitches most used in solar energy storage: (i) norbornadiene–quadricyclane, (ii) E/Z–azobenzene, and (iii) dihydroazulene–vinylheptafulvene.
Along the years, a large number of systems have been proposed as MOST candidates. There are at least six major requirements for a practical system, which makes the design of successful candidates a challenging task; this will be described further in the next section. Until now, none of the previously proposed compounds completely fulfils this list of requirements. Thus, the design and preparation of new alternatives to MOST technology is still a hot topic. In the following sections, the most relevant efforts to prepare suitable compounds according to these requirements will be discussed. For the scope of this review, we will describe the three types of systems that have been mainly used within the current framework of MOST technology: norbornadiene, azobenzene, and dihydroazulene derivatives [15,16,17] (Figure 1b). In addition, we will focus on the central role of the optical properties in the storage of solar energy.
2. Requisites for MOST Systems: Optical Properties
The development of new MOST candidates is a challenging task, which has gained more attention and visibility in the last decades owing to the expectations raised by this technology. As mentioned above, the ideal compound for a MOST device is still unknown, so many different strategies combining experimental and computational tools are being used to assist in the molecular design.
In order to maximize the efficiency of MOST systems, their optimization has received a great deal of attention; in light of this, the design of a suitable photoswitch must meet a large number of objectives to fulfil the ideal MOST system. Thus, the design criterion of a MOST system is subjected to several parameters involving both engineering and chemical challenges [18].
The first key step in the molecular solar thermal energy storage system is the absorption of light by the parent molecule, which undergoes a reversible photoisomerization reaction to its corresponding metastable isomer. This photoisomer should be stable enough to store the chemical potential for varying periods of time, depending on the envisioned application. Then, this stored energy should be released when and where required, in the form of heat. For the initial photoisomerization part of the MOST cycle to be successful, the photoswitch pair needs to fulfil a long list of features, in some cases even contradictory [3]:
- A large difference in the free energies of the parent molecule and its photoisomer, with a minimal increase in the molecular weight to maximize energy density [19,20].
- A moderately large kinetic barrier for back conversion [21,22].
- An absorption spectrum of the photoactive molecule matching the solar spectrum [22,23].
- A high quantum yield (Φ = 1) for the photogeneration of the metastable photoisomer.
- A photochemically inactive (or non-absorbing) photoisomer.
- Negligible degradation of both the photoactive molecule and its photoisomer after multiple cycles, especially moving towards higher temperatures.
These are the major requirements that should be optimized to improve the performance of any potential candidate. Furthermore, when considering a MOST molecule in an integrated device, the use of environmentally friendly compounds and solvents is desired to minimize the risks in case of leaching or losses to its surroundings. In this review, we will focus on the optical properties. Thus, a more detailed explanation of some of the requirements related to the photochemistry of these compounds will be presented: the absorption spectrum (solar match), photoreaction efficiency (quantum yield), and energy storage capacity.
2.1. Solar Match
The photochemical reaction from a low (parent molecule, photoactive molecule) to a high energy configuration (photoisomer) is the central focus of the photochemical part of the MOST cycle. For this transformation to take place, the main requirement is the absorption of energy supply from the sun. Hence, the ideal MOST systems should absorb the maximum number of photons in the UV and visible range of the solar spectrum, 300–700 nm, which corresponds to the maximum intensity of sunlight. Ideally, the absorption spectrum of the lower-energy isomer should overlap with the most intense region of the solar energy window (solar match) and preferably in the solar spectrum range between 340–540 nm, where the solar radiation is relatively high [22]. Moreover, it is desirable that no absorption overlap between the initial isomer and photoisomer exists to avoid a non-desirable photon absorption competition between the two states.
Most of the basic cores of the photoswitches reported to date do not show wavelengths going far beyond 350 nm (for instance, parent norbornadiene has a maximum at 236 nm and some ruthenium derivatives absorb at 350) [24]. This is a significant drawback as the solar photons’ flux at wavelengths below 330 nm is quite low. In this regard, both experimental [25,26] and computational [27] progress has been made in providing functionalized photoswitches that absorb at larger wavelengths. The most used and successful chemical strategy to shift the absorption of MOST compounds toward higher wavelengths is by creating a ‘push–pull’ effect through the introduction of donor–acceptor substituents and increasing the molecule’s conjugated π-system. Preferably, low molecular weight electron donor and acceptor groups are prominent targets for generating relevant photoswitches, as they cause a lower impact on the energy density (affected by both the difference in energy between isomers and the molecular weight). However, beyond the stored energy, the chemical modification of photoswitches may also negatively affect other relevant properties, making it clear that the optimization of a set of molecules for MOST applications is extremely challenging.
2.2. Quantum Yield
Once the parent molecule absorbs a photon from sunlight, the excitation from the ground state (S0) to the excited state (Sn) takes place. Subsequently, a certain number of molecules will undergo photo-conversion, but a fraction could undergo relaxation, returning to their initial state. To quantify the fraction of molecules effectively performing the photoconversion from the photoactive molecule to the photoisomer, the quantum yield is measured. This dimensionless number provides the probability of a parent molecule to furnish the metastable high-energy photoisomer per absorbed photon. From an efficiency perspective, the photo-conversion that leads to the high isomer needs to be as high as possible, being close to unity if possible. This should allow for an efficient conversion of solar energy into chemical energy, hence avoiding other competitive processes such as radiative, non-radiative, or quenching.
2.3. Storage Energy Density
While it is not strictly a photochemical property, another crucial concern in MOST systems is the energy storage. MOST technology is designed for generating the greatest possible increase in temperature after releasing the stored chemical energy in the photoisomer as heat. In this way, the key property to achieve this goal is the enthalpy difference (ΔH) between photoisomers. This means that, the bigger the energy difference between the (not charged) metastable photoisomer and its parent state, the larger the energy storage density that will be accumulated in the system. As a rule of thumb, MOST systems should provide at least 0.3 MJ/kg to be of practical use, prior to the subsequent heat release. Then, heat could be released using an external stimulus like a thermal increment or via a catalyst. Thus, the photoisomer should not undergo back-conversion quickly at room temperature in order to store the energy for hours, days, or months (storage time) depending on the target application. Even if this review is focused on the photochemical aspects of the MOST technology, it is also relevant to mention that alternative cooling and heating methods are available. For instance, the use of phase transition materials and water adsorption in zeolites has been already commercialized [28,29].
3. Photoswitches Used in MOST Technology
As a brief introduction to the state-of-the-art of the historic development of MOST candidates, very different sets of families were considered at some point. However, most of them were discarded at a relatively early stage because of some practical reasons or flaws. Considering the most explored molecular systems, the main parts of them are photoswitches, as explained above. This is partly because of the considerable overlap between the requirements for photoswitches in general and the compounds used in the MOST concept (absorption, high quantum yields, photostability, and robustness). Historically, the compounds designed to be used in MOST systems can be grouped into two main types [2,16], depending on the photochemical transformation that takes place. In this sense, the mechanism of the photochemical process transforming sunlight into chemical energy can be an isomerization or a cycloaddition. Other types of photochemically induced intramolecular rearrangements were also studied. As an example of some more complex rearrangements, organometallic diruthenium fulvalene’s have also been considered [30].
According to the photochemical transformation involved, the systems based on an isomerization are typical examples of molecular photoswitches, like stilbenes [31], azobenzenes, retinal-based photoswitches [32], or other less-known families like hydantoins [33]. The main problem behind using traditional cis-trans photoswitches is the typical small energy gap between the two isomers, producing a small amount of energy storage. This problem has been overcome by two different strategies. Firstly, stabilization of the E-isomer usually occurs when increasing electronic delocalization. Secondly, with a destabilization of the Z-isomer attributed to vicinal groups, steric interactions are incurred. Combining those strategies, some stilbene derivatives could be designed to reach an energy storage of 100 kJ/mol higher than the original unsubstituted stilbene molecule, reaching 105 kJ/mol [34]. Comparably, following the same strategies, retinal-like systems were postulated for this application too, with more modest energy storage capacities [35].
The employment of systems based on a photochemical cycloaddition typically has better properties in terms of energy storage, but their optical properties (absorption spectra) are usually less tuneable as absorption usually lies in the high-energy region of the UV spectra. The main exponent of this approach is the norbornadiene (NBD)–quadricyclane (QC) couple [36], which has been studied since the 1980s and nowadays is a focus of most of the efforts from the community. One of the first proposals using cycloaddition reactions was the use of anthracene derivatives, thanks to their well-known intermolecular [4 + 4] cycloaddition. These compounds also present some problems, as the absorption usually occurs below 300 nm, meaning a low efficiency exposed to solar radiation. This was partially solved by adding (a) bridge group(s) to link two anthracene moieties, but in this case, the efficiency decreased drastically [37]. Another cycloaddition system used is the pair based on dihydroazulene (DHA) and vinylheptafulvene (VHF) [38,39]. Unfortunately, the parent compounds in this couple present a small energy difference between isomers, plus the tunability of the optical properties has already been exhaustively explored [40].
Other systems such as ruthenium fulvalene complexes have also been proposed and studied but have been discarded for practical applications because of the low efficiency and high preparation costs [30,41].
In summary, many molecular systems have been studied along the years as potential MOST candidates. In the following, we will focus our attention on the three most promising families of MOST molecules to date, namely, norbornadiene, azobenzenes, and dihydroazulenes. These three families combine relatively good (or tuneable) properties and are synthetically attainable.
3.1. Norbornadiene/Quadricyclane Couple
Among the previously mentioned MOST systems under investigation, the most profoundly explored is without a doubt the NBD to QC isomerization. Even if the foundations of the MOST concept did not begin with the NBD/QC photoswitch, it is nowadays the main area of research in the field. These molecules have reached energy density values close to the maximum energy density limit of a solar thermal battery at 1 MJ/kg [42]. In contrast, the absorption of unsubstituted NBD is within the UVC range (less than 267 nm) and does not overlap with the solar spectrum, which begins at 340 nm [26].
The ideal absorption scenario for molecular solar thermal energy storage systems is to use solar radiation, which reaches the Earth’s surface at high intensities [43]. Thus, targeting a photoisomerization induced reaction in the 350–450 nm range is highly desirable. In designing new NBD/QC molecules, the difference in the absorption maxima between the NBD and QC molecules needs to be large enough to minimize spectral overlap [25,44], which could diminish the incident radiation flux reaching the photoactive isomer. A significant advantage of NBDs over other photoswitches (i.e., azobenzenes) is that, upon absorption, a blue-shift usually occurs. Thus, the QC molecule tends to absorb outside of the visible spectrum, which purposely mismatches the solar spectrum and, consequently, the presence of photostationary states is circumvented. In turn, this avoids the need for engineering adjustments such as band pass filters.
The long list of requirements for an optimal molecule for MOST applications has caused the design of new molecules with increased complexity. For instance, two principal chemical strategies have been applied in the shifting of the absorption of the parent molecule to higher wavelengths [2]. One of them involves adding electron-donating or electron-withdrawing substituent groups to create a ‘push–pull’ effect, while the other involves increasing the system’s extent of π-conjugation [45]. Some representative examples are shown in Table 1 and Figure 2.

Table 1.
Molar mass, λmax; enthalpy of isomerization (ΔHisomerization); and energy density of NBD derivatives, which are shown in Figure 2 [2].
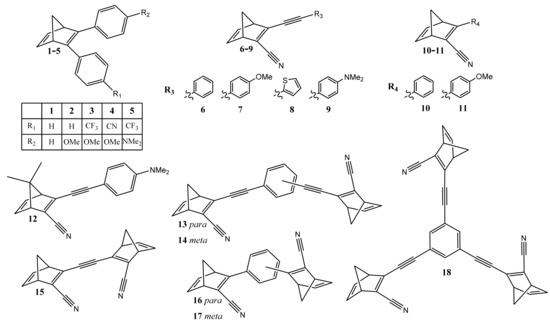
Figure 2.
List of NBD derivatives with absorption maxima in the 300–400 nm range.
The first method is typically performed by adding substituents with electron-rich phenyl rings to red shift the absorption to higher wavelengths. A potential drawback caused by this modification is an increased spectral overlap between the NBD and QC isomers [46]. Concordantly, solely changing the acceptor group from a cyano to a trifluoroacetyl group red shifts the absorbance by 100 nm, although it considerably shortens the lifetimes of QC [47].
In the second strategy, the use of NBD dimers in MOST systems is being explored, although the origin of this molecular cooperativity remains to be fully understood. Extending the π-conjugation by linking two NBD units through an electron-rich aromatic unit minimizes the impact of molecular weight increase as two units are considered and two photons may be absorbed. Unfortunately, shorter wavelengths of absorption are required for the second photoisomerization process from QC/NBD to QC/QC, ultimately decreasing the quantum yield. Moreover, both NBD moieties should be photoisomerized as NBD/QC is far more labile than QC/QC [24].
Another method to try to improve the performance of the NBD/QC couple is the use of alternative deactivation channels. In this sense, thermally activated delayed fluorescent (TADF) molecules undergo an excitation to the lowest-lying singlet state, relax to the triplet state, and finally can be thermally converted back to the singlet state (otherwise known as a reversible intersystem crossing) and relax from this singlet excited state. TADF molecules have the lowest-lying excitation band ideally situated in the perfect range of the solar spectrum required for MOST systems, at lower wavelengths than typical phosphorescent molecules (480–550 nm) and higher wavelengths than fluorescent molecules (330–380 nm). TADF molecules were originally built around a benzophenone core structure, with the addition of carbazole and diphenylamine moieties being common, wherein keto groups are in a co-planar position relative to aromatic moieties [48]. This co-planarity effect enhances the overlap between n and π* states, creating a very small singlet–triplet energy gap. A new promising approach was attempted linking thermally activated delayed fluorescence molecules like phenoxazine–triphenyltriazine (PXZ-TRZ) to NBD moieties, where red-shifted absorption peaks were in the 400–430 nm range [49]. Moreover, NBD molecules were substituted onto these structures with an increased conjugation as a possible solution to maximize the low-energy storage density of a single NBD unit.
Despite the copious publications using the NBD/QC photoswitch, the optimal system has not been devised yet. The prime-substituted NBD has a red-shifted absorption of 59 nm, yet it has an increase in molar mass of 131 g/mol, inevitably reducing the energy density by 13.3 kJ/mol. The energy density of unsubstituted NBDs to the present date still outcompetes the prime-substituted NBD by a margin of 13% [50]. To implement an NBD/QC MOST photoswitch that absorbs in the 350–450 nm region into practical applications will entail a compromise of 20 kJ/mol of storage energy for a better fitting with the solar spectrum, especially considering that, when moving to highly absorbing materials like NBD, a major fraction of light will nonetheless be converted to heat. Controlling thermal fluxes at the surface is not only required to keep the photoisomer stable, but it is also a safety mechanism essential in energy storage devices like batteries to prohibit overheating under working conditions. Utilizing waste heat by coupling a heat exchanger to the final device will prevent local temperature extremes and maintain a truly closed MOST system [44].
As mentioned previously, it is also crucial to control the efficiency of the photochemical process, which starts with the absorption of a photon by NBD. The incoming photon can induce an electronic transition from the minimum in the ground state to the S1 state, causing a [2 + 2] intramolecular cycloaddition [51]. Under natural solar irradiation conditions, this photoconversion does not take place in the parent NBD, as just a few photons below 300 nm arrive at sea level. Therefore, the unsubstituted NBD/QC couple is chemically inactive to sunlight. Furthermore, in the event that the NBD absorbs a proper solar radiation, just a few molecules will undergo photoisomerization because of the quantum yield of this system being quite low (ϕ = 0.05). In consideration of these requirements, the NBD/QC system has been chemically modified in order to increase the quantum yield [18] as well as red-shift the wavelength of absorption.
As shown in Figure 3, the introduction of donor–acceptor groups increases the quantum yield and makes the system capable of absorbing natural solar irradiation above 400 nm. Thus, when the incoming photon is absorbed by NBD, the photoisomerization transformation starts through a S0–S1 transition. The molecules will then undergo relaxation in the excited state potential energy surface to reach the minimum energy conical intersection point (MECI) S0/S1, leading to the photoisomer QC [24]. In the heat release step, the thermodynamic driving force of the process (ΔHisom = 372 kJ/mol) pushes QC in forming the less-strained geometry (NBD), based on the cleavage of two single bonds of the four-membered ring and the transformation of the remaining bonds to double bonds, as seen in Figure 4. In this stage, the reaction releases a high amount of energy because of the high-standard enthalpy of the reversion of QC to NBD. This energy also depends on the gravimetric energy density (MJ/kg), hence systems with smaller molecular weights will hold comparatively greater energy densities. Thus, cyano-substituted NBD derivatives have been computational and experimentally studied as they show attractive energy densities (0.4–0.6 MJ/kg) [25].
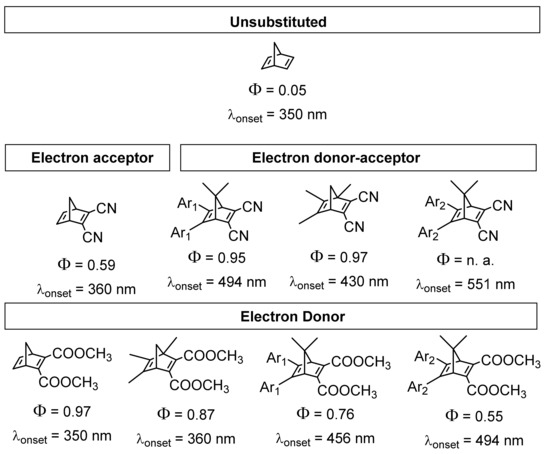
Figure 3.
Summary of the effect of electron donor–acceptor groups on the quantum yield and on the onset of the absorption wavelength. Ar1 means a phenyl group and Ar2 means a p-methoxyphenyl group. Data collected from [18,52,53,54].
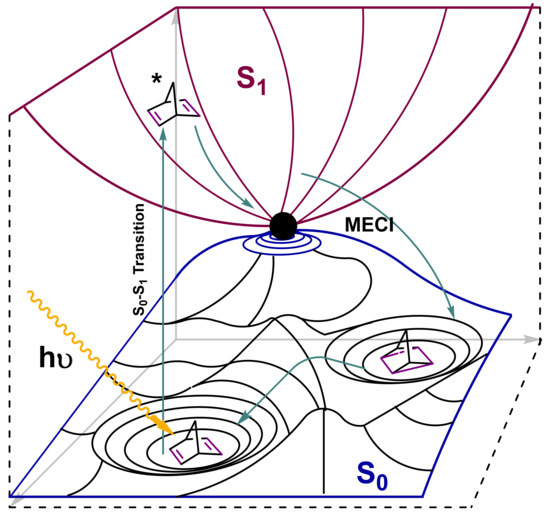
Figure 4.
Approximate 3D illustration of a ground-state (S0, blue) and excited (S1, red) energy landscape depicting the whole photoisomerization mechanism.
Control of the back conversion reaction is crucial in providing energy at the right time. It is well known in these systems that the optimization of solar match and storage lifetime at the same time is challenging, because one property is improved at the expense of the other. However, a recent novel strategy defies this, wherein NBD molecules have an improved solar match, yet the storage time remains untouched. Chiefly, via the introduction of substituents in the ortho position, a substantial increase in back conversion ΔS* occurs, which comes from the steric interference of the side group of the parent molecule [55].
3.2. Azobenzene Photoswitches
Azobenzene is a chemical compound based on diazene (HN=NH), where both hydrogens are substituted by a phenyl ring [56,57]. Azobenzene can be found either in a cis or trans conformation [58]. The trans to cis isomerization can be triggered by different stimuli such as irradiation with ultraviolet (UV) light [59], mechanical force [60], or an electrostatic stimulus [61,62]. Contrastingly, the cis to trans isomerization can be observed in dark conditions when applying heat owing to the thermodynamic stability of the trans isomer, although it can also be driven by visible light (see Figure 5a). The trans conformation assumes a planar structure with C2h symmetry [63,64], with a maximum distance of 9.0 Å between the most distant positions of the aromatic rings. On the other hand, the cis one adopts a non-planar conformation with C2 symmetry [65,66] and the end-to-end distance is reduced to 5.5 Å. The cis-isomer is not planar as a result of steric hindrance, and this causes the π-electron clouds of the aromatic rings to face each other, leading to a small dipolar moment in the molecule (µ ~3D) [67]. This ring’s disposition is also reflected in the proton nuclear magnetic resonance (1H-NMR) spectra. The shielding effect produced by the anisotropy of the π-electron cloud in the cis-isomer affects the signals, thus making them appear at a higher shift compared with the trans-isomer.
The absorbance spectrum of the trans-azobenzene molecule typically presents two absorption bands (see Figure 5b), corresponding to the electronic transitions n–π* and π–π*. The electronic transition π–π* produces a strong band in the UV region and is also present in analogous-carbon systems such as stilbene [68]. The n–π* electronic transition presents a different band, which is far less intense, in the visible region. This latter transition is produced by the nitrogen’s lone pair of electrons [69], which generates a completely different photoisomerization process compared with its analogous carbon system, the stilbene. The trans–cis isomerization process is usually followed by a color change towards more intense tonalities. The absorbance spectra of both isomers are differentiated by the following aspects: (i) trans-isomer: the absorption band related to the π–π* electronic transition is very intense, with a molar extinction coefficient (ε) around 2–3 × 104 M−1 cm−1. On the other hand, the band corresponding to the n–π* electronic transition is much weaker (ε ~400 M−1 cm−1), as this transition is forbidden by the symmetry rules in this isomer. (ii) Cis-isomer: the absorption band related to the π–π* electronic transition is hipsochromically shifted and its intensity diminishes greatly (ε ~7–10 × 10−3 M−1 cm−1) with respect to the trans-isomer. On the other hand, the band corresponding to the n–π* electronic transition (380–520 nm) is allowed in this isomer, thus significantly increasing the intensity of this band (ε ~1500 M−1 cm−1). These optical properties can be greatly modified upon substitution and this fact has been exploited to match the requirements of MOST technology.
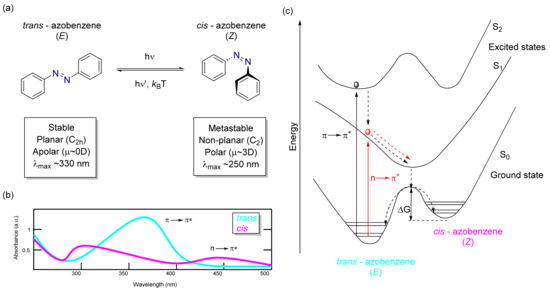
Figure 5.
(a) Azobenzene isomer structures and an overview of properties. (b) Absorbance spectrum scheme of trans and cis-azobenzene isomers with its main bands assigned to electronic transitions. (c) Schematic view of lower energy levels and pathways of the azobenzene isomers, adapted from Georgiev et al. [70].
Figure 5.
(a) Azobenzene isomer structures and an overview of properties. (b) Absorbance spectrum scheme of trans and cis-azobenzene isomers with its main bands assigned to electronic transitions. (c) Schematic view of lower energy levels and pathways of the azobenzene isomers, adapted from Georgiev et al. [70].

Although azobenzene can adopt trans and cis conformations, the former is more stable at the electronic ground state by roughly 58.6 kJ/mol (0.6 eV) [71]; this is explained by the lack of delocalization and a distorted configuration of the cis-form in comparison with the trans-isomer. The experimentally measured energy barrier between trans and cis conformations is about 96.2 kJ/mol (1.0 eV) [72]; thus, in dark conditions and at room temperature, the predominant species is the trans one (Figure 5c). As explained above, the energy difference between isomers will have a strong effect on the stored energy density. This value could be also affected by substitution, but in general, smaller values of energy density could be obtained using azobenzene compared with other MOST molecules such as NBD/QC.
The differences in spectroscopical properties for the cis and trans isomers and the distribution of excited state electronic levels allow them to undergo isomerization upon irradiation. This photochemical interconversion usually ends up providing a varying mixture of cis and trans isomers on the photostationary states (PSS). The ratio of isomers in the mixture depends on the substitution of the azobenzene core and the wavelength of irradiation. In turn, the excitation wavelength at which this process takes place depends on the nature of the substituents on the aryl groups of the azobenzene; although, usually, in the trans to cis isomerization, this process is promoted by wavelengths around 320–380 nm, while exposition to 400–450 nm wavelengths typically favors cis to trans isomerization. This reversion can also be induced thermally. Although both photochemical conversions take place in the order of picoseconds, the cis to trans thermal relaxation is in the order of seconds or even hours and can be tuned by substitution.
Although several mechanistic studies have focused on the isomerization mechanisms of azobenzene, in fact, the effect of the substituents on the phenyl rings as well as the influence of other parameters yields a complex mechanism [73,74,75]. In fact, several mechanisms of the isomerization of the azobenzene and its derivatives can take place depending on the nature of the substituents and the reaction conditions (see Figure 6): (i) The inversion of one of the N–C bonds corresponds to an in-plane inversion of the NNC angle between the azo bond and the first carbon of the benzene ring (angle Φ), reaching 180°, while the angle of CNNC remains fixed at 0°. (ii) Through the rotation of the N=N double bond. This mechanism is similar to the one in the stilbene isomerization [73]. The rotational pathway starts with the breakage of the N=N p-bond, thus becoming an N–N bond. After that, there is an out-of-plane rotation of the CNNC dihedral (φ) angle, while the CNN angle remains at 120°. (iii) Through the concerted inversion [76], where there is a simultaneous increase in the NNC angles up to 180°. (iv) Lastly, through inversion-assisted rotation mechanism, which implies changes in both CNNC and NNC angles at the same time.
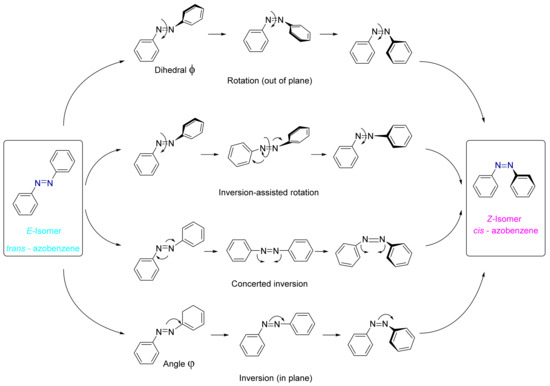
Figure 6.
Schematic view of four different isomerization mechanisms of azobenzene described in the literature.
Nevertheless, recent computational studies [75] using ab initio methodology including dynamic electron correlation have precisely depicted the topography of the potential energy surfaces of the S1 (n–π*) and S2 (π–π*) energy levels. The inclusion of the dynamic correlation makes this study more reliable as the topography of the PES can be highly altered when it is not included, thus providing contradictory data because of changes in the location of the minima and shape of the reaction paths. A high-level computational study [75] at the CASSCF/CASPT2 (Complete Active Space Self Consistent Field/CAS second-order perturbation theory) level, reoptimizing some of the critical points of the excited states at the MS-CASPT2 level, was performed. The mechanisms described fit into the different reaction pathways depending on the isomer excited and the excited state reached (see Figure 7). The inversion mechanism is the most likely pathway for the cis to trans thermal-back isomerization, but for the trans to cis case, depending on the excitation, different paths are available.
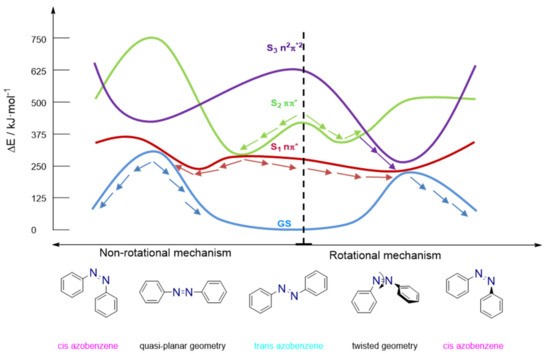
Figure 7.
Different electronic states and paths involved in the free-rotation and restricted rotation of azobenzene derivatives. Adapted from Reguero et al. [75].
When an azobenzene is promoted to the S1 state and rotation is not hindered by substituents, a CNNC rotation will take place, reaching the S1 potential energy surface minimum. An available conical intersection between this excited state and the ground state is located close in terms of geometrical distortion and energy, usually above by just 4–8 kJ/mol. Therefore, the population is funneled to the ground-state surface, yielding photoisomerization. For the excitation to S2, an energy degenerated region is usually present, thus leading to the same rotational pathway discussed for S1. However, alternative pathways can lead to photoisomerization back to the starting material. All of these processes produce differences in the quantum yield observed [76] depending on the excited state reached and the geometries allowed. Consequently, the PSS composition and the photoisomerization quantum yield become largely dependent on the substitution of the phenyl rings of the azobenzene and the specific irradiation wavelength used.
As shown in previous paragraphs, substitution has a major impact on the properties related to the MOST concept. Azobenzene derivatives can be classified into three different groups depending on the energetic order of its electronic states (π–π* and n–π*). This order is mainly dependent on the electronic nature of the azobenzene aromatic rings and their substitution pattern. The three classes are usually labelled as follows (see Figure 8a): (i) Azobenzene-type molecules, in which the electronic nature of the phenyl rings is analogous to that existing in the unsubstituted azobenzene (Ph-N=N-Ph). They present a strong π–π* band in the UV region and a weak n–π* band in the yellow visible region. (ii) Aminoazobenzenes, which can be ortho- or para-substituted with an electron-donating group (EDG), [(para- or ortho-(EDG)-C6H4-N=N-Ar)]. The π–π* and n–π* bands are very close or even overlap in the UV/Vis region; they are typically orange. (iii) Pseudo stilbenes, which have an electron-donating and an electron-withdrawing group (EWG) at the para position of the phenyl rings, thus being a push–pull system [(p-(EDG)-C6H4-N=N-C6H4-p-(EWG)]. They are typically red and the absorption band corresponding to the π–π* electronic transition undergoes a bathochromic shift, overlapping bands or even changing the order of appearance with the corresponding band of the n–π* electronic transition.
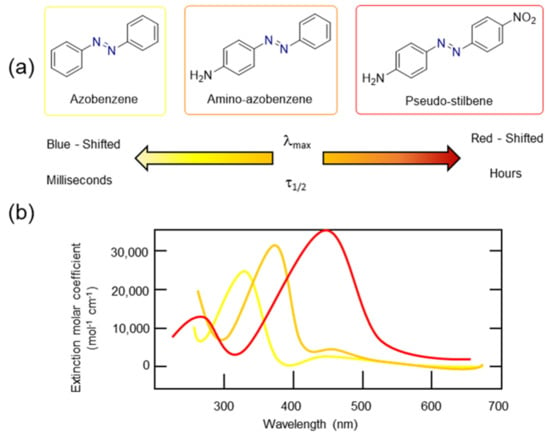
Figure 8.
(a) Azobenzene classification by Rau et al. [77] helps describe the optical and physical properties of this family of compounds (i.e., their color). (b) Typical example of absorption spectra of the different azobenzene derivatives.
Pseudo-stilbenes have a highly asymmetric electron distribution alongside the molecule, which turns into a large molecular dipole and anisotropic optical properties. In addition, this class also presents the best photo-response, which is mainly caused by an overlapped absorption spectrum of the E and Z forms, thus reaching a mixed photo-stationary state where the trans and cis isomers are continuously interconverting. Therefore, for pseudo-stilbenes, a single visible light wavelength can induce both forward and reverse isomerization. Thermal relaxation from cis to trans spans from milliseconds to seconds.
On the other hand, in the other two classes of compounds, the absorption spectra usually do not overlap, meaning that two different wavelengths are needed to switch between the cis and trans forms, which is ideal for photoswitchable materials. Particularly, azobenzene-type molecules are proven to isomerize back from cis into trans isomers very slowly through thermal relaxation when bulky substituents are introduced into the structure, thus providing a thermal relaxation time from cis to trans isomers of hours for the azobenzene-type molecules and minutes for the amino-azobenzene-type molecules. Recently, it has even been proven that some stable cis-isomers can be isolated using the proper solvent, which leads to a stable two-state photoswitchable system.
Even if the optical properties of azobenzenes discussed in previous paragraphs are quite interesting for MOST applications, the applicability of these compounds is somewhat limited by the energy density. The energy difference of a typical azobenzene is usually around 40 kJ/mol, which does not outcompete other systems. In addition, spectral overlap and the increased molecular weight to obtain excellent optical properties have led to a relative decrease in the interest of these compounds [16]. However, the broad tunability and the synthetic availability of different compounds achieved in the last two decades have reactivated their use as MOST candidates. Additionally, the wider scope obtained by azoheteroarenes, in which one of two phenyl rings are modified by a heterocyclic ring, can exponentially increase its possibilities by tuning some of the relevant properties.
For instance, we have already discussed that the photosiomerization quantum yield should be as high as possible [78,79]. Thus, this parameter has been subject to extensive research. The effect of various solvents, temperatures, and rigid environments was explored. Zimmerman et al. reported that n–π* quantum yield is more efficient than π–π* in both directions of conversion (cis-trans/trans-cis) and, in accordance with Birnbaum and Style, the absorption in the cis-trans conversion is more efficient (cis-trans: 0.39, while trans-cis: 0.33) [80,81,82]. This should be carefully considered when designing MOST applications based on azobenzenes. On the other hand, the ability of azobenzenes to absorb light in the visible region to induce the photoisomerization has been considered a clear advantage of these compounds [83].
The poor performance of azobenzenes with respect to energy storage has been addressed differently. In 2014, Grossman and Kolpak performed a series of density functional theory (DFT) computational studies of the azobenzene coupled with carbon nanotubes (CNTs) (Figure 9a). The addition of nanotubes to azobenzene increases the rigidity and conformational hindrance of the structures, so the energy storage per azobenzene increased up to 30% and the storage lifetimes almost reached the unit, in addition to increased fatigue resistance (a half-life (t1/2) of 33 h was observed for the cis isomer) [84].
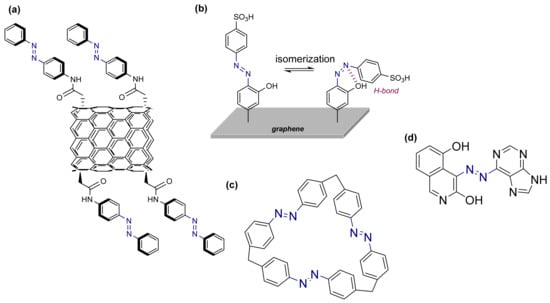
Figure 9.
(a) Azobenzene coupled with carbon nanotubes. (b) (Z)-isomer stabilized by the substitution of the azobenzene. (c) Azobenzene macrocycles. (d) Azoheteroarene derivative.
Later, the same authors extended their DFT study, incorporating azobenzenes functionalized with carbon-based templates such as graphene, fullerene, and β-carotene, identifying various potential molecules that have improved properties. Upon modification, the cis-isomer is stabilized by intramolecular hydrogen bonds (Figure 9b), leading to long-term storage lifetimes (t1/2 = 5408 h) [85,86]. In view of these results, different experimental groups carried out the synthesis of the photoswitches, obtaining good results [87,88]. Specifically, they obtained molecules with 11–12 carbon atoms per azobenzene and the hydrogen bond stabilization of the azobenzenes was confirmed by NMR, FT-IR, and DFT studies.
In a different approach, the addition of azobenzene to macrocycles [89] has also been suggested to improve energy storage capabilities [90]. The use of rings formed by azobenzenes connected through a suitable linker agent increase the barrier of the back reaction (Figure 9c). In this approach, the formation of the macrocycle contributes to increasing the rigidity of the system. In addition, it is possible to add functional groups to the macrocycle, as done in the graphene, allowing to establish hydrogen bonds with the aim of obtaining longer lifetimes for the energy storage.
Even if azobenzene-functionalized CNTs have increased the energy density, these systems cannot be deposited into uniform films. To avoid this problem, an azopolymer was prepared to act as solid-state solar-thermal fuel (STF) [91]. This novel design allows to prepare uniform films. This device allowed to increase the temperature of the heat generated in the back conversion by up to 180 °C [86]. Following this breakthrough, different studies have been conducted on azopolymers [92,93] because of the ease of application of the photoswitches in large areas. Additionally, different studies attempted to combine the rigidity obtained by coupling a polymer and the addition of functional groups to generate non-covalent interactions to the azobenzene, with the aim of stabilizing the azopolymers. Non-covalent interactions have also demonstrated an increase in the lifetime of the stored energy, such as π–π stacking and hydrogen bonds. Taking advantage of these interactions, a new mechanism to synthesize azopolymers controlling the thickness and film morphology by electrodeposition was described. Unfortunately, when the substitution of the polymers is modified, the energy storage is greatly affected [94].
It is known that preparing azobenzenes with very bulky substituents increases the rigidity and, therefore, the lifetime of energy storage. However, different and simpler systems continued to be sought in parallel. In 2017, Grossman and co-workers demonstrated that it was not necessary to use templates or even polymers to achieve SFT materials with high-energy density and thermal stability. In this study, they synthesized various azobenzenes substituted with bulky aromatic rings, obtaining an enthalpy difference between the cis and trans isomers comparable to the unsubstituted azobenzene. A critical factor to improve the azobenzene properties was to distort the molecule to avoid planarity, demonstrating that using small molecules made it possible to generate thin films with excellent charging and cycling properties [95].
Lately, a novel approach implying the use of azoheteroarene photoswitches has been explored. In various experimental and computational studies, it was discovered that changing the type of heteroaromatic ring or the position of the heteroatom with respect to the azo group had a crucial effect on the photoswitching properties [35,96]. Using this approach, it was possible to prepare molecules with a half-life of days or even years, providing an excellent alternative to use these compounds in a MOST system. The increase in the half-lifetime in the photoswitches is due to the formation of an intramolecular hydrogen bond affecting the energy difference between isomers, and thus the energy storage (Figure 9d). However, not all studied azoheteroarenes absorb in the visible region, making their use for energy storage problematic. Again, the long list of features that a system should fulfil to be of practical use in the MOST technology makes it extremely difficult to select the ideal candidate. However, the ever-growing list of available azoheteroarenes turns these compounds into excellent candidates to absorb and store sunlight in MOST devices.
3.3. Dihdroazulene–Vinylheptafluvene (DHA–VHF)
Another system widely studied in the context of MOST applications is the dihdroazulene/vinylheptafulvene (DHA/VHF) couple. While these compounds have some practical issues that hamper their applied use, they also feature some interesting properties. The optical properties of this system imply a DHA absorption of ca. 350 nm and a band at 480 nm for VHF, which also includes an isosbestic point at 400 nm with a very low overlap between both species. The photoisomerization quantum yield is around 0.6, which could be considered a good value for practical applications, although clearly lower than the better examples of NBD/QC, but by far better in comparison with NBD (0.05) or azobenzene (0.33). However, the main drawback for this couple is the short half-life time of parent VHF, being only 10 h at 25 °C. This should rule out its use for a long-term storage application, but others implying daily cycles (as charging during the day and releasing at night) can be considered as an alternative. On the other hand, it has shown some promising features in certain application tests. For instance, it has been used on solar and fluidic devices to provide an efficiency of up to 0.13% in non-laboratory experiments, considering the total solar income harvested. In addition, DHA/VHF also has a very high robustness against degradation, featuring less than 0.01% degradation in 70 irradiation back conversion cycles in toluene solutions [2,17]. Another drawback of this system is the low-energy storage capacity of DHA/VHF, which present an energy difference of ca. 28 kJ/mol, which is quite modest in comparison with NBD derivatives.
The general energy landscape of this couple can be seen in Figure 10. Upon light absorption, DHA isomerizes to VHF through a photoinduced carbon–carbon ring-opening reaction. In the excited state, DHA planarizes, allowing the stereochemical conditions to open the ring. This is favored by an increased steric hindrance due to the electronic reorganization. After the photochemical step, two possible isomers can be obtained. The initially formed cis-VHF can further evolve to the thermodynamically stable trans-VHF [38], with both species being in equilibrium through a small energy barrier [97]. This behaviour can facilitate an early back reversion to DHA, decreasing the lifetime of the photoisomer and affecting the energy density at the same time. In addition, the enthalpy of the photoreaction was measured to be ca. 35.2 kJ/mol in a vacuum [2].
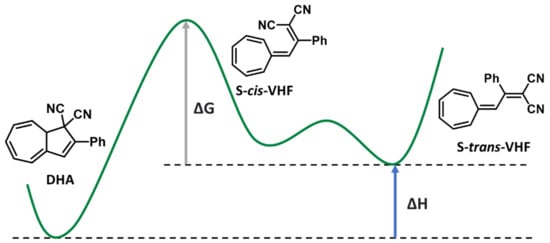
Figure 10.
Dihydroazulene (DHA)/vinylhetafulvene (VHF) couple and potential energy landscape. Adapted from Nielsen et al. [40].
Another relevant point is the large solvent effect that has been observed for the DHA/VHF couple. The photoreaction from DHA to VHF is faster in polar solvents than in apolar ones [98]. In contrast, the robustness through different irradiation conditions decreases notably in polar solvents (0.18–0.28%) compared with in apolar ones like toluene (0.01%). From these studies, some differences in the kinetic parameters have been invoked to explain the stabilization of charge-separated species, which acts as an intermediate during the photochemical transformation [39,98]. On the other hand, DHA is 10 times more soluble in toluene (apolar and aprotic) than in ethanol (polar and protic) and the photoisomerization quantum yield is larger (0.6) in toluene than in ethanol (0.5), being intermediate in acetonitrile (0.55). To sum up these effects, the more polar the solvent, the better the kinetic parameters, but, in contrast, an additional enhancement of system robustness is found with apolar solvents.
Along the years, many different studies were performed to better understand and improve the DHA capabilities, aiming at a better performance. The effect of some vicinal chemical modifications over the photoisomerization from DHA to VHF was improved in the 90s, usually by adding some donor or acceptor electronic groups on para positions of the phenyl ring. This yields in a moderate modification of the photoreaction quantum yield, ranging from 0.6 to 0.3 when increasing the electron-withdrawing strength of the substituent in the para position [98].
More recently, some extensive computational analysis was performed for the screening of interesting new candidates. In this work [40], an impressively wide number of chemical modifications has been performed on DHA systems to tune some of their properties. The most relevant and effective ones are summarized in Figure 11, whereby the main interest was the substitutions in positions 1–3 and 7, according to the numbering seen below for DHA systems.
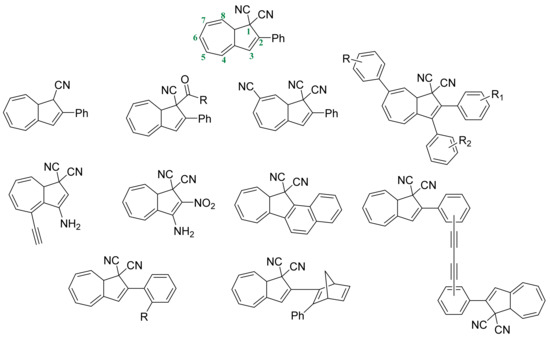
Figure 11.
Numbered positions and more relevant modifications in DHA structure, yielding some type of improvement.
One of the first modifications was to check the importance of the CN groups in position 1 and, as can be expected according to the mechanism, the elimination of this strong electron-withdrawing group causes a lack of photoswitching capacity, proving that the strong electrophilic behavior of C1 is crucial in the charge transfer intermediate on the photochemical transformation. As the elimination lacks the photoisomerization, some interesting attempts to replace one of the CN with other EWGs were carried out [99]. To finalize, the substitution of one CN by an ethoxycarbonyl or carbamoyl group can maintain the photoswitching ability and enhance the energy storage capacity, increasing it by 0.05–0.1 MJ/kg on average, being a relevant increment, also considering the higher molecular weight. As counterpoint, the predicted ΔG is lower than that found in base DHA, facilitating the back conversion. Moreover, with these systems, the solvent dependence seems to be slightly lower, even being quite representative.
The modulation on the stability of VHF and the back reversion speed can be controlled by the modification of positions 2, 3, and 7; herein, the effect of donor- and electron-withdrawing groups can drastically change some properties.
The inclusion of a donor group at C2 and an acceptor group at C3 or C7 have some impact, increasing the lifetime of these derivatives [100,101]. The addition of electron-withdrawing groups (EWGs), like cyano, in position 7 can increase the half-life of VHF by up to six times, in some cases reaching an exceptionally long-lived VHF in acetonitrile. This behavior can be explained by the stabilization of the VHF form, increasing the energy needed to initiate the back-reaction, but, in contrast, having a low impact on the energy storage capacity.
The inclusion of an amino group in position 3 yields a hydrogen bond between the amino and the electron-withdrawing groups (CN). This seems to stabilize the DHA system, increasing the storage energy, also blocking VHF in the s-cis-VHF conformer. This means that the s-cis-VHF becomes the most stable isomer of VHF. Thus, geometrically, it is more similar to the intermediate and, as expected, the back-reaction barrier becomes even lower. This behavior reduces its usability because of the fast reversion achieved despite the higher energy stored. In contrast, the addition of a donor group (amino) on position 3 and an acceptor group (NO2) at C2 produces an increase in the ΔG of the back-reaction, also lengthening the lifetime of the VHF form [40].
Some other modifications, like the condensed effect of adding a 9-anthryl group in position 2 and 3, were found to increase the storage density to 0.38 MJ/kg, but this facilitates the VHF-to-DHA ring closure in a few seconds [46].
The addition of bulky groups on the ortho position of the phenyl group in C2 can stabilize the cis conformer of VHF, increasing the energetic barrier of the back-reaction [102]. The quantum yields of photoisomerization for these ortho substituted derivatives range in acetonitrile close to DHA (0.55) values, thus improving the lifetime without perturbing the photoisomerization rate. This effect is only with an iodine atom; if the size of the added group increases, this effect is maximized, reaching a 60 times longer half-life and quantum yields slightly higher than DHA of ca. 0.6–0.7.
The last modification that we will comment about is the preparation of multi-switcher devices; on these, the combination of two DHA moieties, connected through a bisacetylene bridge, can differ too much depending on the substitution in ortho, meta, and para, ranging from an increase in the lifetime from a few hours to a couple of days [102].
In addition, combinations with other MOST candidates were attempted, offering some promising results, especially the increase in storage density due to the charge of two molecules, and in case of DHA-NBD, the combined absorption is notably red-shifted and the spectral overlap is decreased [103]. Moreover, the use of an azobenzene derivative together with phenylene bridge bis-DHA moieties, all included in a macrocyclic structure, can yield a modification of the DHA isomerization because of the predominant azobenzene isomerization found. This can limit the possibilities of combining azobenzenes and DHA moieties [104].
A principal outlook towards the use of DHA systems presents a few advantages, such as efficient isomerization and good optical properties, as well as a series of drawbacks including the modest energy storage and short lifetimes.
4. Conclusions
In this review, we have covered the three more relevant families of compounds that are under investigation as MOST systems, norbornadiene, azobenzene, and dihydroazulene derivatives. Every set of compounds has its own drawbacks and strengths, which should be carefully considered for any specific application. However, it is also relevant to recognize that we are still far from finding the ideal MOST candidate. The mentioned improvements in the molecular design constitute the basis for the future development of these systems. In addition, the use of molecular modelling and machine learning strategies can provide a fast and valuable input in this way [105]. A general increase in the performance of the molecules used for solar energy storage is required before this technology could provide an alternative and efficient way of harvesting and storing solar energy, as well as its use and release on demand. In the near future of MOST devices, the exploitation of hybrid strategies (multijunction devices) is the more promising field to improve the overall performance.
Author Contributions
Bibliographic search and writing—original draft preparation, A.G.-G., L.M., N.S., B.P. and J.S.; writing—review and editing, R.L. and D.S.; supervision, R.L. and D.S.; funding acquisition, D.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Spanish Ministerio de Ciencia e Innovación, grant number PID2021-126075NB-I00.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
A.G.-G., L.M., N.S., and B.P. thank the European Union’s H2020 research and innovation program under grant agreement No 951801. R.L. thanks Universidad de La Rioja and Ministerio de Universidades for his Margarita Salas grant.
Conflicts of Interest
The authors declare no conflict of interest.
References
- de Amorim, W.S.; Valduga, I.B.; Ribeiro, J.M.P.; Williamson, V.G.; Krauser, G.E.; Magtoto, M.K.; de Andrade Guerra, J.B.S.O. The nexus between water, energy, and food in the context of the global risks: An analysis of the interactions between food, water, and energy security. Environ. Impact Assess. Rev. 2018, 72, 1–11. [Google Scholar] [CrossRef]
- Sun, C.-L.; Wang, C.; Boulatov, R. Applications of Photoswitches in the Storage of Solar Energy. ChemPhotoChem 2019, 3, 268–283. [Google Scholar] [CrossRef]
- Wang, Z.; Erhart, P.; Li, T.; Zhang, Z.-Y.; Sampedro, D.; Hu, Z.; Wegner, H.A.; Brummel, O.; Libuda, J.; Nielsen, M.B.; et al. Storing energy with molecular photoisomers. Joule 2021, 5, 3116–3136. [Google Scholar] [CrossRef]
- Lee, D.S.; Fahey, D.W.; Skowron, A.; Allen, M.R.; Burkhardt, U.; Chen, Q.; Doherty, S.J.; Freeman, S.; Forster, P.M.; Fuglestvedt, J.; et al. The contribution of global aviation to anthropogenic climate forcing for 2000 to 2018. Atmos. Environ. 2021, 244, 117834. [Google Scholar] [CrossRef]
- IEA. Global Status Report for Buildings and Construction; International Energy Agency: Paris, France, 2019. [Google Scholar]
- Rabaia, M.K.H.; Abdelkareem, M.A.; Sayed, E.T.; Elsaid, K.; Chae, K.J.; Wilberforce, T.; Olabi, A.G. Environmental impacts of solar energy systems: A review. Sci. Total Environ. 2021, 754, 141989. [Google Scholar] [CrossRef]
- Xu, X.; Wang, G. Molecular Solar Thermal Systems towards Phase Change and Visible Light Photon Energy Storage. Small 2022, 18, 2107473. [Google Scholar] [CrossRef]
- Ma, Q.; Wang, P.; Fan, J.; Klar, A. Underground solar energy storage via energy piles: An experimental study. Appl. Energy 2022, 306, 118042. [Google Scholar] [CrossRef]
- Ma, Q.; Wang, P. Underground solar energy storage via energy piles. Appl. Energy 2020, 261, 114361. [Google Scholar] [CrossRef]
- Wu, D.; Kong, G.; Liu, H.; Jiang, Q.; Yang, Q.; Kong, L. Performance of a full-scale energy pile for underground solar energy storage. Case Stud. Therm. Eng. 2021, 27, 101313. [Google Scholar] [CrossRef]
- Wang, H.; Qi, C. Performance study of underground thermal storage in a solar-ground coupled heat pump system for residential buildings. Energy Build. 2008, 40, 1278–1286. [Google Scholar] [CrossRef]
- Ciamician, G. The Photochemistry of the Future. Science 1912, 36, 385–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moth-Poulsen, K. Organic Synthesis and Molecular Engineering; Nielsen, M.B., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 179–196. [Google Scholar]
- Moth-Poulsen, K.; Ćoso, D.; Börjesson, K.; Vinokurov, N.; Meier, S.K.; Majumdar, A.; Vollhardt, K.P.C.; Segalman, R.A. Molecular solar thermal (MOST) energy storage and release system. Energy Environ. Sci. 2012, 5, 8534–8537. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; He, Y.; Wang, Z.; Xu, J.; Xie, M.; Tao, P.; Ji, D.; Moth-Poulsen, K.; Li, T. Photochemical Phase Transitions Enable Coharvesting of Photon Energy and Ambient Heat for Energetic Molecular Solar Thermal Batteries That Upgrade Thermal Energy. J. Am. Chem. Soc. 2020, 142, 12256–12264. [Google Scholar] [CrossRef] [PubMed]
- Lennartson, A.; Roffey, A.; Moth-Poulsen, K. Designing photoswitches for molecular solar thermal energy storage. Tetrahedron Lett. 2015, 56, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- Vlasceanu, A.; Broman, S.L.; Hansen, A.S.; Skov, A.B.; Cacciarini, M.; Kadziola, A.; Kjaergaard, H.G.; Mikkelsen, K.V.; Nielsen, M.B. Solar Thermal Energy Storage in a Photochromic Macrocycle. Chem.—A Eur. J. 2016, 22, 10796–10800. [Google Scholar] [CrossRef]
- Yoshida, Z.-i. New molecular energy storage systems. J. Photochem. 1985, 29, 27–40. [Google Scholar] [CrossRef]
- Gur, I.; Sawyer, K.; Prasher, R. Searching for a Better Thermal Battery. Science 2012, 335, 1454–1455. [Google Scholar] [CrossRef]
- Bren, V.A.; Dubonosov, A.D.; Minkin, V.I.; Chernoivanov, V.A. Norbornadiene–quadricyclane—An effective molecular system for the storage of solar energy. Russ. Chem. Rev. 1991, 60, 451–469. [Google Scholar] [CrossRef]
- Philippopoulos, C.; Marangozis, J. Kinetics and efficiency of solar energy storage in the photochemical isomerization of norbornadiene to quadricyclane. Ind. Eng. Chem. Prod. Res. Dev. 1984, 23, 458–466. [Google Scholar] [CrossRef]
- Börjesson, K.; Lennartson, A.; Moth-Poulsen, K. Efficiency Limit of Molecular Solar Thermal Energy Collecting Devices. ACS Sustain. Chem. Eng. 2013, 1, 585–590. [Google Scholar] [CrossRef]
- Fei, L.; Yin, Y.; Yang, M.; Zhang, S.; Wang, C. Wearable solar energy management based on visible solar thermal energy storage for full solar spectrum utilization. Energy Storage Mater. 2021, 42, 636–644. [Google Scholar] [CrossRef]
- Mansø, M.; Petersen, A.U.; Wang, Z.; Erhart, P.; Nielsen, M.B.; Moth-Poulsen, K. Molecular solar thermal energy storage in photoswitch oligomers increases energy densities and storage times. Nat. Commun. 2018, 9, 1945. [Google Scholar] [CrossRef] [PubMed]
- Quant, M.; Lennartson, A.; Dreos, A.; Kuisma, M.; Erhart, P.; Börjesson, K.; Moth-Poulsen, K. Low Molecular Weight Norbornadiene Derivatives for Molecular Solar-Thermal Energy Storage. Chem.—A Eur. J. 2016, 22, 13265–13274. [Google Scholar] [CrossRef] [PubMed]
- Gray, V.; Lennartson, A.; Ratanalert, P.; Börjesson, K.; Moth-Poulsen, K. Diaryl-substituted norbornadienes with red-shifted absorption for molecular solar thermal energy storage. Chem. Commun. 2014, 50, 5330–5332. [Google Scholar] [CrossRef]
- Kuisma, M.; Lundin, A.; Moth-Poulsen, K.; Hyldgaard, P.; Erhart, P. Optimization of Norbornadiene Compounds for Solar Thermal Storage by First-Principles Calculations. ChemSusChem 2016, 9, 1786–1794. [Google Scholar] [CrossRef]
- Solmuş, İ.; Kaftanoğlu, B.; Yamalı, C.; Baker, D. Experimental investigation of a natural zeolite–water adsorption cooling unit. Appl. Energy 2011, 88, 4206–4213. [Google Scholar] [CrossRef]
- Liu, Y.; Leong, K.C. Numerical modeling of a zeolite/water adsorption cooling system with non-constant condensing pressure. Int. Commun. Heat Mass Transf. 2008, 35, 618–622. [Google Scholar] [CrossRef]
- Vollhardt, K.P.C.; Weidman, T.W. Synthesis, structure, and photochemistry of tetracarbonyl(fulvalene)diruthenium. Thermally reversible photoisomerization involving carbon-carbon bond activation at a dimetal center. J. Am. Chem. Soc. 1983, 105, 1676–1677. [Google Scholar] [CrossRef]
- Schwerzel, R.E.; Klosterman, N.E.; Kelly, J.R.; Hillenbrand, L.J. Catalytic Extraction of Stored Solar Energy from Photochemicals. U.S. Patent 4105014/1978, 1978. [Google Scholar]
- Blanco-Lomas, M.; Martínez-López, D.; Campos, P.J.; Sampedro, D. Tuning of the properties of rhodopsin-based molecular switches. Tetrahedron Lett. 2014, 55, 3361–3364. [Google Scholar] [CrossRef]
- Martinez-Lopez, D.; Yu, M.L.; Garcia-Iriepa, C.; Campos, P.J.; Frutos, L.M.; Golen, J.A.; Rasapalli, S.; Sampedro, D. Hydantoin-based molecular photoswitches. J. Org. Chem. 2015, 80, 3929–3939. [Google Scholar] [CrossRef]
- Bastianelli, C.; Caia, V.; Cum, G.; Gallo, R.; Mancini, V. Thermal isomerization of photochemically synthesized (Z)-9-styrylacridines. An unusually high enthalpy of Z→E conversion for stilbene-like compounds. J. Chem. Soc. Perkin Trans. 2 1991, 22, 679–683. [Google Scholar] [CrossRef]
- Losantos, R.; Sampedro, D. Design and Tuning of Photoswitches for Solar Energy Storage. Molecules 2021, 26, 3796. [Google Scholar] [CrossRef] [PubMed]
- Orrego-Hernández, J.; Hölzel, H.; Wang, Z.; Quant, M.; Moth-Poulsen, K. Norbornadiene/Quadricyclane (NBD/QC) and Conversion of Solar Energy. In Molecular Photoswitches; Wiley: Hoboken, NJ, USA, 2022; pp. 351–378. [Google Scholar]
- Jones, G.; Reinhardt, T.E.; Bergmark, W.R. Photon energy storage in organic materials—The case of linked anthracenes. Sol. Energy 1978, 20, 241–248. [Google Scholar] [CrossRef]
- Wang, Z.; Udmark, J.; Börjesson, K.; Rodrigues, R.; Roffey, A.; Abrahamsson, M.; Nielsen, M.B.; Moth-Poulsen, K. Evaluating Dihydroazulene/Vinylheptafulvene Photoswitches for Solar Energy Storage Applications. ChemSusChem 2017, 10, 3049–3055. [Google Scholar] [CrossRef] [PubMed]
- Broman, S.L.; Brand, S.L.; Parker, C.R.; Petersen, M.Å.; Tortzen, C.G.; Kadziola, A.; Kilså, K.; Nielsen, M.B. Optimized synthesis and detailed NMR spectroscopic characterization of the 1,8a-dihydroazulene-1,1-dicarbonitrile photoswitch. ARKIVOC 2011, 2011, 51–67. [Google Scholar] [CrossRef] [Green Version]
- Koerstz, M.; Christensen, A.S.; Mikkelsen, K.V.; Nielsen, M.B.; Jensen, J.H. High throughput virtual screening of 230 billion molecular solar heat battery candidates. PeerJ Phys. Chem. 2021, 3, e16. [Google Scholar] [CrossRef]
- Börjesson, K.; Ćoso, D.; Gray, V.; Grossman, J.C.; Guan, J.; Harris, C.B.; Hertkorn, N.; Hou, Z.; Kanai, Y.; Lee , D.; et al. Exploring the Potential of Fulvalene Dimetals as Platforms for Molecular Solar Thermal Energy Storage: Computations, Syntheses, Structures, Kinetics, and Catalysis. Chem. Eur. J. 2014, 20, 15587–15604. [Google Scholar] [CrossRef]
- Kucharski, T.J.; Tian, Y.; Akbulatov, S.; Boulatov, R. Chemical solutions for the closed-cycle storage of solar energy. Energy Environ. Sci. 2011, 4, 4449–4472. [Google Scholar] [CrossRef]
- International, A. A International in Standard Tables for Reference Solar Spectral Irradiances: Direct Normal and Hemispherical on 37° Tilted Surface; ASTM International: West Conshohocken, PA, USA, 2012. [Google Scholar]
- Jorner, K.; Dreos, A.; Emanuelsson, R.; El Bakouri, O.; Galván, I.F.; Börjesson, K.; Feixas, F.; Lindh, R.; Zietz, B.; Moth-Poulsen, K.; et al. Unraveling factors leading to efficient norbornadiene–quadricyclane molecular solar-thermal energy storage systems. J. Mater. Chem. A 2017, 5, 12369–12378. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Z.; Cole, J.M. Molecular Design of UV–vis Absorption and Emission Properties in Organic Fluorophores: Toward Larger Bathochromic Shifts, Enhanced Molar Extinction Coefficients, and Greater Stokes Shifts. J. Phys. Chem. C 2013, 117, 16584–16595. [Google Scholar] [CrossRef]
- Skov, A.B.; Broman, S.L.; Gertsen, A.S.; Elm, J.; Jevric, M.; Cacciarini, M.; Kadziola, A.; Mikkelsen, K.V.; Nielsen, M.B. Aromaticity-Controlled Energy Storage Capacity of the Dihydroazulene-Vinylheptafulvene Photochromic System. Chem.—A Eur. J. 2016, 22, 14567–14575. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.U.; Hofmann, A.I.; Fillols, M.; Mansø, M.; Jevric, M.; Wang, Z.; Sumby, C.J.; Müller, C.; Moth-Poulsen, K. Solar Energy Storage by Molecular Norbornadiene–Quadricyclane Photoswitches: Polymer Film Devices. Adv. Sci. 2019, 6, 1900367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bas, E.E.; Ulukan, P.; Monari, A.; Aviyente, V.; Catak, S. Photophysical Properties of Benzophenone-Based TADF Emitters in Relation to Their Molecular Structure. J. Phys. Chem. A 2022, 4, 473–484. [Google Scholar] [CrossRef]
- Meng, F.-Y.; Chen, I.-H.; Shen, J.-Y.; Chang, K.-H.; Chou, T.-C.; Chen, Y.-A.; Chen, Y.-T.; Chen, C.-L.; Chou, P.-T. A new approach exploiting thermally activated delayed fluorescence molecules to optimize solar thermal energy storage. Nat. Commun. 2022, 13, 797. [Google Scholar] [CrossRef]
- Wang, Z.; Roffey, A.; Losantos, R.; Lennartson, A.; Jevric, M.; Petersen, A.U.; Quant, M.; Dreos, A.; Wen, X.; Sampedro, D.; et al. Macroscopic heat release in a molecular solar thermal energy storage system. Energy Environ. Sci. 2019, 12, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Dubonosov, A.D.; Bren, V.A.; Chernoivanov, V.A. Norbornadiene–quadricyclane as an abiotic system for the storage of solar energy. Russ. Chem. Rev. 2002, 71, 917–927. [Google Scholar] [CrossRef]
- Harel, Y.; Adamson, A.W.; Kutal, C.; Grutsch, P.A.; Yasufuku, K. Photocalorimetry. 6. Enthalpies of isomerization of norbornadiene and of substituted norbornadienes to corresponding quadricyclenes. J. Phys. Chem. 1987, 91, 901–904. [Google Scholar] [CrossRef]
- Dilling, W.L. Intramolecular Photochemical Cycloaddition of Nonconjugated Olefins. Chem. Rev. 1966, 66, 373–393. [Google Scholar] [CrossRef]
- Sadao, M.; Yoshinobu, A.; Zen-ichi, Y. Photochromic Solid Films Prepared by Doping with Donor–Acceptor Norbornadienes. Chem. Lett. 1987, 16, 195–198. [Google Scholar]
- Jevric, M.; Petersen, A.U.; Mansø, M.; Kumar Singh, S.; Wang, Z.; Dreos, A.; Sumby, C.; Nielsen, M.B.; Börjesson, K.; Erhart, P.; et al. Norbornadiene-Based Photoswitches with Exceptional Combination of Solar Spectrum Match and Long-Term Energy Storage. Chem.—A Eur. J. 2018, 24, 12767–12772. [Google Scholar] [CrossRef]
- Mitscherlich, E. Ueber das Stickstoffbenzid. Ann. Der Phys. 1834, 108, 225–227. [Google Scholar] [CrossRef] [Green Version]
- Noble, A. III. Zur Geschichte des Azobenzols und des Benzidins. Justus Liebigs Ann. Der Chem. 1856, 98, 253–256. [Google Scholar] [CrossRef] [Green Version]
- Hartley, G.S. The Cis-form of Azobenzene. Nature 1937, 140, 281. [Google Scholar] [CrossRef]
- Durr, H.; Bouas-Laurent, H. Photochromism: Molecules and Systems; Elsevier Science: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Turanský, R.; Konôpka, M.; Doltsinis, N.L.; Ŝtich, I.; Marx, D. Switching of functionalized azobenzene suspended between gold tips by mechanochemical, photochemical, and opto-mechanical means. Phys. Chem. Chem. Phys. 2010, 12, 13922–13932. [Google Scholar] [CrossRef] [PubMed]
- Henzl, J.; Mehlhorn, M.; Gawronski, H.; Rieder, K.H.; Morgenstern, K. Reversible cis-trans isomerization of a single azobenzene molecule. Angew. Chem.-Int. Ed. 2006, 45, 603–606. [Google Scholar] [CrossRef]
- Tong, X.; Pelletier, M.; Lasia, A.; Zhao, Y. Fast cis-trans isomerization of an azobenzene derivative in liquids and liquid crystals under a low electric field. Angew. Chem.-Int. Ed. 2008, 47, 3596–3599. [Google Scholar] [CrossRef]
- Crecca, C.R.; Roitberg, A.E. Theoretical study of the isomerization mechanism of azobenzene and disubstituted azobenzene derivatives. J. Phys. Chem. A 2006, 110, 8188–8203. [Google Scholar] [CrossRef]
- Brown, C.J. A refinement of the crystal structure of azobenzene. Acta Crystallogr. 1966, 21, 146–152. [Google Scholar] [CrossRef]
- Mostad, A.; Romming, C. A refinement of the crystal structure of cis-azobenzene. Acta Chem. Scand. 1971, 25, 3561–3568. [Google Scholar] [CrossRef]
- Gagliardi, L.; Orlandi, G.; Bernardi, F.; Cembran, A.; Garavelli, M. A theoretical study of the lowest electronic states of azobenzene: The role of torsion coordinate in the cis–trans photoisomerization. Theor. Chem. Acc. 2004, 111, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Sudesh, G.; Road, T.B.; Sciences, P.; Green, B. Photochemistry of Azobenzene-Containing Polymers. Chem. Rev. 1989, 89, 1915–1925. [Google Scholar]
- Sension, R.J.; Repinec, S.T.; Szarka, A.Z.; Hochstrasser, R.M. Femtosecond laser studies of the cis-stilbene photoisomerization reactions. J. Chem. Phys. 1993, 98, 6291–6315. [Google Scholar] [CrossRef]
- Hamm, P.; Ohline, S.M.; Zinth, W. Vibrational cooling after ultrafast photoisomerization of azobenzene measured by femtosecond infrared spectroscopy. J. Chem. Phys. 1997, 106, 519–529. [Google Scholar] [CrossRef]
- Georgiev, A.; Bubev, E.; Dimov, D.; Yancheva, D.; Zhivkov, I.; Krajčovič, J.; Vala, M.; Weiter, M.; Machkova, M. Synthesis, structure, spectral properties and DFT quantum chemical calculations of 4-aminoazobenzene dyes. Effect of intramolecular hydrogen bonding on photoisomerization. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 175, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Schulze, F.W.; Petrick, H.J.; Cammenga, H.K.; Klinge, H.Z. Thermodynamic properties of the structural analogues benzo[c]-cinnoline, trans-azobenzene and cis-azobenzene. Z. Phys. Chem. Neue Fol. 1997, 1, 107. [Google Scholar] [CrossRef]
- Brown, E.V.; Granneman, G.R. Cis-Trans Isomerism in the Pyridyl Analogs of Azobenzene. A Kinetic and Molecular Orbital Analysis. J. Am. Chem. Soc. 1975, 97, 621–627. [Google Scholar] [CrossRef]
- Tamai, N.; Miyasaka, H. Ultrafast Dynamics of Photochromic Systems. Chem. Rev. 2000, 100, 1875–1890. [Google Scholar] [CrossRef]
- Casellas, J.; Bearpark, M.J.; Reguero, M. Excited-State Decay in the Photoisomerisation of Azobenzene: A New Balance between Mechanisms. ChemPhysChem 2016, 17, 3068–3079. [Google Scholar] [CrossRef] [Green Version]
- Rau, H. Further evidence for rotation in the π,π* and inversion in the n,π* photoisomerization of azobenzenes. J. Photochem. 1984, 26, 221–225. [Google Scholar] [CrossRef]
- Fujino, T.; Arzhantsev, S.Y.; Tahara, T. Femtosecond Time-Resolved Fluorescence Study of Photoisomerization of trans-Azobenzene. J. Phys. Chem. A 2001, 105, 8123–8129. [Google Scholar] [CrossRef]
- Rau, H. Photochromism: Molecules and Systems, 1st ed.; Elsevier: Amsterdam, The Netherlands, 1990; pp. 165–192. [Google Scholar]
- Kunz, A.; Heindl, A.H.; Dreos, A.; Wang, Z.; Moth-Poulsen, K.; Becker, J.; Wegner, H.A. Intermolecular London Dispersion Interactions of Azobenzene Switches for Tuning Molecular Solar Thermal Energy Storage Systems. ChemPlusChem 2019, 84, 1145–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Losantos, R.; Sampedro, D.; Morikawa, M.-a.; Börjesson, K.; Kimizuka, N.; Moth-Poulsen, K. Demonstration of an azobenzene derivative based solar thermal energy storage system. J. Mater. Chem. A 2019, 7, 15042–15047. [Google Scholar] [CrossRef] [Green Version]
- Shigeru, Y.; Hiroshi, O.; Osamu, T. The cis-trans Photoisomerization of Azobenzene. Bull. Chem. Soc. Jpn. 1962, 35, 1849–1853. [Google Scholar]
- Zimmerman, G.; Chow, L.-Y.; Paik, U.-J. The Photochemical Isomerization of Azobenzene1. J. Am. Chem. Soc. 1958, 80, 3528–3531. [Google Scholar] [CrossRef]
- Ladányi, V.; Dvořák, P.; Al Anshori, J.; Vetráková, Ľ.; Wirz, J.; Heger, D. Azobenzene photoisomerization quantum yields in methanol redetermined. Photochem. Photobiol. Sci. 2017, 16, 1757–1761. [Google Scholar] [CrossRef]
- Mahimwalla, Z.; Yager, K.G.; Mamiya, J.-i.; Shishido, A.; Priimagi, A.; Barrett, C.J. Azobenzene photomechanics: Prospects and potential applications. Polym. Bull. 2012, 69, 967–1006. [Google Scholar] [CrossRef]
- Kolpak, A.M.; Grossman, J.C. Azobenzene-Functionalized Carbon Nanotubes As High-Energy Density Solar Thermal Fuels. Nano Lett. 2011, 11, 3156–3162. [Google Scholar] [CrossRef]
- Kucharski, T.J.; Ferralis, N.; Kolpak, A.M.; Zheng, J.O.; Nocera, D.G.; Grossman, J.C. Templated assembly of photoswitches significantly increases the energy-storage capacity of solar thermal fuels. Nat. Chem. 2014, 6, 441–447. [Google Scholar] [CrossRef]
- Wu, S.; Butt, H.-J. Solar-Thermal Energy Conversion and Storage Using Photoresponsive Azobenzene-Containing Polymers. Macromol. Rapid Commun. 2020, 41, 1900413. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Luo, W.; Feng, Y. Photo-responsive carbon nanomaterials functionalized by azobenzene moieties: Structures, properties and application. Nanoscale 2012, 4, 6118–6134. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, H.; Luo, W.; Liu, E.; Zhao, N.; Yoshino, K.; Feng, W. Covalent functionalization of graphene by azobenzene with molecular hydrogen bonds for long-term solar thermal storage. Sci. Rep. 2013, 3, 3260. [Google Scholar] [CrossRef] [PubMed]
- Norikane, Y.; Kitamoto, K.; Tamaoki, N. Novel crystal structure, cis-trans isomerization, and host property of meta-substituted macrocyclic azobenzenes with the shortest linkers. J. Org. Chem. 2003, 68, 8291–8304. [Google Scholar] [CrossRef]
- Durgun, E.; Grossman, J.C. Photoswitchable Molecular Rings for Solar-Thermal Energy Storage. J. Phys. Chem. Lett. 2013, 4, 854–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhitomirsky, D.; Cho, E.; Grossman, J.C. Solid-State Solar Thermal Fuels for Heat Release Applications. Adv. Energy Mater. 2016, 6, 1502006. [Google Scholar] [CrossRef]
- Lv, J.-A.; Liu, Y.; Wei, J.; Chen, E.; Qin, L.; Yu, Y. Photocontrol of fluid slugs in liquid crystal polymer microactuators. Nature 2016, 537, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.P.; Renna, L.A.; Boyle, C.J.; Kwak, H.S.; Harder, E.; Damm, W.; Venkataraman, D. High Energy Density in Azobenzene-based Materials for Photo-Thermal Batteries via Controlled Polymer Architecture and Polymer-Solvent Interactions. Sci. Rep. 2017, 7, 17773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhitomirsky, D.; Grossman, J.C. Conformal Electroplating of Azobenzene-Based Solar Thermal Fuels onto Large-Area and Fiber Geometries. ACS Appl. Mater. Interfaces 2016, 8, 26319–26325. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.N.; Zhitomirsky, D.; Han, G.G.D.; Liu, Y.; Grossman, J.C. Molecularly Engineered Azobenzene Derivatives for High Energy Density Solid-State Solar Thermal Fuels. ACS Appl. Mater. Interfaces 2017, 9, 8679–8687. [Google Scholar] [CrossRef]
- Calbo, J.; Weston, C.E.; White, A.J.P.; Rzepa, H.S.; Contreras-García, J.; Fuchter, M.J. Tuning Azoheteroarene Photoswitch Performance through Heteroaryl Design. J. Am. Chem. Soc. 2017, 139, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Saritas, K.; Grossman, J.C. Accurate Isomerization Enthalpy and Investigation of the Errors in Density Functional Theory for Dihydroazulene/Vinylheptafulvene Photochromism Using Diffusion Monte Carlo. J. Phys. Chem. C 2017, 121, 26677–26685. [Google Scholar] [CrossRef]
- Goerner, H.; Fischer, C.; Gierisch, S.; Daub, J. Dihydroazulene/vinylheptafulvene photochromism: Effects of substituents, solvent, and temperature in the photorearrangement of dihydroazulenes to vinylheptafulvenes. J. Phys. Chem. 1993, 97, 4110–4117. [Google Scholar] [CrossRef]
- Christensen, O.; Nielsen, L.E.; Johansen, J.; Holk, K.; Udmark, J.; Nielsen, M.B.; Cacciarini, M.; Mikkelsen, K.V. Density Functional Theory Study of Carbamoyl-Substituted Dihydroazulene/Vinylheptafulvene Derivatives and Solvent Effects. J. Phys. Chem. C 2022, 126, 4815–4825. [Google Scholar] [CrossRef]
- Broman, S.L.; Jevric, M.; Nielsen, M.B. Linear Free-Energy Correlations for the Vinylheptafulvene Ring Closure: A Probe for Hammett σ Values. Chem.—A Eur. J. 2013, 19, 9542–9548. [Google Scholar] [CrossRef] [PubMed]
- Kilde, M.D.; Hansen, M.H.; Broman, S.L.; Mikkelsen, K.V.; Nielsen, M.B. Expanding the Hammett Correlations for the Vinylheptafulvene Ring-Closure Reaction. Eur. J. Org. Chem. 2017, 2017, 1052–1062. [Google Scholar] [CrossRef]
- Ranzenigo, A.; Cordero, F.M.; Cacciarini, M.; Nielsen, M.B. ortho-Substituted 2-Phenyldihydroazulene Photoswitches: Enhancing the Lifetime of the Photoisomer by ortho-Aryl Interactions. Molecules 2021, 26, 6462. [Google Scholar] [CrossRef]
- Schøttler, C.; Vegge, S.K.; Cacciarini, M.; Nielsen, M.B. Long-Term Energy Storage Systems Based on the Dihydroazulene/Vinylheptafulvene Photo-/Thermoswitch. ChemPhotoChem 2022, 6, e202200037. [Google Scholar] [CrossRef]
- Abedi, M.; Pápai, M.; Henriksen, N.E.; Møller, K.B.; Nielsen, M.B.; Mikkelsen, K.V. Theoretical Investigation on the Control of Macrocyclic Dihydroazulene/Azobenzene Photoswitches. J. Phys. Chem. C 2019, 123, 25579–25584. [Google Scholar] [CrossRef]
- Wang, Z.; Hölzel, H.; Moth-Poulsen, K. Status and challenges for molecular solar thermal energy storage system based devices. Chem. Soc. Rev. 2022. Advance Article. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).