


Advances in Selective Photocatalytic Oxidation of p-Xylene to Terephthalic Acid as a Sustainable Route: A Short Review on Photocatalyst Formulation and Related Reaction Mechanisms




Abstract
:1. Introduction
1.1. The Urgency of TPA Production
1.2. Alternative Routes for TPA Production
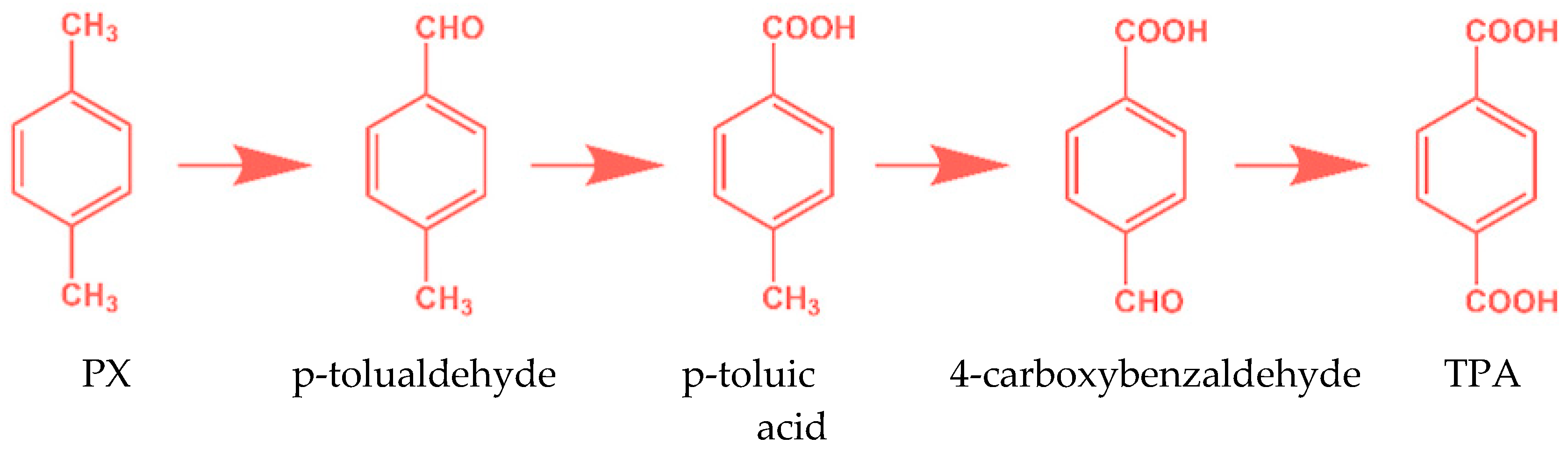
2. Catalytic Oxidation of p-Xylene
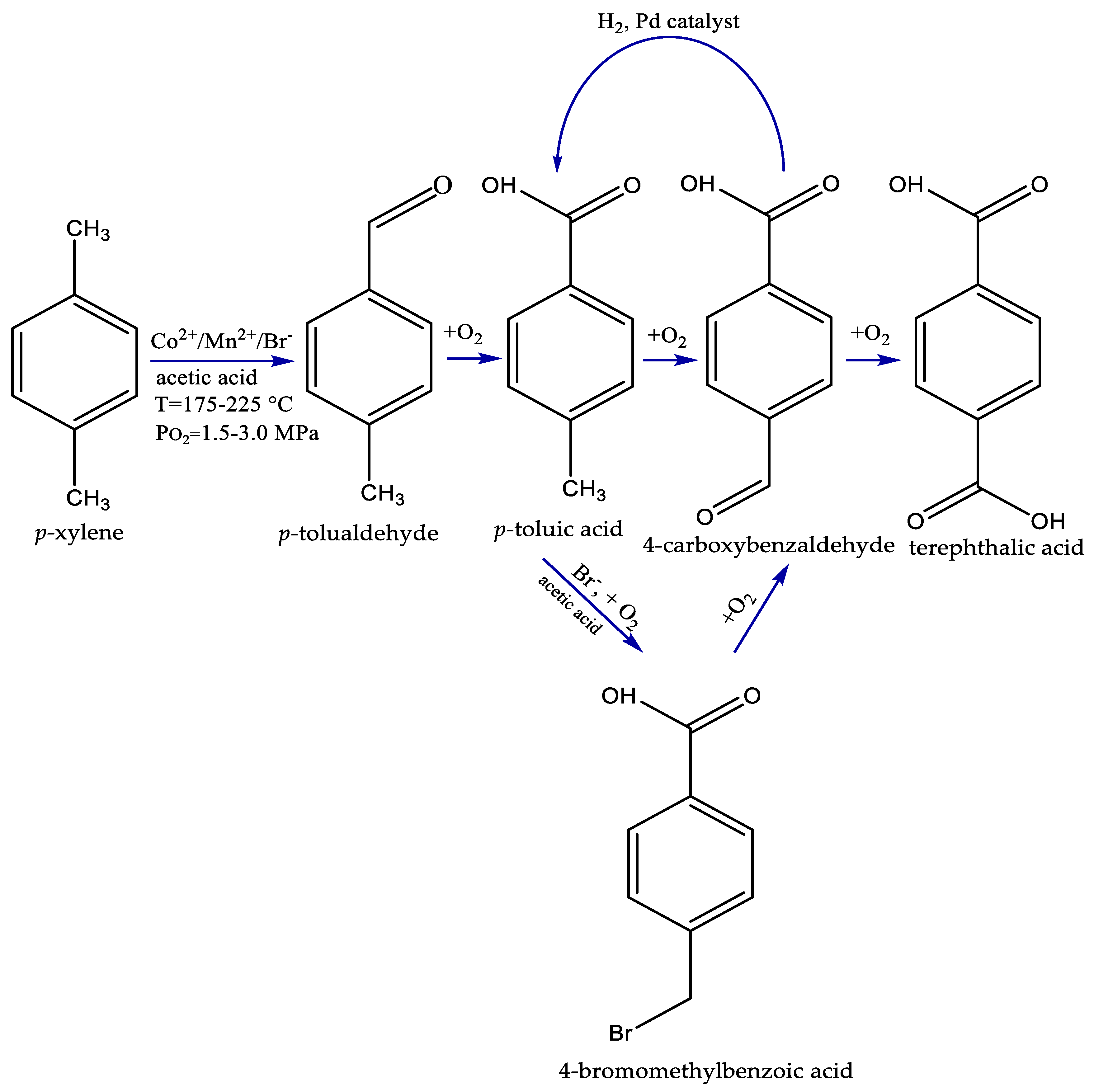
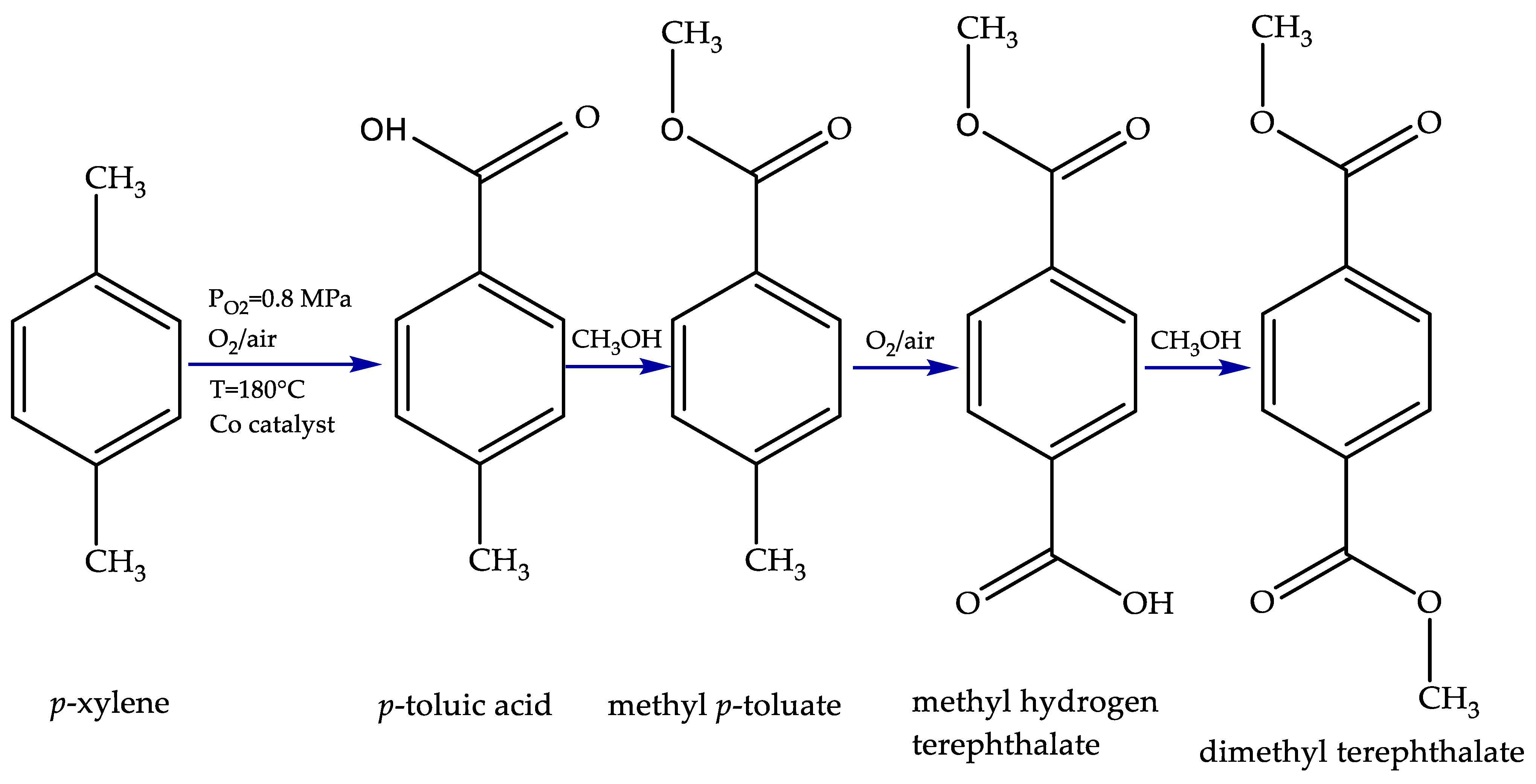
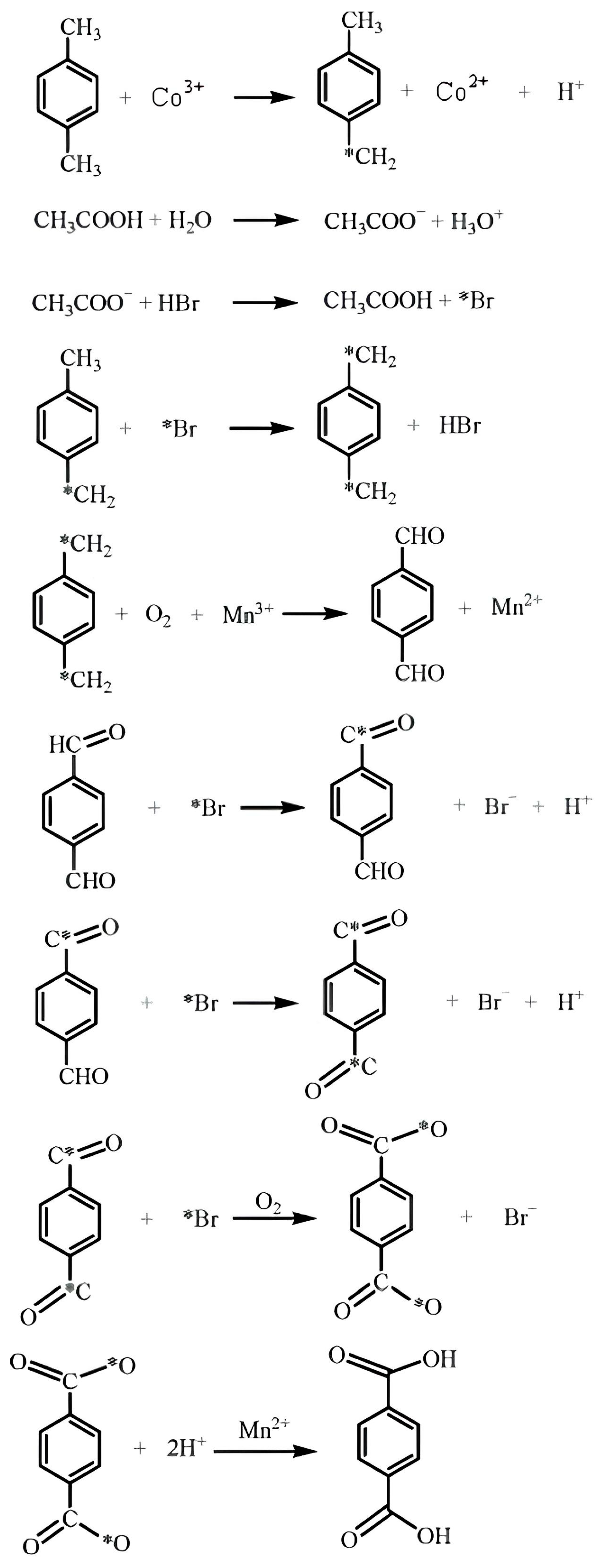
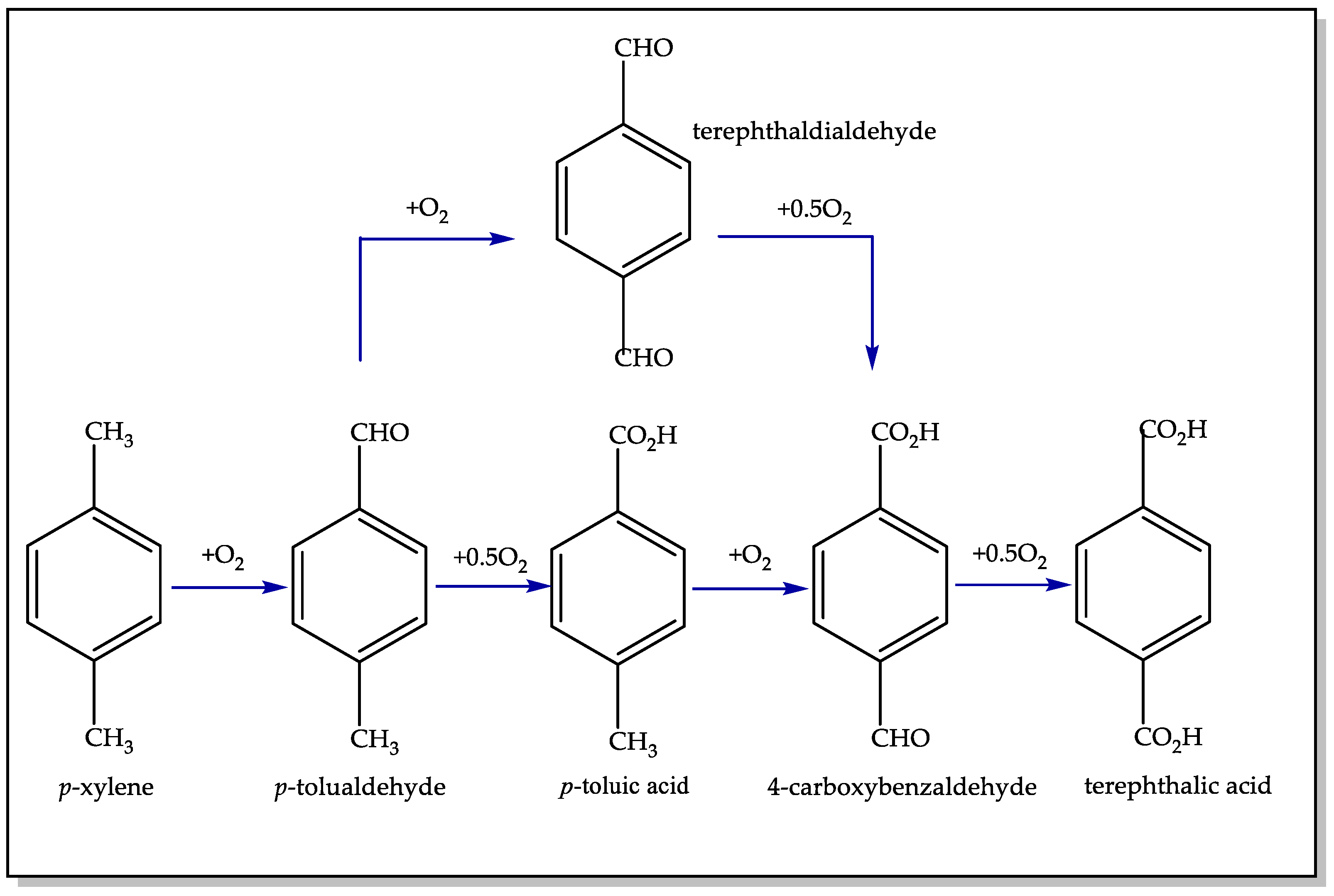
2.1. Homogeneous Catalytic Oxidation
2.2. Heterogeneous Catalytic Oxidation
3. Photocatalytic Oxidation of p-Xylene as a Sustainable Alternative for TPA Production
4. Comparison Between Catalytic and Photocatalytic Systems
5. Conclusions, Challenges and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vaidya, U.R.; Nadkarni, V.M. Unsaturated polyester resins from poly (ethylene terephthalate) waste. 1. Synthesis and characterization. Ind. Eng. Chem. Res. 1987, 26, 194–198. [Google Scholar] [CrossRef]
- Cui, Y.; Deng, C.; Fan, L.; Qiu, Y.; Zhao, L. Progress in the biosynthesis of bio-based PET and PEF polyester monomers. Green Chem. 2023, 25, 5836–5857. [Google Scholar] [CrossRef]
- Wang, K.; Guo, C.; Li, J.; Wang, K.; Cao, X.; Liang, S.; Wang, J. High value-added conversion and functional recycling of waste polyethylene terephthalate (PET) plastics: A comprehensive review. J. Environ. Chem. Eng. 2024, 12, 113539. [Google Scholar] [CrossRef]
- Toufaili, F.-A.E. Catalytic and Mechanistic Studies of Polyethylene Terephthalate Synthesis. Ph.D. Thesis, Technische Universität Berlin, Berlin, Germany, 2006. [Google Scholar]
- Benyathiar, P.; Kumar, P.; Carpenter, G.; Brace, J.; Mishra, D.K. Polyethylene terephthalate (PET) bottle-to-bottle recycling for the beverage industry: A review. Polymers 2022, 14, 2366. [Google Scholar] [CrossRef] [PubMed]
- Karakhanov, E.; Maksimov, A.; Zolotukhina, A.; Vinokurov, V. Oxidation of p-Xylene. Russ. J. Appl. Chem. 2018, 91, 707–727. [Google Scholar] [CrossRef]
- Fadzil, N.A.M.; Rahim, M.H.A.; Maniam, G.P. A brief review of para-xylene oxidation to terephthalic acid as a model of primary C–H bond activation. Chin. J. Catal. 2014, 35, 1641–1652. [Google Scholar] [CrossRef]
- Tomas, R.A.; Bordado, J.o.C.; Gomes, J.F. p-Xylene oxidation to terephthalic acid: A literature review oriented toward process optimization and development. Chem. Rev. 2013, 113, 7421–7469. [Google Scholar] [CrossRef]
- Li, M.; Niu, F.; Zuo, X.; Metelski, P.D.; Busch, D.H.; Subramaniam, B. A spray reactor concept for catalytic oxidation of p-xylene to produce high-purity terephthalic acid. Chem. Eng. Sci. 2013, 104, 93–102. [Google Scholar] [CrossRef]
- Li, M.; Niu, F.; Busch, D.H.; Subramaniam, B. Kinetic investigations of p-Xylene oxidation to terephthalic acid with a Co/Mn/Br catalyst in a homogeneous liquid phase. Ind. Eng. Chem. Res. 2014, 53, 9017–9026. [Google Scholar] [CrossRef]
- Slapnik, J.; Kraft, G.; Wilhelm, T.; Lobnik, A. Purification of recycled terephthalic acid and synthesis of polyethylene terephthalate. In Proceedings of the 1st International Conference on Circular Packaging, Ljubljana, Slovenia, 26–27 September 2019; pp. 151–159. [Google Scholar]
- Mu, S.; Su, H.; Jia, T.; Gu, Y.; Chu, J. Scalable multi-objective optimization of industrial purified terephthalic acid (PTA) oxidation process. Comput. Chem. Eng. 2004, 28, 2219–2231. [Google Scholar] [CrossRef]
- Bigdeli, S.; Khosravi-Nikou, M.R.; Shariati, A.; Jafari, N.A. Purification of crude terephthalic acid using phosphomolybdic acid supported on MCM-41 and TiO2 photocatalysts. J. Photochem. Photobiol. A Chem. 2023, 444, 114940. [Google Scholar] [CrossRef]
- Kwon, E.E.; Lee, J. Polyethylene terephthalate production from a carbon neutral resource. J. Clean. Prod. 2024, 469, 143210. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, S.H. Poly (ethylene terephthalate) recycling for high value added textiles. Fash. Text. 2014, 1, 1. [Google Scholar] [CrossRef]
- Pan, H.; Li, S.; Shu, M.; Ye, Y.; Cui, Q.; Zhao, Z. p-Xylene catalytic oxidation to terephthalic acid by ozone. ScienceAsia 2018, 44, 212–217. [Google Scholar] [CrossRef]
- Usman, M.; Cheng, S.; Boonyubol, S.; Cross, J.S. From biomass to biocrude: Innovations in hydrothermal liquefaction and upgrading. Energy Convers. Manag. 2024, 302, 118093. [Google Scholar] [CrossRef]
- Jing, Y.; Guo, Y.; Xia, Q.; Liu, X.; Wang, Y. Catalytic production of value-added chemicals and liquid fuels from lignocellulosic biomass. Chem 2019, 5, 2520–2546. [Google Scholar] [CrossRef]
- Pacheco, J.J.; Davis, M.E. Synthesis of terephthalic acid via Diels-Alder reactions with ethylene and oxidized variants of 5-hydroxymethylfurfural. Proc. Natl. Acad. Sci. USA 2014, 111, 8363–8367. [Google Scholar] [CrossRef]
- Pang, J.; Zheng, M.; Sun, R.; Wang, A.; Wang, X.; Zhang, T. Synthesis of ethylene glycol and terephthalic acid from biomass for producing PET. Green Chem. 2016, 18, 342–359. [Google Scholar] [CrossRef]
- Smith, P.B. Bio-based sources for terephthalic acid. In Green Polymer Chemistry: Biobased Materials And Biocatalysis; ACS Publications: Washington, DC, USA, 2015; pp. 453–469. [Google Scholar]
- Jassal, U.A.K. Advanced Solid Catalysis for Biomass Conversion into High Value-Added Chemicals. In Solid Base Catalysts: Synthesis, Characterization, and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2024; pp. 97–127. [Google Scholar]
- Kataya, G.; Cornu, D.; Bechelany, M.; Hijazi, A.; Issa, M. Biomass waste conversion technologies and its application for sustainable environmental development—A review. Agronomy 2023, 13, 2833. [Google Scholar] [CrossRef]
- Landau, R.; Saffer, A. Development of MC process. Chem. Eng. Prog. 1968, 64, 20. [Google Scholar]
- Cheng, Y.; Li, X.; Wang, L.; Wang, Q. Optimum ratio of Co/Mn in the liquid-phase catalytic oxidation of p-xylene to terephthalic acid. Ind. Eng. Chem. Res. 2006, 45, 4156–4162. [Google Scholar] [CrossRef]
- Li, K.-T.; Li, S.-W. CoBr2-MnBr2 containing catalysts for catalytic oxidation of p-xylene to terephthalic acid. Appl. Catal. A Gen. 2008, 340, 271–277. [Google Scholar] [CrossRef]
- Ishii, Y.; Sakaguchi, S.; Iwahama, T. Innovation of hydrocarbon oxidation with molecular oxygen and related reactions. Adv. Synth. Catal. 2001, 343, 393–427. [Google Scholar] [CrossRef]
- Chavan, S.; Srinivas, D.; Ratnasamy, P. Selective oxidation of para-xylene to terephthalic acid by μ3-oxo-bridged Co/Mn cluster complexes encapsulated in zeolite–Y. J. Catal. 2001, 204, 409–419. [Google Scholar] [CrossRef]
- Jacob, C.R.; Varkey, S.P.; Ratnasamy, P. Oxidation of para-xylene over zeolite-encapsulated copper and manganese complexes. Appl. Catal. A Gen. 1999, 182, 91–96. [Google Scholar] [CrossRef]
- Caloyannis, A.; Graydon, W. Heterogeneous catalysis in the oxidation of p-xylene in the liquid phase. J. Catal. 1971, 22, 287–296. [Google Scholar] [CrossRef]
- Karakhanov, E.; Maximov, A.; Zolotukhina, A.; Vinokurov, V.; Ivanov, E.; Glotov, A. Manganese and cobalt doped hierarchical mesoporous halloysite-based catalysts for selective oxidation of p-xylene to terephthalic acid. Catalysts 2019, 10, 7. [Google Scholar] [CrossRef]
- Li, Y.; Duan, D.; Wu, M.; Li, J.; Yan, Z.; Wang, W.; Zi, G.; Wang, J. One-step synthesis of 2, 5-dihydroxyterephthalic acid by the oxidation of p-xylene over M-MCM-41 (M = Fe, Fe/Cu, Cu) catalysts. Chem. Eng. J. 2016, 306, 777–783. [Google Scholar] [CrossRef]
- Liu, H.; Li, Y.; Jiang, H.; Vargas, C.; Luque, R. Significant promoting effects of Lewis acidity on Au–Pd systems in the selective oxidation of aromatic hydrocarbons. Chem. Commun. 2012, 48, 8431–8433. [Google Scholar] [CrossRef]
- Li, Y.; Wu, M.; Chen, D.; Jiang, L.; He, J.; Luo, Z.; Wang, W.; Wang, J. One-step highly selective oxidation of p-xylene to 4-hydroxymethylbenzoic acid over Cu-MOF catalysts under mild conditions. Mol. Catal. 2019, 477, 110542. [Google Scholar] [CrossRef]
- Deori, K.; Gupta, D.; Saha, B.; Awasthi, S.K.; Deka, S. Introducing nanocrystalline CeO2 as heterogeneous environmental friendly catalyst for the aerobic oxidation of para-xylene to terephthalic acid in water. J. Mater. Chem. A 2013, 1, 7091–7099. [Google Scholar] [CrossRef]
- Hronec, M.; Hrabe, Z. Liquid-phase oxidation of p-xylene catalyzed by metal oxides. Ind. Eng. Chem. Prod. Res. Dev. 1986, 25, 257–261. [Google Scholar] [CrossRef]
- Lapa, H.M.; Martins, L.M. p-xylene oxidation to terephthalic acid: New trends. Molecules 2023, 28, 1922. [Google Scholar] [CrossRef]
- Wang, C.C.; Zhang, G.X.; Zuo, Z.W.; Zeng, R.; Zhai, D.D.; Liu, F.; Shi, Z.-J. Photo-induced deep aerobic oxidation of alkyl aromatics. Sci. China Chem. 2021, 64, 1487–1492. [Google Scholar] [CrossRef]
- Xiong, L.; Tang, J. Strategies and challenges on selectivity of photocatalytic oxidation of organic substances. Adv. Energy Mater. 2021, 11, 2003216. [Google Scholar] [CrossRef]
- Toe, C.Y.; Tsounis, C.; Zhang, J.; Masood, H.; Gunawan, D.; Scott, J.; Amal, R. Advancing photoreforming of organics: Highlights on photocatalyst and system designs for selective oxidation reactions. Energy Environ. Sci. 2021, 14, 1140–1175. [Google Scholar] [CrossRef]
- Sannino, D.; Vaiano, V.; Ciambelli, P. Developments and new frontiers in gas-solid photocatalytic partial oxidation of hydrocarbons. Curr. Org. Chem. 2013, 17, 2420–2426. [Google Scholar] [CrossRef]
- Mohamadpour, F.; Amani, A.M. Photocatalytic systems: Reactions, mechanism, and applications. RSC Adv. 2024, 14, 20609–20645. [Google Scholar] [CrossRef]
- Tee, S.Y.; Kong, J.; Koh, J.J.; Teng, C.P.; Wang, X.Z.; Wang, X.; Teo, S.L.; Thitsartarn, W.; Han, M.-Y.; Seh, Z.W. Structurally and surficially activated TiO2 nanomaterials for photochemical reactions. Nanoscale 2024, 16, 18165–18212. [Google Scholar] [CrossRef]
- Kisch, H. Semiconductor photocatalysis—Mechanistic and synthetic aspects. Angew. Chem. Int. Ed. 2013, 52, 812–847. [Google Scholar] [CrossRef]
- Muhmood, T.; Ahmad, I.; Haider, Z.; Haider, S.K.; Shahzadi, N.; Aftab, A.; Ahmed, S.; Ahmad, F. Graphene-like graphitic carbon nitride (g-C3N4) as a semiconductor photocatalyst: Properties, classification, and defects engineering approaches. Mater. Today Sustain. 2024, 25, 100633. [Google Scholar] [CrossRef]
- Sun, N.; Si, X.; He, L.; Zhang, J.; Sun, Y. Strategies for enhancing the photocatalytic activity of semiconductors. Int. J. Hydrogen Energy 2024, 58, 1249–1265. [Google Scholar] [CrossRef]
- Hou, J.-C.; Cai, W.; Ji, H.-T.; Ou, L.-J.; He, W.-M. Recent advances in semi-heterogenous photocatalysis in organic synthesis. Chin. Chem. Lett. 2024, 36, 110469. [Google Scholar] [CrossRef]
- Guo, Q.; Zhou, C.; Ma, Z.; Yang, X. Fundamentals of TiO2 photocatalysis: Concepts, mechanisms, and challenges. Adv. Mater. 2019, 31, 1901997. [Google Scholar] [CrossRef]
- Maxsimov, A.L.v.; Beletskaya, I.P. Carbon dioxide and “methanol” economy: Advances in the catalytic synthesis of methanol from CO2. Uspekhi Khimii 2024, 93, 1–41. [Google Scholar]
- Mancuso, A.; Vaiano, V.; Antico, P.; Sacco, O.; Venditto, V. Photoreactive polymer composite for selective oxidation of benzene to phenol. Catal. Today 2023, 413, 113914. [Google Scholar] [CrossRef]
- Sannino, D.; Vaiano, V.; Ciambelli, P. Innovative structured VOx/TiO2 photocatalysts supported on phosphors for the selective photocatalytic oxidation of ethanol to acetaldehyde. Catal. Today 2013, 205, 159–167. [Google Scholar] [CrossRef]
- Ciambelli, P.; Sannino, D.; Palma, V.; Vaiano, V.; Bickley, R.I. Reaction mechanism of cyclohexane selective photo-oxidation to benzene on molybdena/titania catalysts. Appl. Catal. A Gen. 2008, 349, 140–147. [Google Scholar] [CrossRef]
- Mancuso, A.; Gottuso, A.; Parrino, F.; Picca, R.A.; Venditto, V.; Sacco, O.; Vaiano, V. Tuning the selectivity of visible light-driven hydroxylation of benzene to phenol by using Cu, Fe and V oxides supported on N-doped TiO2. Green Chem. 2023, 25, 10664–10677. [Google Scholar] [CrossRef]
- Mancuso, A.; Picca, R.A.; Izzi, M.; Di Franco, C.; Sacco, O.; Vaiano, V.; Venditto, V. An innovative biphasic reaction system for highly selective benzene hydroxylation based on photocatalytic polymer composites. ChemPhotoChem 2024, 8, e202400203. [Google Scholar] [CrossRef]
- Dharma, H.N.C.; Jaafar, J.; Widiastuti, N.; Matsuyama, H.; Rajabsadeh, S.; Othman, M.H.D.; Rahman, M.A.; Jafri, N.N.M.; Suhaimin, N.S.; Nasir, A.M. A review of titanium dioxide (TiO2)-based photocatalyst for oilfield-produced water treatment. Membranes 2022, 12, 345. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Feng, Z.; Wang, S.; Wang, Q.-N.; Zhang, P.; Li, R.; Li, C. Insight into the role of TiO2 facets in photocatalytic selective oxidation of p-xylene. ACS Catal. 2024, 14, 5356–5365. [Google Scholar] [CrossRef]
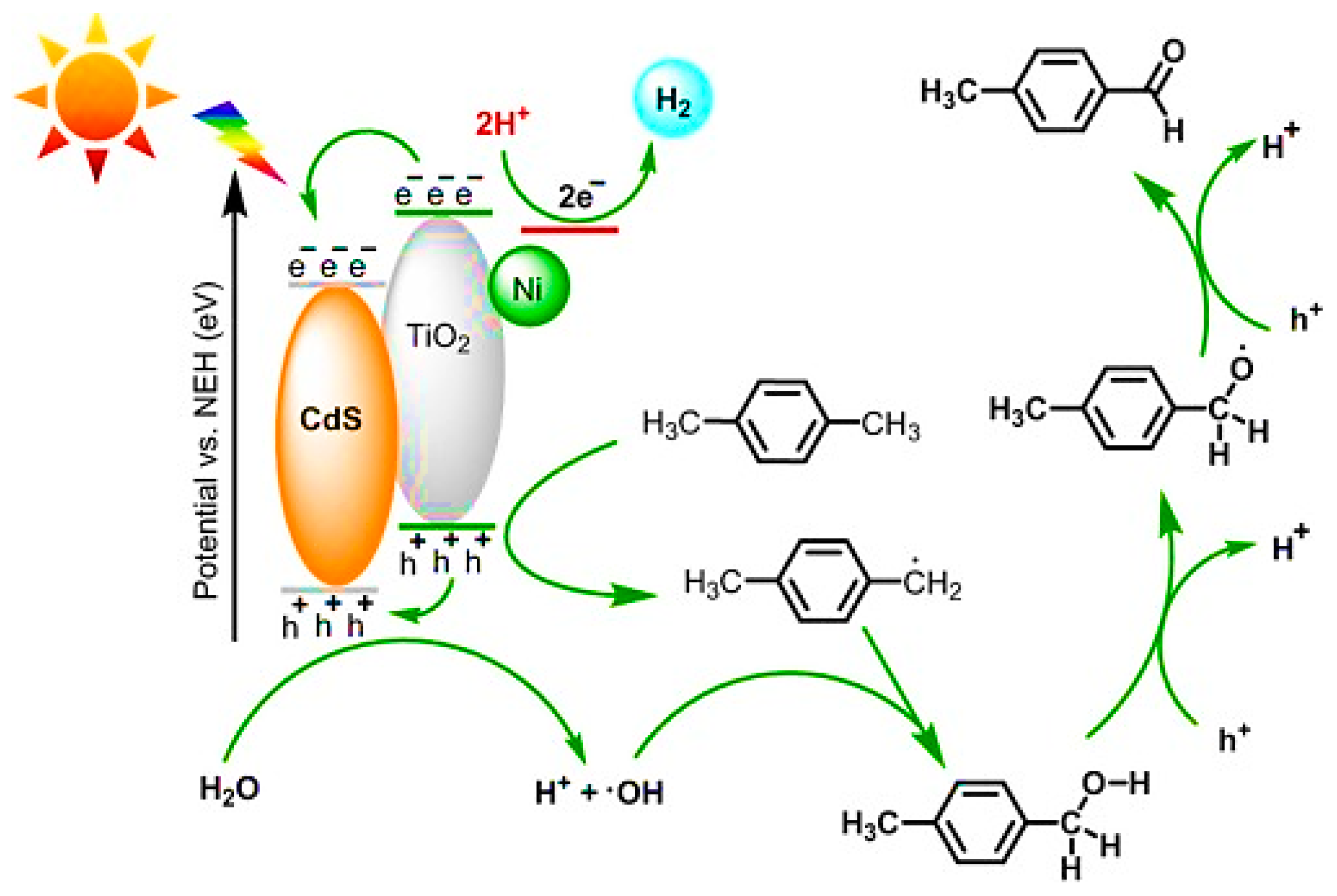
- Guo, C.; Guo, C.; Zhang, Y.; Chen, T.; Zhang, L.; Zou, Y.; Wang, J. Photocatalytic oxidation of p-xylene coupled with hydrogen evolution over MOFs-based bifunctional catalyst. J. Environ. Chem. Eng. 2022, 10, 108079. [Google Scholar] [CrossRef]
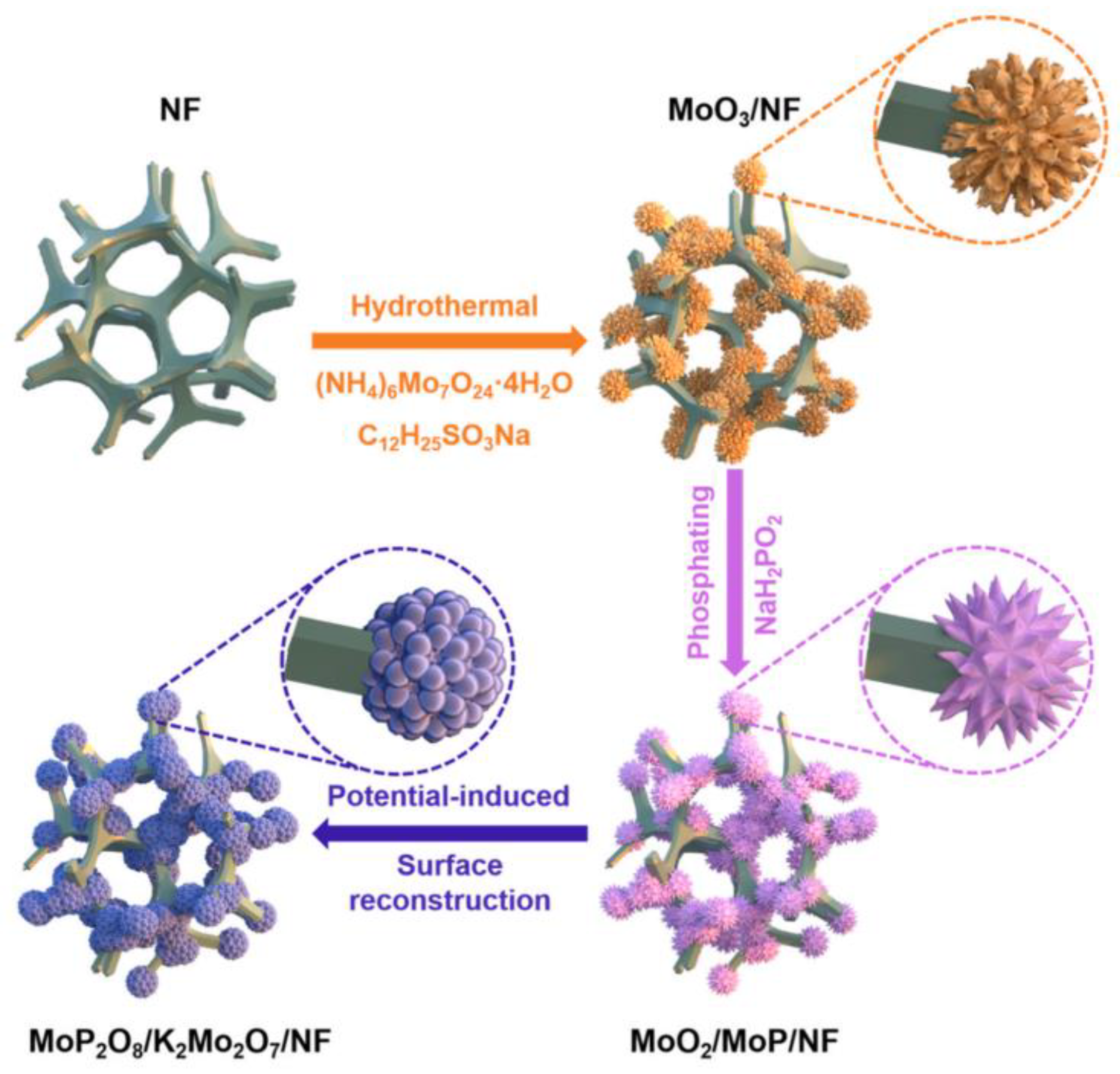
- Lv, Y.; Peng, M.; Yang, W.; Liu, M.; Kong, A.; Fu, Y.; Li, W.; Zhang, J. Surface reconstruction enabling MoO2/MoP hybrid for efficient electrocatalytic oxidation of p-xylene to terephthalic acid. Appl. Catal. B Environ. 2024, 340, 123229. [Google Scholar] [CrossRef]
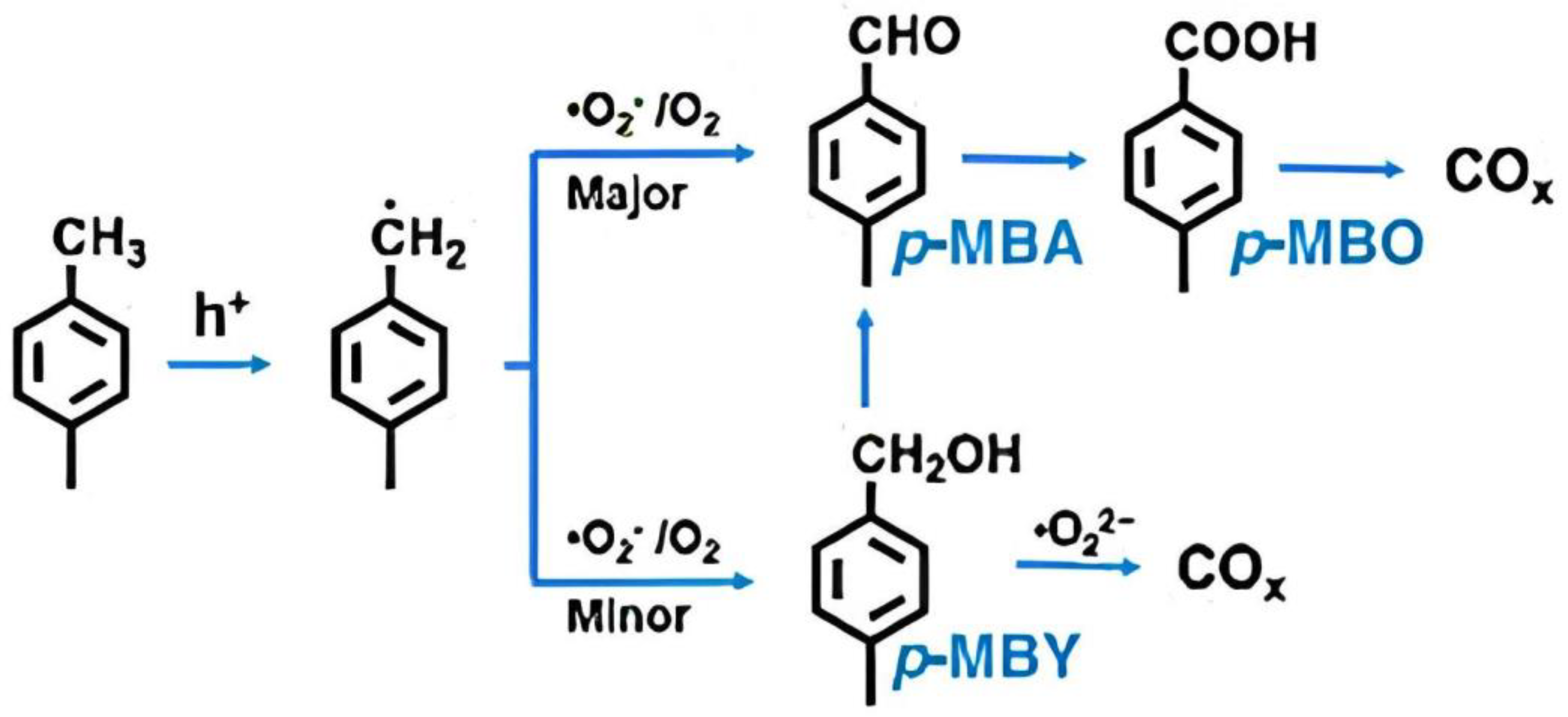
- Sun, X.; Wang, Q.-N.; Wang, S.; Zhang, P.; Feng, Z.; Zhang, X.; Feng, Z.; Li, C. Inhibiting COx formation on WOx-loaded Au/TiO2 photocatalyst for selective oxidation of p-xylene to p-methyl benzaldehyde. J. Catal. 2022, 416, 11–17. [Google Scholar] [CrossRef]
- Li, Z.; Dong, Y.; Zeng, Y.; Zhang, M.; Lv, H.; Yang, G.-Y. A continuous-flow photocatalytic system for highly selective oxidation of p-xylene to terephthalic acid by decatungstate catalyst. Chin. J. Catal. 2024, 66, 282–291. [Google Scholar] [CrossRef]
- Samanta, S.; Srivastava, R. Thermal catalysis vs. photocatalysis: A case study with FeVO4/g-C3N4 nanocomposites for the efficient activation of aromatic and benzylic CH bonds to oxygenated products. Appl. Catal. B Environ. 2017, 218, 621–636. [Google Scholar] [CrossRef]
- Yuan, F.; Cao, P.; Sun, W.; Zhao, L. A Green Oxidation Process of p-Xylene to Terephthalic Acid through the UV Irradiation-Enhanced Ozonation Process. Ind. Eng. Chem. Res. 2024, 63, 8208–8215. [Google Scholar] [CrossRef]
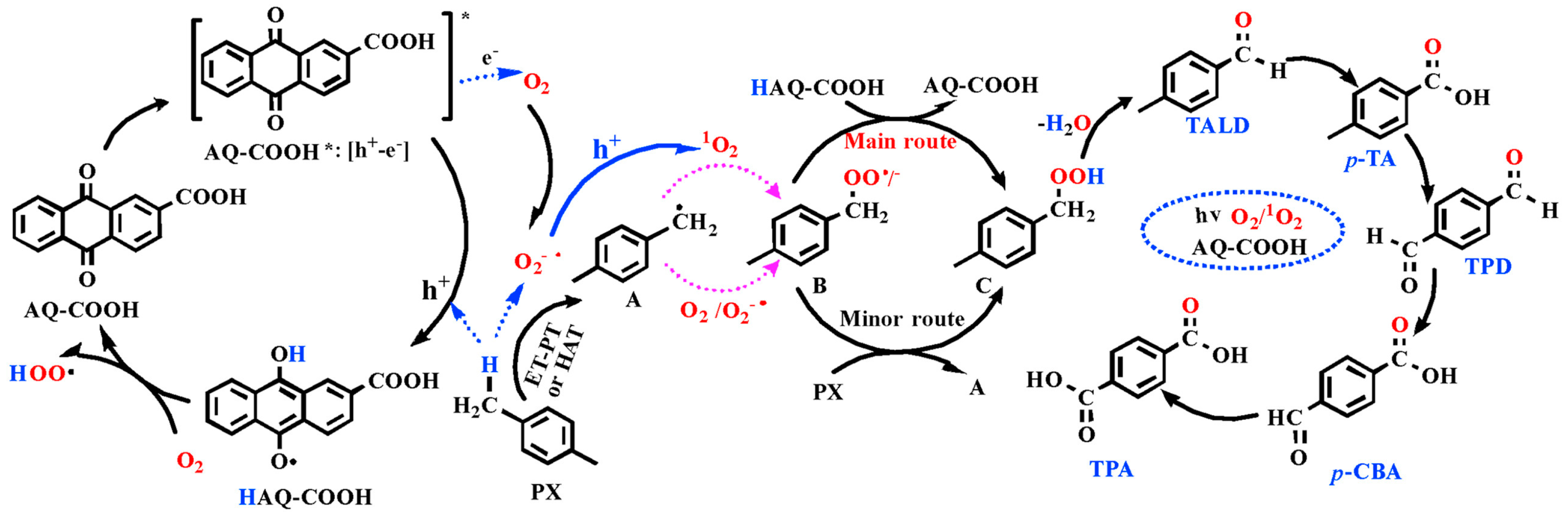
- Jiang, D.; Zhang, Q.; Yang, L.; Deng, Y.; Yang, B.; Liu, Y.; Zhang, C.; Fu, Z. Regulating effects of anthraquinone substituents and additives in photo-catalytic oxygenation of p-xylene by molecular oxygen under visible light irradiation. Renew. Energy 2021, 174, 928–938. [Google Scholar] [CrossRef]
- Miao, X.-L.; Jia, M.-Z.; Yao, X.-R.; Chen, Y.-R.; Pan, J.-Q.; Yu, S.-K.; Zhang, J. Multistage Transformation of Xylene Compounds Enabled by Pyridinium Photocatalyst through Solvent and Light Manipulation. ACS Sustain. Chem. Eng. 2024, 12, 12938–12947. [Google Scholar] [CrossRef]
- Amoneit, M.; Weckowska, D.; Spahr, S.; Wagner, O.; Adeli, M.; Mai, I.; Haag, R. Green chemistry and responsible research and innovation: Moving beyond the 12 principles. J. Clean. Prod. 2024, 484, 144011. [Google Scholar] [CrossRef]
- Xu, L.; Chen, D.; Jiang, H.; Yuan, X. Efficient oxidation of p-xylene to terephthalic acid by using N, N-dihydroxypyromellitimide in conjunction with Co-benzenetricarboxylate. Appl. Catal. A Gen. 2020, 599, 117569. [Google Scholar] [CrossRef]
- Falcon, H.; Campos-Martin, J.M.; Al-Zahrani, S.M.; Fierro, J. Liquid-phase oxidation of p-xylene using N-hydroxyimides. Catal. Commun. 2010, 12, 5–8. [Google Scholar] [CrossRef]
- Heidari, M.; Sedrpoushan, A.; Mohannazadeh, F. Selective oxidation of benzylic C–H using nanoscale graphene oxide as highly efficient carbocatalyst: Direct synthesis of terephthalic acid. Org. Process Res. Dev. 2017, 21, 641–647. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalytic System | Type Of Catalyst | Operating Condition | PX Conversion | Selectivity to TPA | References |
|---|---|---|---|---|---|
| Cu-MOF | heterogeneous | mcatalyst = 20 mg VPX = 1.0 mL (8.1 mmol) Vacetonitrile = 10 mL VH2O2 (30%) = 5.0 mL treaction = 5 h | 86.1%mol | <20% mol | [34] |
| CeO2 | heterogeneous | Vwater = 6 mL PX concentration = 100 mM and mcatalyst = 10 mg Treaction= 95 °C P02 = 1 bar treaction = 8 h | - | 35% | [35] |
| Cobalt acetate | homogeneous | dosagecatalyst = 0.10 mol/mol PX, dosageKBr = 0.545 mmol/mol PX, Treaction = 110 °C, treaction = 6 h, O3 concentration = 20 mg/L gas flow rate = 0.8 L/min. V perchlorethylene = 1 mL | 97.0% ± 0.2 | 81.9% ± 0.1 | [16] |
| N,N -dihydroxypyromellitimide (NDHPI) in conjunction with Co-benzenetricarboxylate (Co-BTC) | homogeneous | Treaction = 150 °C treaction = 12 h macetonitrile = 0.54 g mPX = 25 g, PO2 = 3.0 MPa. m Co-BTC = 0.02 g mNDHPI = 0.372 g | 100% | 96.2% | [66] |
| Nhydroxyphthalimide (NHPI) | homogeneous | treaction = 24 h. Treaction = 100 °C PO2 = 1 bar VPX = 5 mL (40 mmol) dosageNHPI = 8 mmol dosageCo(OAc)2 = 0.2 mmol dosage Mn(OAc)2. = 0.2 mmol Vglacial acetic acid = 100 mL | 100% | 61.9% | [67] |
| N-hydroxysuccinimide (NHSI) | homogeneous | treaction = 24 h. Treaction = 100 °C; PO2 = 1 bar VPX = 5 mL (40 mmol) dosageNHSI = 8 mmol dosageCo(OAc)2 = 0.2 mmol Vglacial acetic acid = 100 mL | 100% | 98.6% | [67] |
| N-hydroxy-1,8-naphthalimide (NHNI) | homogeneous | treaction = 24 h. Treaction = 100 °C; PO2 = 1 bar VPX = 5 mL (40 mmol) dosageNHNI = 8 mmol dosageCo(OAc)2 = 0.2 mmol Vglacial acetic acid = 100 mL | 100% | 84.6% | [67] |
| Mn(OAc)2/Co(OAc)2 | homogeneous | VPX = 0.5 mL (4.055 mmol) VAcOH = 5 mL, dosage KBr = 4.5 mg (0.038 mmol), treaction = 200 C PO2 = 20 atm, mCo(OAc)2 = 32 mg (0.127 mmol) m Mn(OAc)2 = 3.1 mg (0.13 mmol) treaction = 3 h molar ratio PX/acetic acid = 0.08 molar ratio Mn/Co = 1:10 | 98% | 95.2% | [31] |
| Mn2+1Co2+10@MCM-41/HNT | heterogeneous | VPX = 0.5 mL (4.055 mmol) VAcOH = 5 mL dosage KBr = 4.5 mg (0.038 mmol), treaction = 200 C PO2 = 20 atm treaction = 3 h molar ratio PX/acetic acid = 0.08 molar ratio Mn/Co = 1:10 treaction = 3 h dosageMnII1CoII10@MCM-41/HNT = 150 mg | 99% | 93.8% | [31] |
| Nanoscale graphene oxide sheets (NGO) | heterogeneous | H2O/Acetone = 5/1 concentrationNGO = 200 wt % treaction= 24 h T = 100 °C concentrationH2O2 = 7 eq | 70% | 99% | [68] |
| Photocatalytic System | Type of Photocatalyst | Operating Conditions | PX Conversion | Selectivity to TPA | References |
|---|---|---|---|---|---|
| TiO2 | heterogeneous | PX = 0.4 mmol, mTiO2 = 20 mg Vacetonitrile = 4 mL, VH3PO4 = 4 μL, O2 T = 25 °C, treaction = 1 h. light source = UV LED strip λ = 365 nm (15 W) | 15.2% | _ | [56] |
| Au/WOx/TiO2 | heterogeneous | VPX = 2 mL, mAu/WOx/TiO2 =10 mg treaction 1 h, T = 25 °C light source = 300 W Xe-lamp | 1% | _ | [59] |
| tetrabutylammonium decatungstate (TBADT) | CPX = 10 mmol L−1 CTBADT = 0.5 mmol L−1 Vacetonitrile = 20 mL CHCl = 1 mol L−1 (37.5%) light source = 365 nm LED light intensity = 360 mW cm−2 T = 20 °C treaction =19 h. | 100% | 93.4% | [60] | |
| MoO2/MoP/NF | heterogeneous | Electrocatalytic oxidation of PX: Canodic electrolyte = 25 mM Ccathodic electrolyte solution = 1 M KOH with 30% acetonitrile (VCH3CN: VH2O = 3/7) | 71.6% | 94.8% | [58] |
| O(1D) by decomposition of O3 | heterogeneous | Gas-liquid reaction Acetonitrile as solvent Light source = UV lamp (Hg light, 500 W = CO3 = 120 mg/L, gas flow rate = 250 mL/min; P = 1 atm; CPX = 5 wt % treaction = 16 h | 100% | 78% | [62] |
| 2-carboxyanthraquinone (AQCOOH) | homogeneous | PX = 0.5 mmol AQ-COOH = 7.5 mol% O2 P = 1 atm Vacetone = 5 mL light source = 35 W tungsten-bromine lamp treaction = 12 h. | 70.9%, | 2.9% | [63] |
| 4,4′,4″-(1,3,5-triazine-2,4,6-triyl)tris(1-benzylpyridinium)bromide (TPT-3XB) | heterogeneous | PX = 0.10 mmol TPT-3XB = 2.4 mol % Vacetonitrile = 5 mL light source = 365 nm treaction = 3 h in an open-air vessel | 96% | <1% | [64] |
| 4,4′,4″-(1,3,5-triazine-2,4,6-triyl)tris(1-benzylpyridin-1-ium)bromide (TPT-3XB) | heterogeneous | PX = 0.10 mmol TPT-3XB = 2.4 mol % Vacetonitrile = 5 mL light source = 365 nm treaction = 9 h Acetonitrile/H2O = 2:3 v/v in an open-air vessel | 81% | <1% | [64] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancuso, A.; Sacco, O.; Vaiano, V. Advances in Selective Photocatalytic Oxidation of p-Xylene to Terephthalic Acid as a Sustainable Route: A Short Review on Photocatalyst Formulation and Related Reaction Mechanisms. Photochem 2025, 5, 11. https://doi.org/10.3390/photochem5020011
Mancuso A, Sacco O, Vaiano V. Advances in Selective Photocatalytic Oxidation of p-Xylene to Terephthalic Acid as a Sustainable Route: A Short Review on Photocatalyst Formulation and Related Reaction Mechanisms. Photochem. 2025; 5(2):11. https://doi.org/10.3390/photochem5020011
Chicago/Turabian StyleMancuso, Antonietta, Olga Sacco, and Vincenzo Vaiano. 2025. "Advances in Selective Photocatalytic Oxidation of p-Xylene to Terephthalic Acid as a Sustainable Route: A Short Review on Photocatalyst Formulation and Related Reaction Mechanisms" Photochem 5, no. 2: 11. https://doi.org/10.3390/photochem5020011
APA StyleMancuso, A., Sacco, O., & Vaiano, V. (2025). Advances in Selective Photocatalytic Oxidation of p-Xylene to Terephthalic Acid as a Sustainable Route: A Short Review on Photocatalyst Formulation and Related Reaction Mechanisms. Photochem, 5(2), 11. https://doi.org/10.3390/photochem5020011