
Efficient Access of Phenyl-Spaced 5,5′-Bridged Dinuclear Ruthenium Metal Complexes and the Effect of Dynamic Ligand Exchange on Catalysis
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Steady-State UV/vis Absorption Spectroscopy
2.2. Steady-State Emission Spectroscopy
2.3. Time-Resolved Emission Studies
2.4. Electrochemistry
2.5. Mass Spectrometry
2.6. NMR Experiments
2.7. Hydrogen Determination
2.8. Preparation of Catalytic Mixture
2.9. Reactor Design
3. Results and Discussion
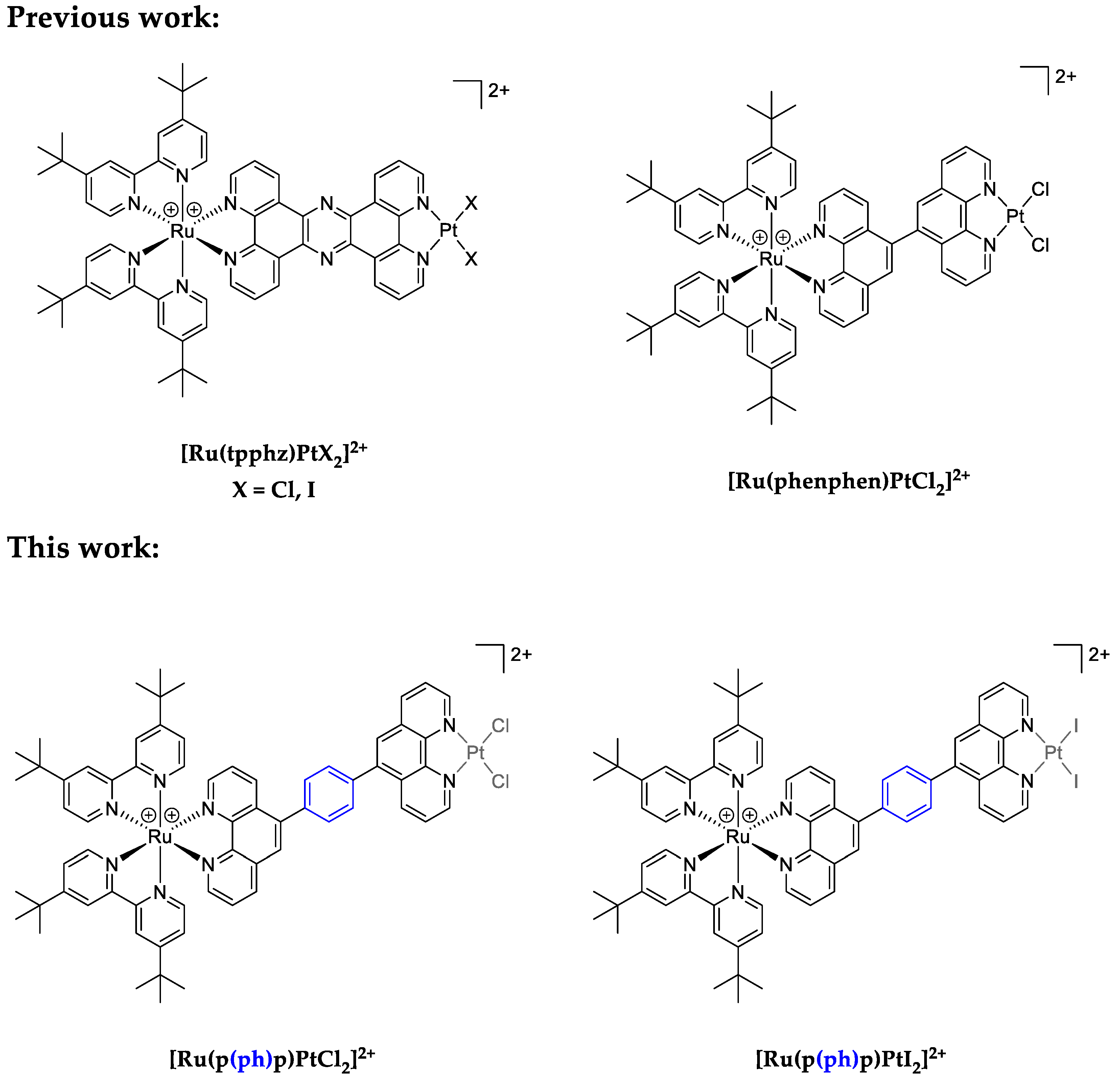
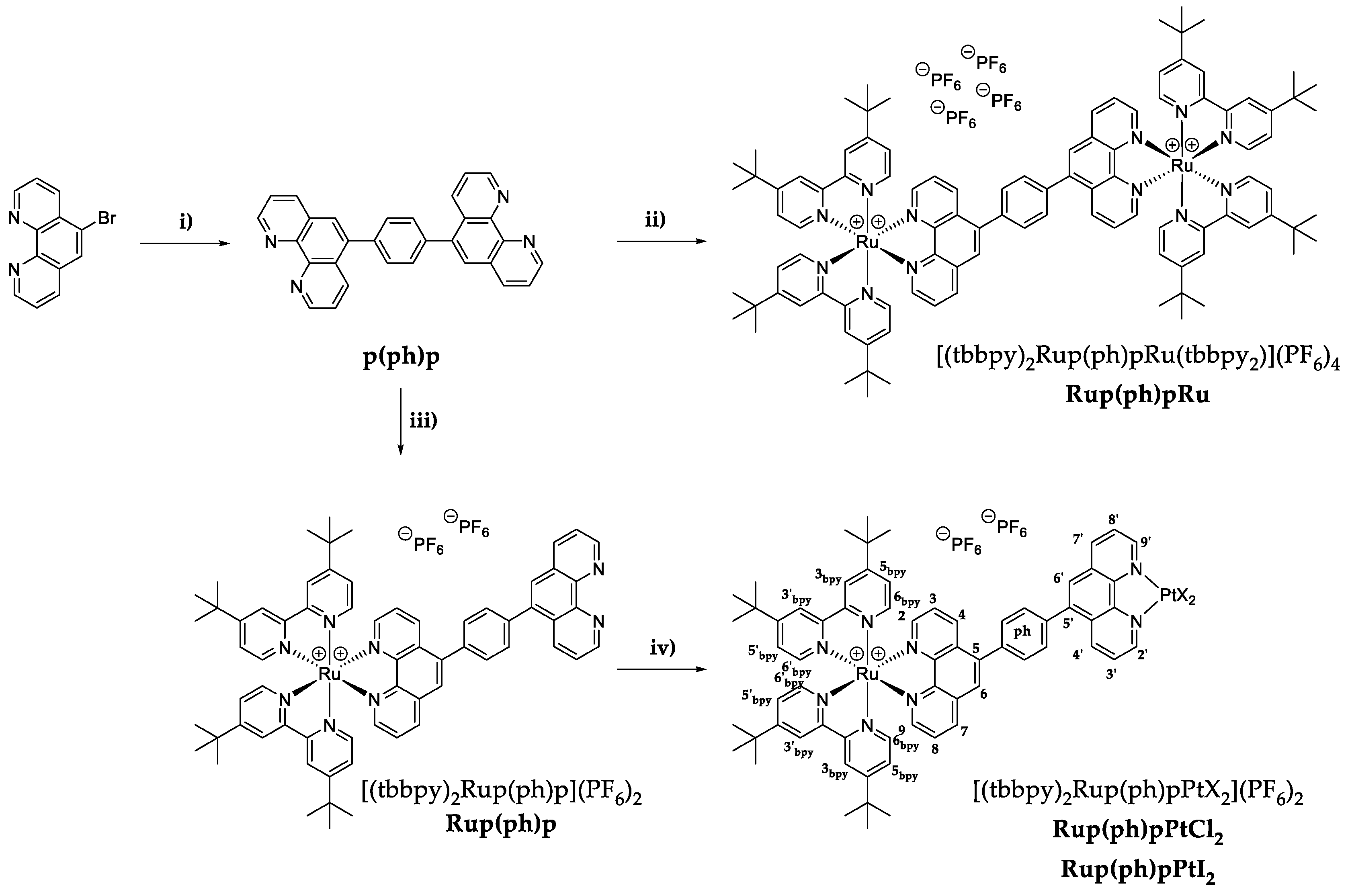
3.1. Synthesis of the Ligand and the Complexes
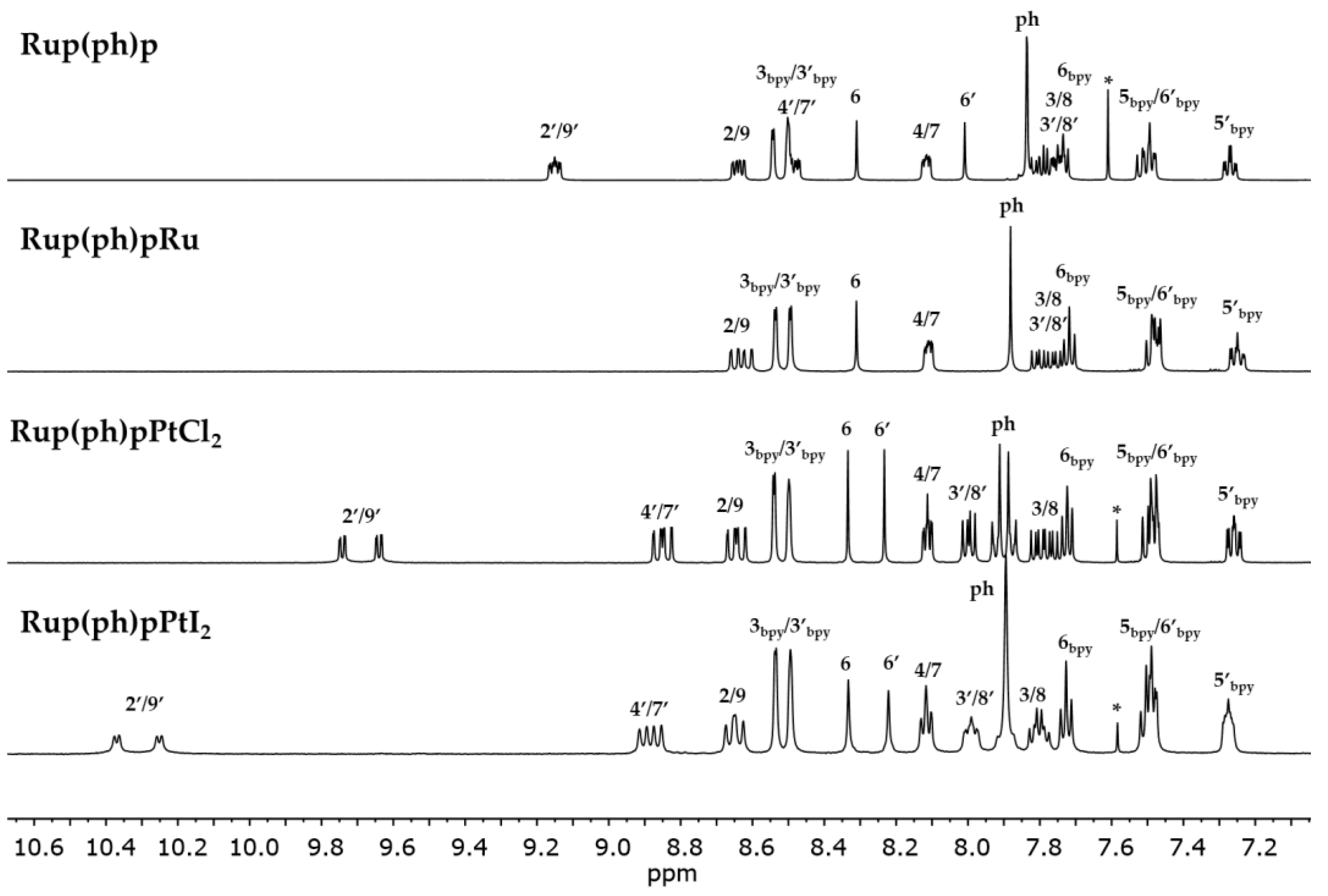
3.2. Structural Characterization
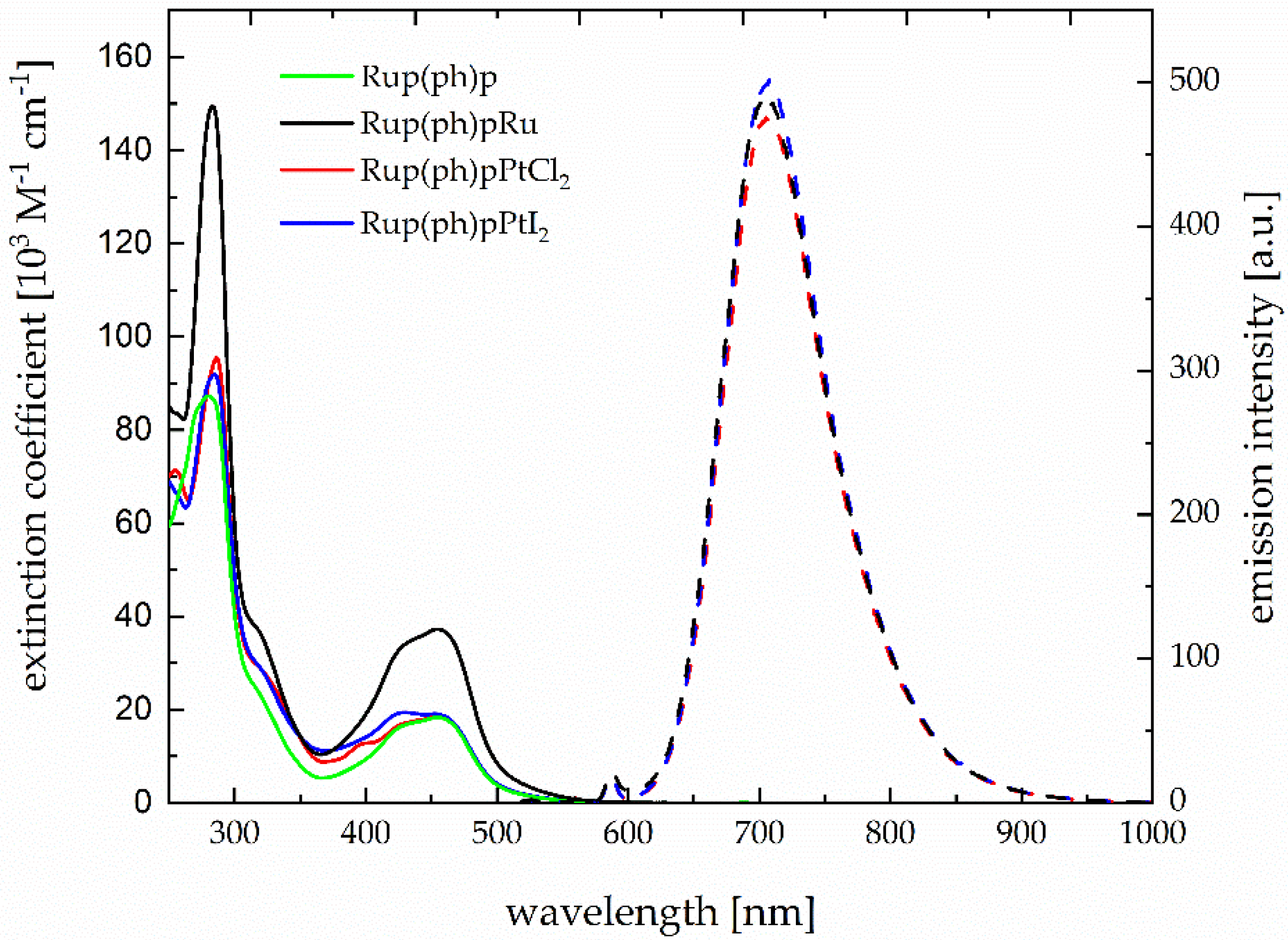
3.3. Photophysical Characterization
3.4. Photochemical Characterization
3.5. Electrochemical Characterization
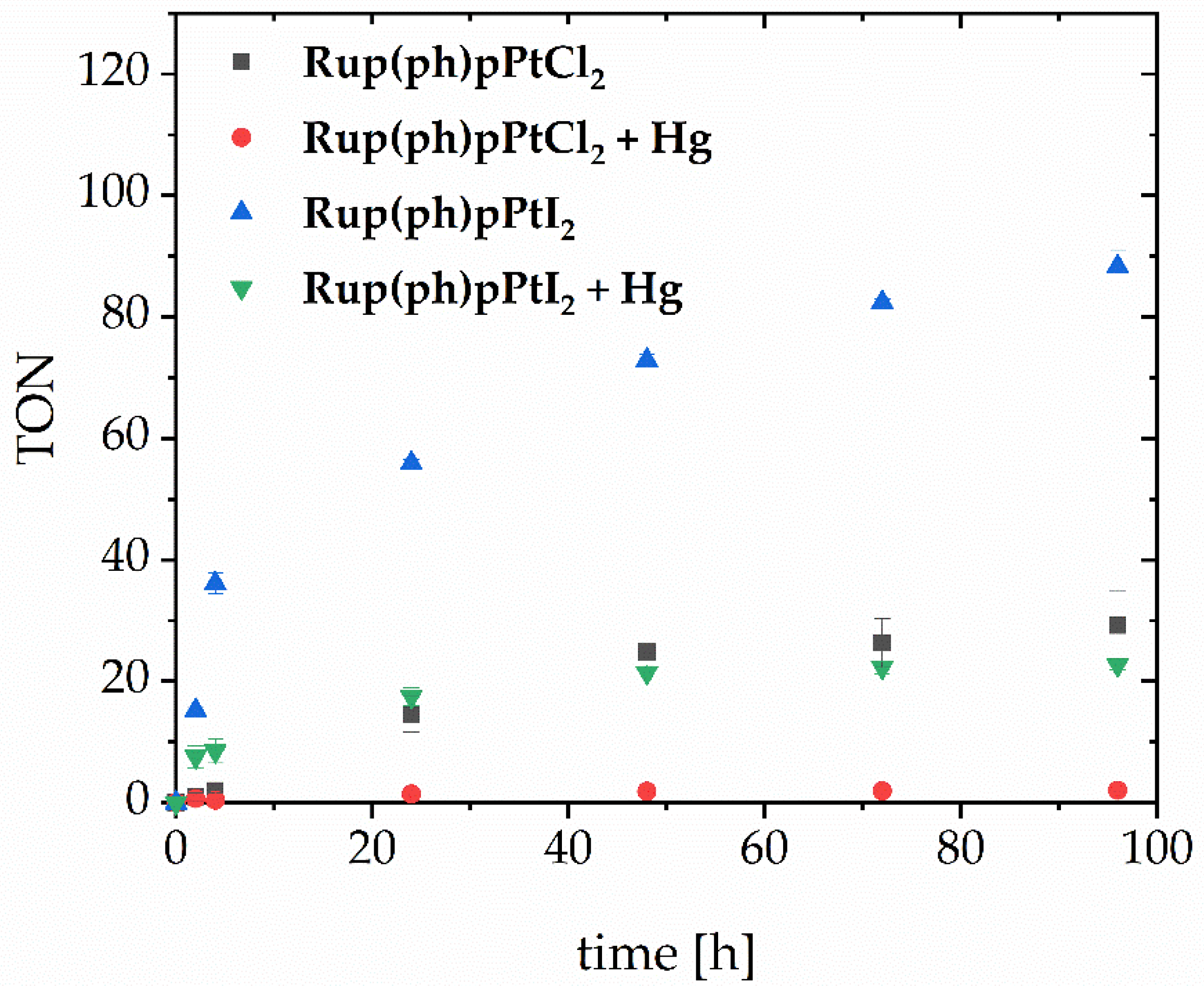
3.6. Light-Induced Evolution of Molecular Hydrogen
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Donnelly, C.; Greuell, W.; Andersson, J.; Gerten, D.; Pisacane, G.; Roudier, P.; Ludwig, F. Impacts of climate change on European hydrology at 1.5, 2 and 3 degrees mean global warming above preindustrial level. Clim. Chang. 2017, 143, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Miao, C.; Hanel, M.; Borthwick, A.G.L.; Duan, Q.; Ji, D.; Li, H. Global heat stress on health, wildfires, and agricultural crops under different levels of climate warming. Environ. Int. 2019, 128, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Armaroli, N.; Balzani, V. The future of energy supply: Challenges and opportunities. Angew. Chem. Int. Ed. 2007, 46, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Vos, J.G.; Kelly, J.M. Ruthenium polypyridyl chemistry; from basic research to applications and back again. Dalton Trans. 2006, 41, 4869–4883. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Na, Y.; Gorlov, M.; Sun, L. Light-driven hydrogen production catalysed by transition metal complexes in homogeneous systems. Dalton Trans. 2009, 33, 6458–6467. [Google Scholar] [CrossRef]
- Goldsmith, J.I.; Hudson, W.R.; Lowry, M.S.; Anderson, T.H.; Bernhard, S.J. Discovery and high-throughput screening of heteroleptic iridium complexes for photoinduced hydrogen production. Am. Chem. Soc. 2005, 127, 7502–7510. [Google Scholar] [CrossRef]
- Pfeffer, M.G.; Schäfer, B.; Smolentsev, G.; Uhlig, J.; Nazarenko, E.; Guthmuller, J.; Kuhnt, C.; Wächtler, M.; Dietzek, B.; Sundström, V.; et al. Palladium versus platinum: The metal in the catalytic center of a molecular photocatalyst determines the mechanism of the hydrogen production with visible light. Angew. Chem. Int. Ed. 2015, 54, 5044–5048. [Google Scholar] [CrossRef]
- Lämmle, M.; Pilz, T.D.; Kutta, R.J.; Müßler, M.; Mengele, A.K.; Görls, H.; Heinemann, F.W.; Rau, S. Insights into the different mechanistic stages of light-induced hydrogen evolution of a 5, 5′-bisphenanthroline linked RuPt complex. Dalton Trans. 2022. [Google Scholar] [CrossRef]
- Mengele, A.K.; Kaufhold, S.; Streb, C.; Rau, S. Generation of a stable supramolecular hydrogen evolving photocatalyst by alteration of the catalytic center. Dalton Trans. 2016, 45, 6612–6618. [Google Scholar] [CrossRef]
- Ozawa, H.; Kobayashi, M.; Balan, B.; Masaoka, S.; Sakai, K. Photo-Hydrogen-Evolving Molecular Catalysts Consisting of Polypyridyl Ruthenium (II) Photosensitizers and Platinum (II) Catalysts: Insights into the Reaction Mechanism. Chem. Asian J. 2010, 5, 1860–1869. [Google Scholar] [CrossRef]
- Ozawa, H.; Haga, M.A.; Sakai, K.J. A photo-hydrogen-evolving molecular device driving visible-light-induced EDTA-reduction of water into molecular hydrogen. Am. Chem. Soc. 2006, 128, 4926–4927. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.G.; Kowacs, T.; Wächtler, M.; Guthmuller, J.; Dietzek, B.; Vos, J.G.; Rau, S. Optimization of hydrogen-evolving photochemical molecular devices. Angew. Chem. I. Edt. 2015, 54, 6627–6631. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, H.; Yokoyama, Y.; Haga, M.-A.; Sakai, K.J. Syntheses, characterization, and photo-hydrogen-evolving properties of tris(2,2′-bipyridine)ruthenium(ii) derivatives tethered to a cis-Pt(ii)Cl2 unit: Insights into the structure–activity relationship. Chem. Soc. Dalton Trans. 2006, 12, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, M.; Li, X.Q.; Cui, H.G.; Dong, J.F.; Sun, L.C. Photochemical hydrogen production with molecular devices comprising a zinc porphyrin and a cobaloxime catalyst. Sci. China Chem. 2012, 55, 1274–1282. [Google Scholar] [CrossRef]
- Li, C.; Wang, M.; Pan, J.; Zhang, P.; Zhang, R.; Sun, L.J. Photochemical hydrogen production catalyzed by polypyridyl ruthenium–cobaloxime heterobinuclear complexes with different bridges. Organomet. Chem. 2009, 694, 2814–2819. [Google Scholar] [CrossRef]
- Petermann, L.; Staehle, R.; Pfeifer, M.; Reichardt, C.; Sorsche, D.; Wächtler, M.; Popp, J.; Dietzek, B.; Rau, S. Oxygen-Dependent Photocatalytic Water Reduction with a Ruthenium (imidazolium) Chromophore and a Cobaloxime Catalyst. Chem. Eur. J. 2016, 22, 8240–8253. [Google Scholar] [CrossRef]
- Karnahl, M.; Kuhnt, C.; Ma, F.; Yartsev, A.; Schmitt, M.; Dietzek, B.; Rau, S.; Popp, J. Tuning of photocatalytic hydrogen production and photoinduced intramolecular electron transfer rates by regioselective bridging ligand substitution. ChemPhysChem 2011, 12, 2101–2109. [Google Scholar] [CrossRef]
- Zedler, L.; Wintergerst, P.; Mengele, A.K.; Müller, C.; Li, C.; Dietzek-Ivanšić, B.; Rau, S. Outpacing conventional nicotinamide hydrogenation catalysis by a strongly communicating heterodinuclear photocatalyst. Nat. Commun. 2022, 13, 2538. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Takeda, H.; Ishitani, O.J. Photocatalytic reduction of CO2 using metal complexes. Photochem. Photobiol. C 2015, 25, 106–137. [Google Scholar] [CrossRef]
- Lei, P.; Hedlund, M.; Lomoth, R.; Rensmo, H.; Johansson, O.; Hammarström, L.J. The role of colloid formation in the photoinduced H2 production with a RuII-PdII supramolecular complex: A study by GC, XPS, and TEM. Am. Chem. Soc. 2008, 130, 26–27. [Google Scholar] [CrossRef]
- Pfeffer, M.G.; Müller, C.; Kastl, E.T.E.; Mengele, A.K.; Bagemihl, B.; Fauth, S.S.; Habermehl, J.; Petermann, L.; Wächtler, M.; Schulz, M.; et al. Active repair of a dinuclear photocatalyst for visible-light-driven hydrogen production. Nat. Chem. 2022, 14, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.M.; Loiseau, F.; Puntoriero, F.; Serroni, S.; Campagna, S. New luminescent and redox-active homometallic dinuclear iridium (iii), ruthenium (ii) and osmium (ii) complexes prepared by metal-catalyzed coupling reactions Electronic supplementary information (ESI) available: Spectral data for 1–4, cyclic voltammogram for 3 and absorption and emission spectra for 1–3. Chem. Commun. 2000, 23, 2297–2298. [Google Scholar]
- Gong, Z.J.; Narayana, Y.S.L.V.; Lin, Y.-C.; Huang, W.-H.; Su, W.-N.; Li, Y.-P.; Higuchi, M.; Yu, W.-Y. Rational synthesis of ruthenium-based metallo-supramolecular polymers as heterogeneous catalysts for catalytic transfer hydrogenation of carbonyl compounds. Appl. Catal. B Environ. 2022, 312, 121383. [Google Scholar] [CrossRef]
- Stumper, A.; Pilz, T.D.; Schaub, M.; Görls, H.; Sorsche, D.; Peuntinger, K.; Guldi, D.; Rau, S. Efficient Access to 5-Bromo-and 5, 6-Dibromophenanthroline Ligands. Eur. J. Inorg. Chem. 2017, 32, 3799–3810. [Google Scholar] [CrossRef]
- Rau, S.; Schäfer, B.; Grüßing, A.; Schebesta, S.; Lamm, K.; Vieth, J.; Görls, H.; Walther, D.; Rudolph, M.; Grummt, U.W.; et al. Efficient synthesis of ruthenium complexes of the type (R-bpy) 2RuCl2 and [(R-bpy) 2Ru (L–L)] Cl2 by microwave-activated reactions (R: H, Me, tert-But) (L–L: Substituted bibenzimidazoles, bipyrimidine, and phenanthroline). Inorg. Chim. Acta 2004, 357, 4496–4503. [Google Scholar] [CrossRef]
- Wu, T.; Liu, J.; Liu, M.; Liu, S.; Zhao, S.; Tian, R.; Wei, D.; Liu, Y.; Zhao, Y.; Xiao, H.; et al. A nanobody-conjugated DNA nanoplatform for targeted platinum-drug delivery. Angew. Chem. Int. Ed. 2019, 58, 14224–14228. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kobayashi, A.; Kaneko, S.; Takehira, K.; Yoshihara, T.; Ishida, H.; Shiina, Y.; Oishi, S.; Tobita, S. Reevaluation of absolute luminescence quantum yields of standard solutions using a spectrometer with an integrating sphere and a back-thinned CCD detector. Phys. Chem. Chem. Phys. 2009, 11, 9850–9860. [Google Scholar] [CrossRef]
- Hossain, M.D.; Higuchi, M. Synthesis of metallo-supramolecular polymers using 5, 5′-linked bis (1, 10-phenanthroline) ligands. Synthesis 2013, 45, 753–758. [Google Scholar]
- Yang, W.; Nakano, T. Synthesis of poly (1, 10-phenanthroline-5, 6-diyl) s having a π-stacked, helical conformation. Chem. Commun. 2015, 51, 17269–17272. [Google Scholar] [CrossRef]
- Hu, Y.Z.; Xiang, Q.; Thummel, R.P. Bi-1, 10-phenanthrolines and their mononuclear Ru (II) complexes. Inorg. Chem. 2002, 41, 3423–3428. [Google Scholar] [CrossRef]
- Rau, S.; Schäfer, B.; Gleich, D.; Anders, E.; Rudolph, M.; Friedrich, M.; Görls, H.; Henry, W.; Vos, J.G. A supramolecular photocatalyst for the production of hydrogen and the selective hydrogenation of tolane. Angew. Chem. Int. Ed. 2006, 45, 6215–6218. [Google Scholar] [CrossRef] [PubMed]
- Karnahl, M.; Tschierlei, S.; Kuhnt, C.; Dietzek, B.; Schmitt, M.; Popp, J.; Schwalbe, M.; Krieck, S.; Görls, H.; Heinemann, F.W.; et al. Synthesis and characterization of regioselective substituted tetrapyridophenazine ligands and their Ru (II) complexes. Dalton Trans. 2010, 39, 2359–2370. [Google Scholar] [CrossRef]
- Toyota, S.; Goto, A.; Kaneko, K.; Umetani, T. Syntheses, spectroscopic properties, and Cu (I) complexes of all possible symmetric Bi-1, 10-phenanthrolines. Heterocycles 2005, 65, 551–562. [Google Scholar] [CrossRef]
- Juris, A.; Balzani, V.; Barigelletti, F.; Campagna, S.; Belser, P.; von Zelewsky, A. Ru (II) polypyridine complexes: Photophysics, photochemistry, eletrochemistry, and chemiluminescence. Coord. Chem. Rev. 1988, 84, 85–277. [Google Scholar] [CrossRef]
- Barker, N.M.; Li, Y.X.; Lee, M.M.; Shen, C.R.; Krause, J.A.; Sun, S.S.; Lu, N.; Connick, W.B.; McMillin, D.R. Synthesis, Luminescence, and Structure of a Polymorphic Polyfluorinated Diiodoplatinum (II) Diimine Complex. Inorg. Chem. 2019, 58, 10716–10724. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.A.; Puzyk, M.V.; Balashev, K.P. Spectroscopic and electrochemical properties of dichlorodiimine complexes of Au (III) and Pt (II) with 1, 4-diazine derivatives of o-phenanthroline. Russ. J. Gen. Chem. 2006, 76, 843–848. [Google Scholar] [CrossRef]
- Schwalbe, M.; Schäfer, B.; Görls, H.; Rau, S.; Tschierlei, S.; Schmitt, M.; Popp, J.; Vaughan, G.; Henry, W.; Vos, J.G. Synthesis and characterization of poly(bipyridine)ruthenium complexes as building blocks for heterosupramolecular arrays. Eur. J. Inorg. Chem. 2008, 3, 3310–3319. [Google Scholar] [CrossRef]
- Petermann, L.; Staehle, R.; Pilz, T.D.; Sorsche, D.; Görls, H.; Rau, S. Synthetic Strategies for Variably Substituted Ruthenium–Imidazophenanthrolinium Complexes. Eur. J. Inorg. Chem. 2015, 2015, 750–762. [Google Scholar] [CrossRef]
- Staehle, R.; Reichardt, C.; Popp, J.; Sorsche, D.; Petermann, L.; Kastner, K.; Streb, C.; Dietzek, B.; Rau, S. Ruthenium Imidazophenanthrolinium Complexes with Prolonged Excited-State Lifetimes. Eur. J. Inorg. Chem. 2015, 2015, 3932–3939. [Google Scholar] [CrossRef]
- Schäfer, B.; Görls, H.; Meyer, S.; Henry, W.; Vos, J.G.; Rau, S. Synthesis and Properties of Tetrasubstituted 1, 10-Phenanthrolines and Their Ruthenium Complexes. Eur. J. Inorg. Chem. 2007, 2007, 4056–4063. [Google Scholar] [CrossRef]
- Robin, M.B.; Day, P. Mixed valence chemistry—A survey and classification. Adv. Inorg. Chem. Radiochem. 1968, 10, 247–422. [Google Scholar]
- Mengele, A.K.; Müller, C.; Nauroozi, D.; Kupfer, S.; Dietzek, B.; Rau, S. Molecular scylla and charybdis: Maneuvering between pH sensitivity and excited-state localization in ruthenium bi (benz) imidazole complexes. Inorg. Chem. 2020, 59, 12097–12110. [Google Scholar] [CrossRef] [PubMed]
- Kamath, S.S.; Uma, V.; Srivastava, T.S. Experimental and quantum-chemical studies of 1H, 13C and 15N NMR coordination shifts in Pd (II) and Pt (II) chloride complexes with methyl and phenyl derivatives of 2, 2′-bipyridine and 1, 10-phenanthroline. Inorg. Chim. Acta 1989, 161, 49–56. [Google Scholar] [CrossRef]
- Pavlishchuk, V.V.; Addison, A.W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]
- Fraser, C.; Bosnich, B. Bimetallic reactivity. Investigation of metal-metal interaction in complexes of a chiral macrocyclic binucleating ligand bearing 6-and 4-coordinate sites. Inorg. Chem. 1994, 33, 338–346. [Google Scholar] [CrossRef]
- Matsumoto, K.; Sakai, K. Structures and reactivities of platinum-blues and the related amidate-bridged platinumIII compounds. Adv. Inorg. Chem. 1999, 49, 375–427. [Google Scholar]
- Sakai, K.; Ozawa, H. Homogeneous catalysis of platinum (II) complexes in photochemical hydrogen production from water. Coord. Chem. Rev. 2007, 251, 2753–2766. [Google Scholar] [CrossRef]
- Kobayashi, M.; Masaoka, S.; Sakai, K. Synthesis, crystal structure, solution and spectroscopic properties, and hydrogen-evolving activity of [K (18-crown-6)] [Pt (ii) (2-phenylpyridinato) Cl 2]. Photochem. Photobiol. Sci. 2009, 8, 196–203. [Google Scholar] [CrossRef]
- Chernyshev, V.M.; Astakhov, A.V.; Chikunov, I.E.; Tyurin, R.V.; Eremin, D.B.; Ranny, G.S.; Khrustalev, V.N.; Ananikov, V.P. Pd and Pt catalyst poisoning in the study of reaction mechanisms: What does the mercury test mean for catalysis? ACS Catal. 2019, 9, 2984–2995. [Google Scholar] [CrossRef]
- Artero, V.; Fontecave, M. Solar fuels generation and molecular systems: Is it homogeneous or heterogeneous catalysis? Chem. Soc. Rev. 2013, 42, 2338–2356. [Google Scholar] [CrossRef]
- Knoll, J.D.; Arachchige, S.M.; Brewer, K.J. A structurally diverse RuII, PtII tetrametallic motif for photoinitiated electron collection and photocatalytic hydrogen production. ChemSusChem 2011, 4, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, R.; Masaoka, S.; Sakai, K. Photo-hydrogen-evolving activity of chloro (terpyridine) platinum (II): A single-component molecular photocatalyst. Dalton Trans. 2009, 31, 6127–6133. [Google Scholar] [CrossRef] [PubMed]
- Amthor, S.; Hernández-Castillo, D.; Maryasin, B.; Seeber, P.; Mengele, A.K.; Gräfe, S.; González, L.; Rau, S. Strong Ligand Stabilization Based on π-Extension in a Series of Ruthenium Terpyridine Water Oxidation Catalysts. Chem. Eur. J. 2021, 27, 16871–16878. [Google Scholar] [CrossRef] [PubMed]
- Heiland, M.; De, R.; Rau, S.; Dietzek-Ivanšić, B.; Streb, C. Not that innocent–ammonium ions boost homogeneous light-driven hydrogen evolution. Chem. Commun. 2022, 58, 4603. [Google Scholar] [CrossRef] [PubMed]
- Whang, D.R.; Park, S.Y. Rational Design of an Electron-Reservoir PtII Complex for Efficient Photocatalytic Hydrogen Production from Water. ChemSusChem 2015, 8, 3204–3207. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | λmax,abs [nm] (ϵ, 103 M−1 cm−1) [a] | λmax,emi [nm][a,b] | [a,b] | [b,c] | [ns][a,d] | [ns][c,d] |
|---|---|---|---|---|---|---|
| Rup(ph)p | 455 (18.33) | 617 | 0.01 | 0.19 | 110 | 1848 |
| Rup(ph)pRu | 455 (37.59) | 615 | 0.01 | 0.20 | 111 | 2046 |
| Rup(ph)pPtCl2 | 455 (17.99) | 614 | 0.01 | 0.19 | 109 | 1647 |
| Rup(ph)pPtI2 | 452 (19.00) | 614 | 0.01 | 0.11 | 107 | 1512 |
| [Ru(tbbpy)3]2+ [37,38,39] | 458 (17.34) | 613 | 0.01 | 0.05 | 107 | 730 |
| [(tbbpy)2Ru(phen)]2+ [40] | 454 (16.00) | 610 | -- | -- | 211 | 1423 |
| [(tbbpy)2Ru(phenphen)]2+ [8] | 454 (18.68) | 618 | 0.01 | 0.20 | 127 | 2000 |
| Complex | L3 [V] | L2 [V] | L1 [V] | L0 [V] | Ru2+/Ru3+ [V] |
|---|---|---|---|---|---|
| Rup(ph)p | −2.24 | −2.00 | −1.73 | -- | 0.79 |
| Rup(ph)pRu | −2.28 | −1.99 | −1.70 | -- | 0.79 |
| Rup(ph)pPtCl2 | −2.30 | −2.03 | −1.73 (irr) | −1.57 | 0.79 |
| Rup(ph)pPtI2 | −2.31 (irr) | −1.97 | −1.74 (irr) | −1.60 (irr) | 0.84 |
| [Ru(tbbpy)3]2+ [42] | −2.28 | −2.01 | −1.80 | -- | 0.73 |
| [Ru(bpy)2(phen)]2+ [40] | −2.25 | −1.98 | −1.77 | -- | 0.74 |
| [Ru(phenphen)]2+ [8] | −2.25 | −1.98 | −1.72 | -- | 0.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lämmle, M.; Volk, S.; Klinkerman, M.; Müßler, M.; Mengele, A.K.; Rau, S. Efficient Access of Phenyl-Spaced 5,5′-Bridged Dinuclear Ruthenium Metal Complexes and the Effect of Dynamic Ligand Exchange on Catalysis. Photochem 2022, 2, 831-848. https://doi.org/10.3390/photochem2040053
Lämmle M, Volk S, Klinkerman M, Müßler M, Mengele AK, Rau S. Efficient Access of Phenyl-Spaced 5,5′-Bridged Dinuclear Ruthenium Metal Complexes and the Effect of Dynamic Ligand Exchange on Catalysis. Photochem. 2022; 2(4):831-848. https://doi.org/10.3390/photochem2040053
Chicago/Turabian StyleLämmle, Martin, Steffen Volk, Madelyn Klinkerman, Marius Müßler, Alexander K. Mengele, and Sven Rau. 2022. "Efficient Access of Phenyl-Spaced 5,5′-Bridged Dinuclear Ruthenium Metal Complexes and the Effect of Dynamic Ligand Exchange on Catalysis" Photochem 2, no. 4: 831-848. https://doi.org/10.3390/photochem2040053