

An Explainable AI System for the Diagnosis of High-Dimensional Biomedical Data


 , , and
, , and
Abstract

1. Introduction
- Algorithmic population descriptions (ALPODS) is a novel supervised explainable AI (XAI) that provides physicians with tailored explanations in biological datasets.
- ALPODS is fast-working and requires for learning only a very low number of cases.
- ALPODS distinguishes normal controls from leukemia and bone marrow from peripheral blood samples with high accuracy.
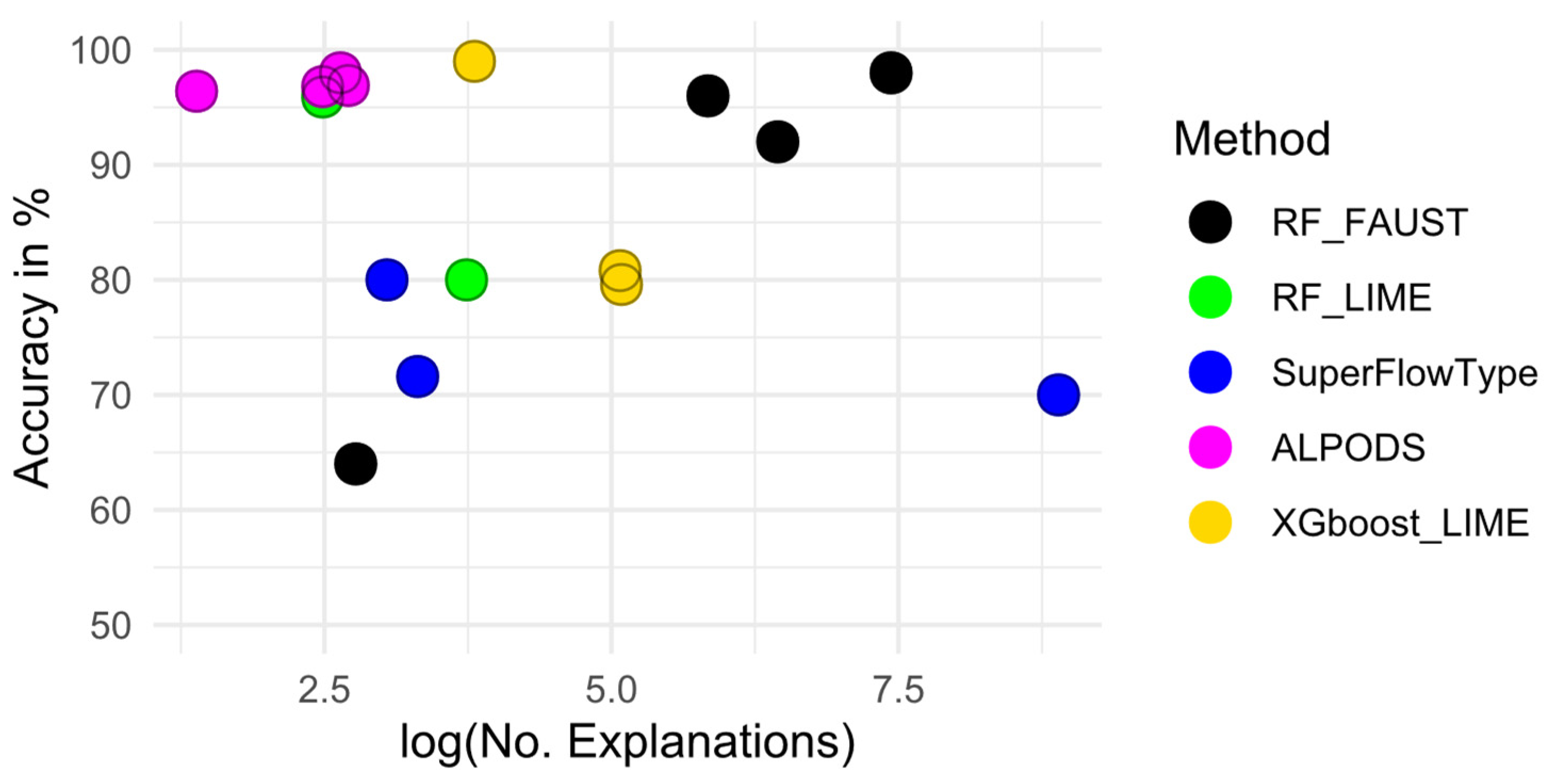
- ALPODS outperforms comparable systems on several datasets in terms of interpretability, performance, and processing time.
- The XAI was already successfully applied for the hemodilution in BM samples to prevent false negative MRD reports and was able to identify to physicians prior unknown and highly predictive cell populations and improved chronic lymphocytic leukemia outcome predictions.
2. Methods
2.1. Supervised and Unsupervised Decision Trees
2.2. Random Forest and Gradient Boosting with LIME (RF-LIME, Xgboost-LIME)
2.3. Comparable XAI Methods Used in Flow Cytometry
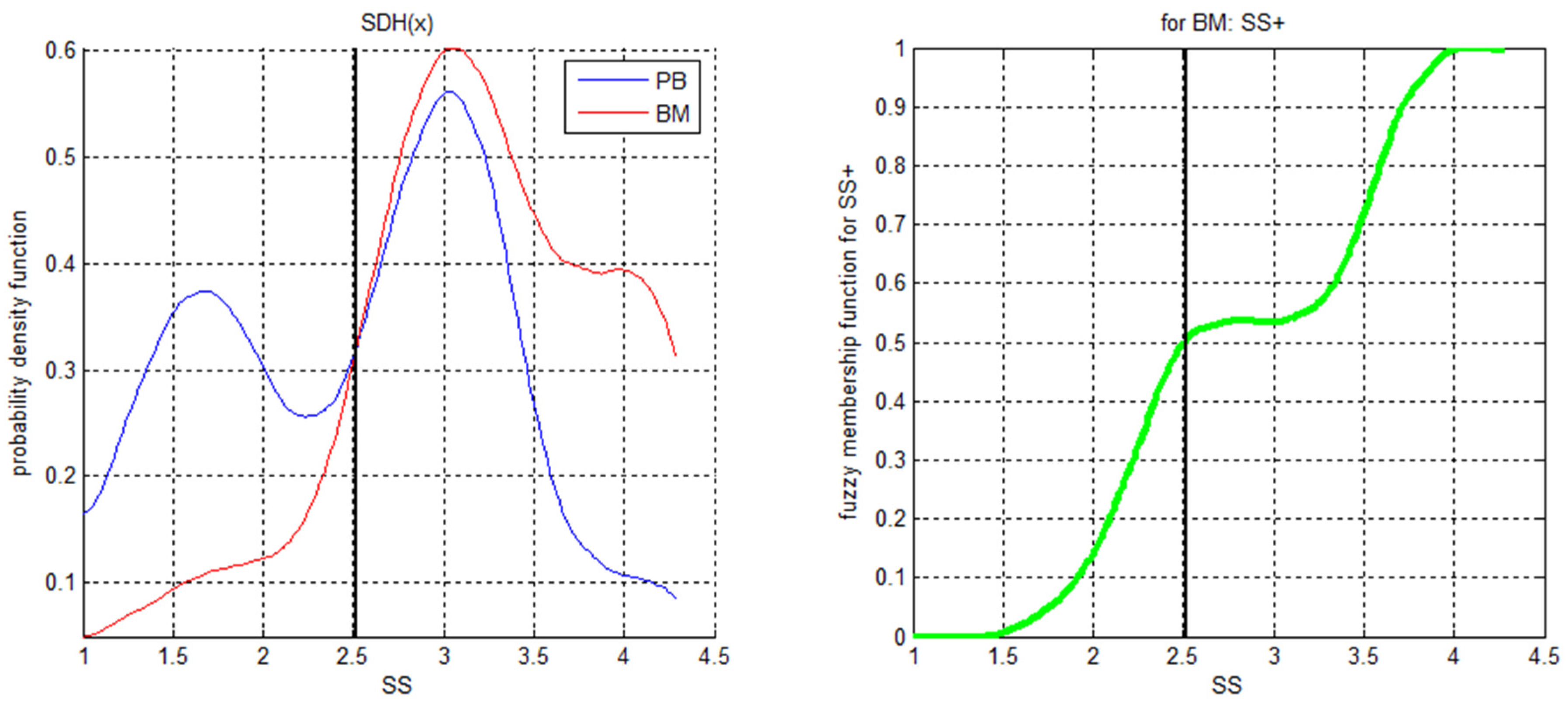
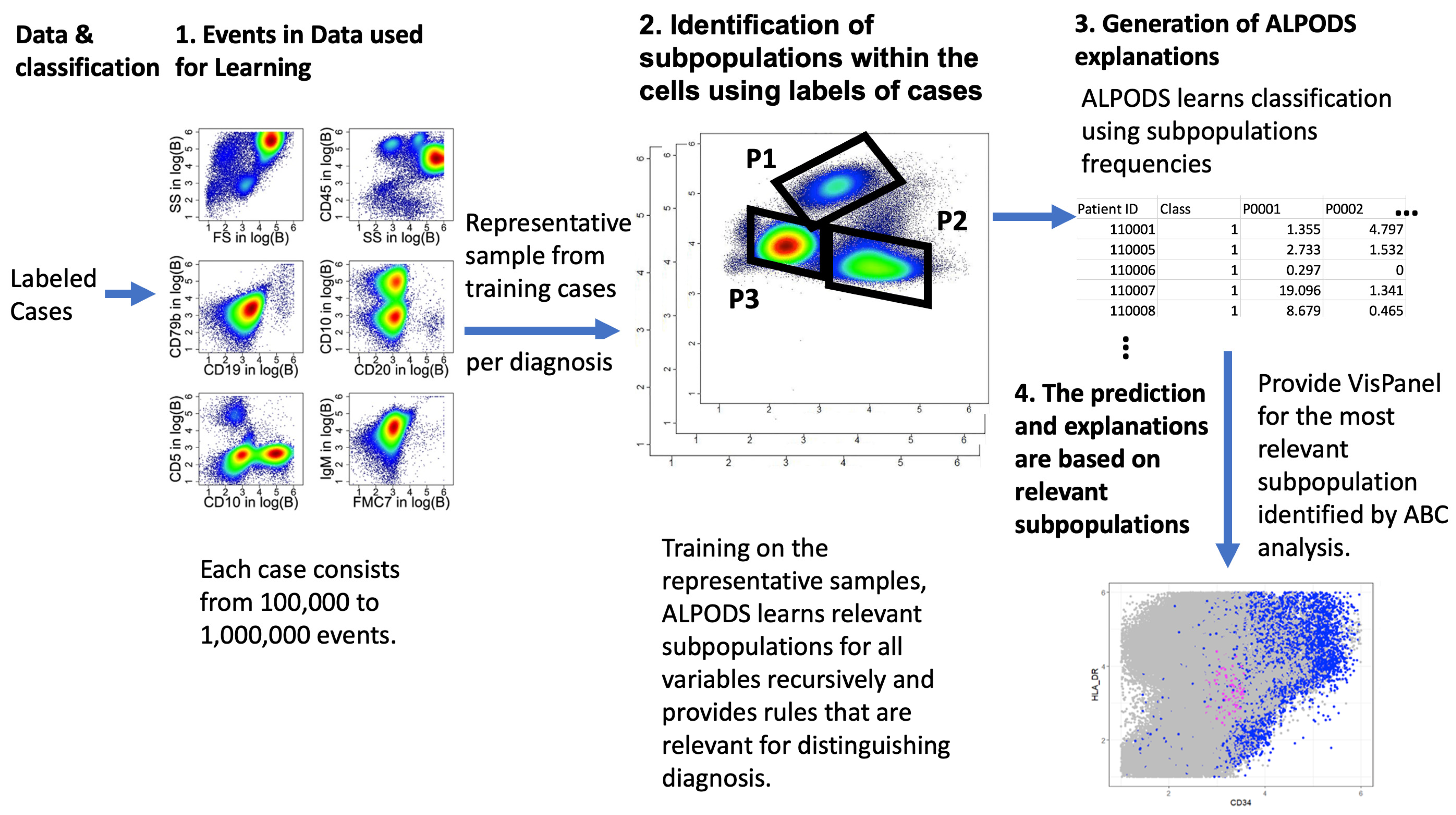
2.4. ALPODS–Algorithmic Population Descriptions
| Algorithm 1: ALPODS | |||||||
| DAG = GrowDAGforALPODS(DAG,CDi,depth,Population,Classification) | |||||||
| Input: | |||||||
| DAG | |||||||
| CDi | |||||||
| depth | |||||||
| Population | |||||||
| Classifcation | |||||||
| 1: | if Termination(depth, size(Population), Classification) then | ||||||
| 2: | leafLabel = classify(Population, Classification) | ▷ Majority vote | |||||
| 3: | DAG = AddLeafToDAG(DAG, leafLabel) | ▷ DAG is complete at this leaf | |||||
| 4: | else | ▷ Termination not reached | |||||
| 5: | for each variable CDi do | ||||||
| 6: | for each plausible condition cond do | ||||||
| 7: | Let cond(Population) be the subpopulation resulting from the application of cond. | ||||||
| 8: | Use the probability distributions PDF(Population) and PDF(cond(Population)) | ||||||
| 9: | to calculate a decision on membership using theorem of Bayes | ||||||
| 10: | ⇒ ClassOfSubpopulation | ||||||
| 11: | S(CDi) = SimpsonIndex(Population, cond(Population)) | ||||||
| 12: | if Significant(S(CDi)) then | ||||||
| 13: | AddEdgeToDAG(cond(Population),Label=”cond on CDi”) | ▷ Note: edge is only added if it is not already descendent of current DAG | |||||
| 14: | DAG = GrowDAGforALPODS(DAG,CDi,depth+1,Subpopulation,ClassOfSubpopulation) | ▷ Expand the DAG recursively | |||||
| 15: | end if | ▷ Only for suitable S | |||||
| 16: | end for | ▷ For all conditions | |||||
| 17: | end for | ▷ For all variables | |||||
| 18: | end if | ▷ Expansion of the DAG | |||||
| Used Procedures: | |||||||
| 1: | Termination() ▷ Is true if depth size or size exceeds limits or classification contains only one class | ||||||
| 2: | AddLeafToDAG() ▷ Adds a leaf node to the DAG | ||||||
| 3: | AddEdgeToDAG() ▷ Adds edge with label, edge is only added if it is not already descendant of current DAG | ||||||
| 4: | Significant() ▷ Threshold for S | ||||||
| 5. | SimpsonIndex() ▷ Computes the Simpson index S | ||||||
3. Data Description
3.1. Synthetic Data with Gaussian Noise Derived from Iris
3.2. Marburg and Dresden Data
4. Quality Assessment
4.1. Evaluation of Randomized Synthetic Data
4.2. Evaluation of Flow Cytometry Data
5. Results
The Applications of ALPODS
6. Discussion
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Keyes, T.J.; Domizi, P.; Lo, Y.C.; Nolan, G.P.; Davis, K.L. A cancer biologist’s primer on machine learning applications in high-dimensional cytometry. Cytom. A 2020, 97, 782–799. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Glicksberg, B.S.; Butte, A.J. Robust prediction of clinical outcomes using cytometry data. Bioinformatics 2019, 35, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Mallesh, N.; Höllein, A.; Schabath, R.; Haferlach, C.; Haferlach, T.; Elsner, F.; Lüling, H.; Krawitz, P.; Kern, W. Hematologist-level classification of mature B-cell neoplasm using deep learning on multiparameter flow cytometry data. Cytom. A 2020, 97, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.P. Machine Learning: A Probabilistic Perspective; MIT Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Wainberg, M.; Alipanahi, B.; Frey, B.J. Are random forests truly the best classifiers? J. Mach. Learn. Res. 2016, 17, 3837–3841. [Google Scholar]
- Delgado, M.F.; García, E.C.; Ameneiro, S.B.; Amorim, D.G. Do we need hundreds of classifiers to solve real world classification problems? J. Mach. Learn. Res. 2014, 15, 3133–3181. [Google Scholar]
- Ultsch, A. The integration of connectionist models with knowledge-based systems: Hybrid systems. In Proceedings of the SMC’98 Conference Proceedings—1998 IEEE International Conference on Systems, Man, and Cybernetics (Cat. No.98CH36218), San Diego, CA, USA, 14 October 1998; pp. 1530–1535. [Google Scholar]
- Tjoa, E.; Guan, C. A survey on explainable artificial intelligence (XAI): Toward medical XAI. IEEE Trans. Neural Netw. Learn. Syst. 2020, 32, 4793–4813. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C. Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nat. Mach. Intell. 2019, 1, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Kahneman, D.; Miller, D.T. Norm theory: Comparing reality to its alternatives. Psychol. Rev. 1986, 93, 136. [Google Scholar] [CrossRef]
- Sen, S.; Knight, L. A genetic prototype learner. IJCAI 1995, 1, 725–733. [Google Scholar]
- Nakamura, E.; Kehtarnavaz, N. Determining number of clusters and prototype locations via multi-scale clustering. Pattern Recognit. Lett. 1998, 19, 1265–1283. [Google Scholar] [CrossRef]
- Vesanto, J. SOM-based data visualization methods. Intell. Data Anal. 1999, 3, 111–126. [Google Scholar] [CrossRef]
- Thrun, M.C.; Ultsch, A. Uncovering High-Dimensional Structures of Projections from Dimensionality Reduction Methods. MethodsX 2020, 7, 101093. [Google Scholar] [CrossRef]
- Angelov, P.; Soares, E. Towards explainable deep neural networks (xDNN). Neural Netw. 2020, 130, 185–194. [Google Scholar] [CrossRef]
- Stöger, K.; Schneeberger, D.; Holzinger, A. Medical artificial intelligence: The European legal perspective. Commun. ACM 2021, 64, 34–36. [Google Scholar] [CrossRef]
- Adadi, A.; Berrada, M. Peeking inside the black-box: A survey on Explainable Artificial Intelligence (XAI). IEEE Access 2018, 6, 52138–52160. [Google Scholar] [CrossRef]
- Hayes-Roth, F.; Waterman, D.A.; Lenat, D.B. Building Expert System; Addison-Wesley Publishing Co.: Reading, MA, USA, 1983. [Google Scholar]
- Breiman, L.; Friedman, J.; Stone, C.J.; Olshen, R.A. Classification and Regression Trees; CRC Press: Boca Raton, FL, USA, 1984. [Google Scholar]
- Salzberg, S.L. C4.5: Programs for machine learning by J. Ross Quinlan. Morgan Kaufmann Publishers, Inc., 1993. Mach. Learn. 1994, 16, 235–240. [Google Scholar] [CrossRef]
- Cohen, W.W. Fast effective rule induction. In Proceedings of the Twelfth International Conference on Machine Learning, Tahoe City, CA, USA, 9–12 July 1995; Prieditis, A., Russell, S., Eds.; Morgan Kaufmann: Tahoe City, CA, USA, 1995; pp. 115–123. [Google Scholar] [CrossRef]
- Dehuri, S.; Mall, R. Predictive and comprehensible rule discovery using a multi-objective genetic algorithm. Knowl.-Based Syst. 2006, 19, 413–421. [Google Scholar] [CrossRef]
- Miller, G.A. The magical number seven, plus or minus two: Some limits on our capacity for processing information. Psychol. Rev. 1956, 63, 81–97. [Google Scholar] [CrossRef]
- Ribeiro, M.T.; Singh, S.; Guestrin, C. “Why should I trust you?” Explaining the predictions of any classifier. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 1135–1144. [Google Scholar]
- Loyola-González, O.; Gutierrez-Rodríguez, A.E.; Medina-Pérez, M.A.; Monroy, R.; Martínez-Trinidad, J.F.; Carrasco-Ochoa, J.A.; García-Borroto, M. An explainable artificial intelligence model for clustering numerical databases. IEEE Access 2020, 8, 52370–52384. [Google Scholar] [CrossRef]
- Aghaeepour, N.; Jalali, A.; O’Neill, K.; Chattopadhyay, P.K.; Roederer, M.; Hoos, H.H.; Brinkman, R.R. RchyOptimyx: Cellular hierarchy optimization for flow cytometry. Cytom. A 2012, 81, 1022–1030. [Google Scholar] [CrossRef]
- O’Neill, K.; Jalali, A.; Aghaeepour, N.; Hoos, H.; Brinkman, R.R. Enhanced flowType/RchyOptimyx: A bioconductor pipeline for discovery in high-dimensional cytometry data. Bioinformatics 2014, 30, 1329–1330. [Google Scholar] [CrossRef]
- Mason, D. Leucocyte Typing VII: White Cell Differentiation Antigens: Proceedings of the Seventh International Workshop and Conference Held in Harrogate, United Kindom; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Shapiro, H.M. Practical Flow Cytometry; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Lipton, Z.C. The mythos of model interpretability. Queue 2018, 16, 31–57. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Lee, S.I. A unified approach to interpreting model predictions. In Advances in Neural Information Processing System; Guyon, I., Luxburg, U., Bengio, S., Wallach, H., Fergus, R., Vishwanathan, S., Garnett, R., Eds.; Curran Associates Inc.: Long Beach, CA, USA, 2017; pp. 4765–4774. [Google Scholar]
- Greene, E.; Finak, G.; D’Amico, L.A.; Bhardwaj, N.; Church, C.D.; Morishima, C.; Ramchurren, N.; Taube, J.M.; Nghiem, P.T.; Cheever, M.A. New interpretable machine-learning method for single-cell data reveals correlates of clinical response to cancer immunotherapy. Patterns 2021, 2, 100372. [Google Scholar] [CrossRef]
- Vick, S.C.; Frutoso, M.; Mair, F.; Konecny, A.J.; Greene, E.; Wolf, C.R.; Logue, J.K.; Franko, N.M.; Boonyaratanakornkit, J.; Gottardo, R. A regulatory T cell signature distinguishes the immune landscape of COVID-19 patients from those with other respiratory infections. Sci. Adv. 2021, 7, eabj0274. [Google Scholar] [CrossRef] [PubMed]
- Ripley, B.D. Pattern Recognition and Neural Networks; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Dasgupta, S.; Frost, N.; Moshkovitz, M.; Rashtchian, C. Explainable k-means and k-medians clustering. In Proceedings of the 37th International Conference on Machine Learning, Vienna, Austria, 13–18 July 2020; Daumé, H., III, Singh, A., Eds.; 2020; pp. 7055–7065. [Google Scholar]
- Thrun, M.C.; Ultsch, A.; Breuer, L. Explainable AI framework for multivariate hydrochemical time series. Mach. Learn. Knowl. Extr. 2021, 3, 170–205. [Google Scholar] [CrossRef]
- Thrun, M.C. Exploiting Distance-Based Structures in Data Using an Explainable AI for Stock Picking. Information 2022, 13, 51. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Grabmeier, J.L.; Lambe, L.A. Decision trees for binary classification variables grow equally with the Gini impurity measure and Pearson’s chi-square test. Int. J. Bus. Intell. Data Min. 2007, 2, 213–226. [Google Scholar] [CrossRef]
- Burkart, N.; Huber, M.F. A survey on the explainability of supervised machine learning. J. Artif. Intell. Res. 2021, 70, 245–317. [Google Scholar] [CrossRef]
- Jesus, S.; Belém, C.; Balayan, V.; Bento, J.; Saleiro, P.; Bizarro, P.; Gama, J. How can I choose an explainer? An application-grounded evaluation of post-hoc explanations. In Proceedings of the 2021 ACM Conference on Fairness, Accountability, and Transparency, Toronto, Canada, 3–10 March 2021; pp. 805–815. [Google Scholar]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM Sigkdd International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Le, N.Q.K.; Do, D.T.; Chiu, F.-Y.; Yapp, E.K.Y.; Yeh, H.-Y.; Chen, C.-Y. XGBoost improves classification of MGMT promoter methylation status in IDH1 wildtype glioblastoma. J. Pers. Med. 2020, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Linde, Y.; Buzo, A.; Gray, R. An algorithm for vector quantizer design. IEEE Trans. Commun. 1980, 28, 84–95. [Google Scholar] [CrossRef]
- Lo, K.; Hahne, F.; Brinkman, R.R.; Gottardo, R. flowClust: A bioconductor package for automated gating of flow cytometry data. BMC Bioinform. 2009, 10, 145. [Google Scholar] [CrossRef] [PubMed]
- Villanova, F.; Di Meglio, P.; Inokuma, M.; Aghaeepour, N.; Perucha, E.; Mollon, J.; Nomura, L.; Hernandez-Fuentes, M.; Cope, A.; Prevost, A.T. Integration of lyoplate based flow cytometry and computational analysis for standardized immunological biomarker discovery. PLoS ONE 2013, 8, e65485. [Google Scholar] [CrossRef] [PubMed]
- Craig, F.E.; Brinkman, R.R.; Eyck, S.T.; Aghaeepour, N. Computational analysis optimizes the flow cytometric evaluation for lymphoma. Cytom. B Clin. Cytom. 2014, 86, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Aghaeepour, N.; Finak, G.; Hoos, H.; Mosmann, T.R.; Brinkman, R.; Gottardo, R.; Scheuermann, R.H. Critical assessment of automated flow cytometry data analysis techniques. Nat. Methods 2013, 10, 228–238. [Google Scholar] [CrossRef]
- Aghaeepour, N.; Chattopadhyay, P.; Chikina, M.; Dhaene, T.; Van Gassen, S.; Kursa, M.; Lambrecht, B.N.; Malek, M.; McLachlan, G.; Qian, Y. A benchmark for evaluation of algorithms for identification of cellular correlates of clinical outcomes. Cytom. A 2016, 89, 16–21. [Google Scholar] [CrossRef]
- Hartigan, J.A.; Hartigan, P.M. The dip test of unimodality. Ann. Stat. 1985, 13, 70–84. [Google Scholar] [CrossRef]
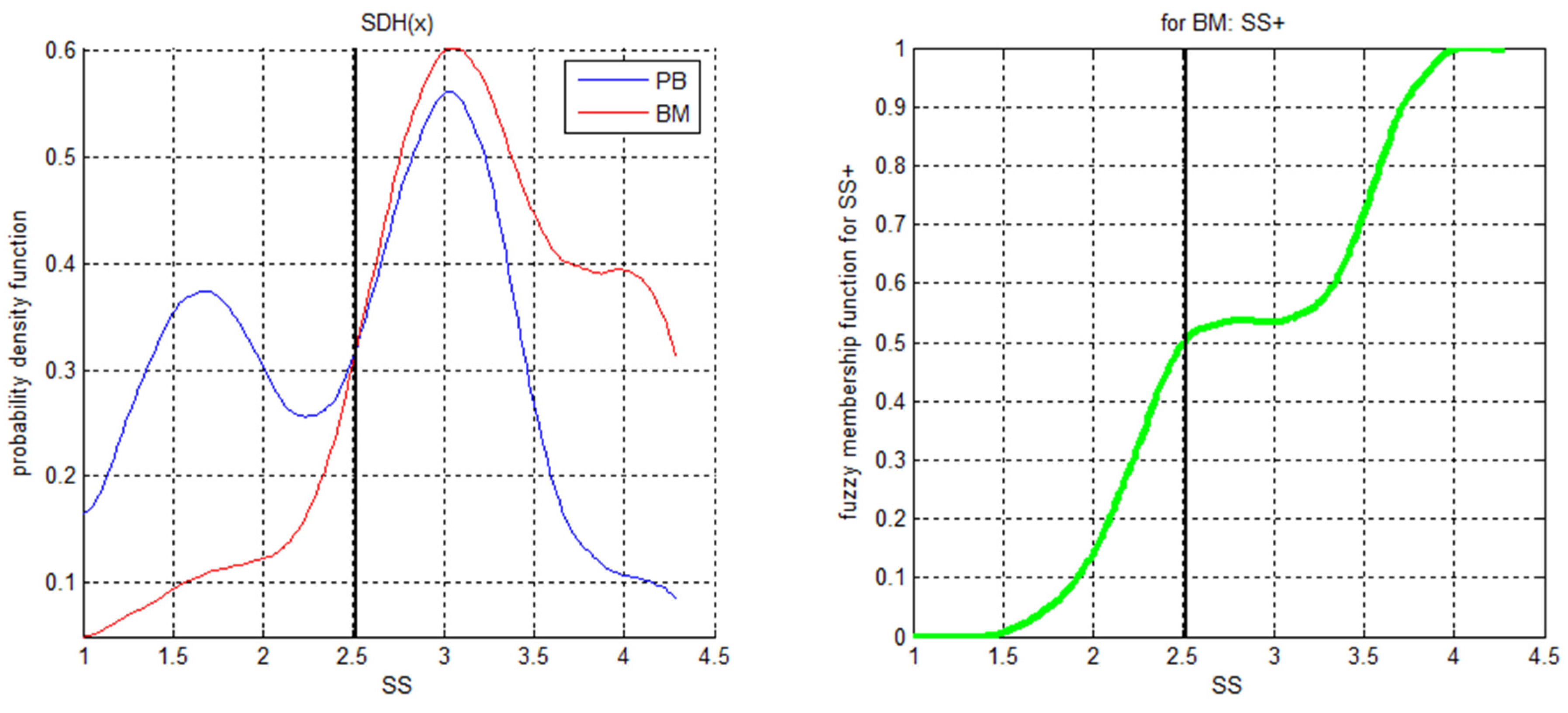
- Hurulbert, S. The nonconcept of species diversity: A critique and alternatives parameters. Ecology 1971, 52, 577–586. [Google Scholar] [CrossRef]
- McGrayne, S.B. The Theory That Would Not Die: How Bayes’ Rule Cracked the Enigma Code, Hunted Down Russian Submarines, & Emerged Triumphant from Two Centuries of Controversy; Yale University Press: New Haven, CT, USA, 2011. [Google Scholar]
- Ruck, D.W.; Rogers, S.K.; Kabrisky, M.; Oxley, M.E.; Suter, B.W. The multilayer perceptron as an approximation to a Bayes optimal discriminant function. IEEE Trans. Neural Netw. 1990, 1, 296–298. [Google Scholar] [CrossRef]
- Freitas, A.A. Comprehensible classification models: A position paper. ACM SIGKDD Explor. Newsl. 2014, 15, 1–10. [Google Scholar] [CrossRef]
- Luan, S.; Schooler, L.J.; Gigerenzer, G. A signal-detection analysis of fast-and-frugal trees. Psychol. Rev. 2011, 118, 316–338. [Google Scholar] [CrossRef]
- Wilson, E.J.; Sherrell, D.L. Source effects in communication and persuasion research: A meta-analysis of effect size. J. Acad. Mark. Sci. 1993, 21, 101–112. [Google Scholar] [CrossRef]
- Cohen, J. Statistical Power Analysis for the Behavioral Sciences; Academic Press: New York, NU, USA, 2013. [Google Scholar]
- Ultsch, A.; Lötsch, J. Computed ABC Analysis for Rational Selection of Most Informative Variables in Multivariate Data. PLoS ONE 2015, 10, e0129767. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.; Howe, P.; Sonenberg, L.; AI, E. Explainable AI: Beware of inmates running the asylum. arXiv 2017, arXiv:1712.00547. [Google Scholar]
- Miller, T. Explanation in artificial intelligence: Insights from the social sciences. Artif. Intell. 2019, 267, 1–38. [Google Scholar] [CrossRef]
- Thrun, M.C. Identification of explainable structures in data with a human-in-the-loop. KI Künstliche Intelligenz 2022, 36, 297–301. [Google Scholar] [CrossRef]
- Thrun, M.C.; Ultsch, A. Effects of the payout system of income taxes to municipalities in Germany. In Proceedings of the 12th Professor Aleksander Zelias International Conference on Modelling and Forecasting of Socio-Economic Phenomena, Kraków, Poland, 8–11 May 2018; pp. 533–542. [Google Scholar]
- Eilers, P.H.; Goeman, J.J. Enhancing scatterplots with smoothed densities. Bioinformatics 2004, 20, 623–628. [Google Scholar] [CrossRef]
- Mamdani, E.H.; Assilian, S. An experiment in linguistic synthesis with a fuzzy logic controller. Int. J. Man Mach. Stud. 1975, 7, 1–13. [Google Scholar] [CrossRef]
- Bodenhofer, U.; Danková, M.; Stepnicka, M.; Novák, V. A plea for the usefulness of the deductive interpretation of fuzzy rules in engineering applications. In Proceedings of the 2007 IEEE International Fuzzy Systems Conference, London, UK, 23–26 July 2007; pp. 1–6. [Google Scholar]
- Thrun, M.C.; Hoffman, J.; Röhnert, M.; Von Bonin, M.; Oelschlägel, U.; Brendel, C.; Ultsch, A. Flow cytometry datasets consisting of peripheral blood and bone marrow samples for the evaluation of explainable artificial intelligence methods. Data Br. 2022, 43, 108382. [Google Scholar] [CrossRef] [PubMed]
- Bacigalupo, A.; Tong, J.; Podesta, M.; Piaggio, G.; Figari, O.; Colombo, P.; Sogno, G.; Tedone, E.; Moro, F.; Van Lint, M. Bone marrow harvest for marrow transplantation: Effect of multiple small (2 mL) or large (20 mL) aspirates. Bone Marrow Transplant. 1992, 9, 467–470. [Google Scholar] [PubMed]
- Muschler, G.F.; Boehm, C.; Easley, K. Aspiration to obtain osteoblast progenitor cells from human bone marrow: The influence of aspiration volume. J. Bone Joint Surg. 1997, 79, 1699–1709. [Google Scholar] [CrossRef]
- Thrun, M.C.; Ultsch, A. Clustering benchmark datasets exploiting the fundamental clustering problems. Data Br. 2020, 30, 105501. [Google Scholar] [CrossRef]
- Thrun, M.C. Distance-based clustering challenges for unbiased benchmarking studies. Nat. Sci. Rep. 2021, 11, 18988. [Google Scholar] [CrossRef]
- Anderson, E. The irises of the gaspé Peninsula. Bull. Am. Iris Soc. 1935, 39, 2–5. [Google Scholar]
- Setzu, M.; Guidotty, R.; Mionreale, A.; Turini, F.; Pedreschie, D.; Gianotti, F. GLocalX—From local to global explanations of black box AI models. Artif. Intell. 2021, 294, 103457. [Google Scholar] [CrossRef]
- Ritter, G. Robust Cluster Analysis and Variable Selection; CRC Press: Passau, Germany, 2014. [Google Scholar]
- Florkowski, C.M. Sensitivity, specificity, receiver-operating characteristic (ROC) curves and likelihood ratios: Communicating the performance of diagnostic tests. Clin. Biochem. Rev. 2008, 29, S83–S87. [Google Scholar]
- Langer, I.; Von Thun, F.S.; Tausch, R.; Höder, J. Sich Verständlich Ausdrücken; Ernst Reinhardt: München, Germany, 1999. [Google Scholar]
- Kane, E.; Howell, D.; Smith, A.; Crouch, S.; Burton, C.; Roman, E.; Patmore, R. Emergency admission and survival from aggressive non-Hodgkin lymphoma: A report from the UK’s population-based haematological malignancy research network. Eur. J. Cancer 2017, 78, 53–60. [Google Scholar] [CrossRef]
- Thrun, M.C.; Stier, Q. Fundamental clustering algorithms suite. SoftwareX 2021, 13, 100642. [Google Scholar] [CrossRef]
- Group, I.C.-I.W. An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): A meta-analysis of individual patient data. Lancet Oncol. 2016, 17, 779–790. [Google Scholar]
- Hoffmann, J.; Eminovic, S.; Wilhelm, C.; Krause, S.W.; Neubauer, A.; Thrun, M.C.; Ultsch, A.; Brendel, C. Prediction of clinical outcomes with explainable artificial intelligence in patients with chronic lymphocytic leukemia. Curr. Oncol. 2023, 30, 1903–1915. [Google Scholar] [CrossRef]
- Short, N.J.; Zhou, S.; Fu, C.; Berry, D.A.; Walter, R.B.; Freeman, S.D.; Hourigan, C.S.; Huang, X.; Gonzalez, G.N.; Hwang, H. Association of measurable residual disease with survival outcomes in patients with acute myeloid leukemia: A systematic review and meta-analysis. JAMA Oncol. 2020, 6, 1890–1899. [Google Scholar] [CrossRef]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; Al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.; Gradowska, P.L.; Meijer, R.; Cloos, J. Molecular minimal residual disease in acute myeloid leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Heuser, M.; Freeman, S.D.; Ossenkoppele, G.J.; Buccisano, F.; Hourigan, C.S.; Ngai, L.L.; Tettero, J.M.; Bachas, C.; Baer, C.; Béné, M.-C. 2021 Update on MRD in acute myeloid leukemia: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2021, 138, 2753–2767. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; Thrun, M.C.; Röhnert, M.; Von Bonin, M.; Oelschlägel, U.; Neubauer, A.; Ultsch, A.; Brendel, C. Identification of critical hemodilution by artificial intelligence in bone marrow assessed for minimal residual disease analysis in acute myeloid leukemia: The Cinderella method. Cytom. Part A 2022, 103, 304–312. [Google Scholar] [CrossRef]
- Holdrinet, R.; Van Egmond, J.; Kessels, J.; Haanen, C. A method for quantification of peripheral blood admixture in bone marrow aspirates. Exp. Hematol. 1980, 8, 103–107. [Google Scholar]
- Delgado, J.A.; Guillén-Grima, F.; Moreno, C.; Panizo, C.; Pérez-Robles, C.; Mata, J.J.; Moreno, L.; Arana, P.; Chocarro, S.; Merino, J. A simple flow-cytometry method to evaluate peripheral blood contamination of bone marrow aspirates. J. Immunol. Methods 2017, 442, 54–58. [Google Scholar] [CrossRef]
- Abrahamsen, J.F.; Lund-Johansen, F.; Laerum, O.D.; Schem, B.C.; Sletvold, O.; Smaaland, R. Flow cytometric assessment of peripheral blood contamination and proliferative activity of human bone marrow cell populations. Cytom. A 1995, 19, 77–85. [Google Scholar] [CrossRef]
- Thrun, M.C.; Ultsch, A. Using projection based clustering to find distance and density based clusters in high-dimensional data. J. Classif. 2020, 38, 280–312. [Google Scholar] [CrossRef]
- Lötsch, J.; Ultsch, A. Exploiting the structures of the U-matrix. In Advances in Self-Organizing Maps and Learning Vector Quantization; Villmann, T., Schleif, F.M., Kaden, M., Lange, M., Eds.; Springer International Publishing: Mittweida, Germany, 2014; pp. 249–257. [Google Scholar]
- Hotelling, H. Analysis of a complex of statistical variables into principal components. J. Educ. Psychol. 1933, 24, 417. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthetic Data with Gaussian Noise | eUD3.5 | FAUST * | RF-LIME | Xgboost-LIME | Super FlowType | ALPODS |
|---|---|---|---|---|---|---|
| Processing Time | <1 min | <1 min | <1 min | <1 min | <1 min | <1 min |
| No. Cross-Validation Steps | 50 | * | 50 | 50 | 50 | 50 |
| Max no. of SP | 15 | 4 | 14 | 3 | 30 | 5 |
| Mean no. of SP | 8 ± 3.4 | 4 | 6 ± 3.2 | 3 | 7 ± 3 | 2 ± 1 |
| Max no. of Conditions for a SP | 4 | 4 | 2 | 2 | 4 | 4 |
| Mean no. of Conditions for a SP | 2 ± 1.1 | 4 | 2 ± 0.5 | 1.5 | 3 ± 0.8 | 2 ± 0.7 |
| Accuracy Performance | 98 ± 0.5 | 64 * | 96.3 ± 1.4 | 99 ± 0.7 | 80 ± 10 | 96 ± 0 |
| Marburg Dataset | RF-FAUST | RF-LIME | Xgboost-LIME | SuperFlowType | ALPODS |
|---|---|---|---|---|---|
| Processing Time | <1 min | 72 h | 15 min | 24 h | 1 min |
| No. Cross-Validation Steps | * | 2 | 50 | 3 | 50 |
| Max no. of SP | 79 | 40 | 2 | 5486 | 5 |
| Mean no. of SP | 79 | 21 ± 11.6 | 2 | 2744 ± 1583 | 3 ± 1 |
| Max no. of Conditions for a SP | 8 | 2 | 80 | 10 | 6 |
| Mean no. of Conditions for a SP | 8 | 2 ± 0.5 | 80 | 10 | 5 ± 0.8 |
| Accuracy Performance | 92 | 80.0 ± 0.0 | 80.8 ± 13.7 | 71.6 ± 15.9 | 96.9 ± 0.9 |
| Dresden Dataset | RF-FAUST | RF-LIME | Xgboost-LIIME | SuperFlowType | ALPODS |
|---|---|---|---|---|---|
| Processing Time | 17 min | >72 h | 36 h | 24 h | 1 min |
| No. Cross-Validation Steps | 50 | - | 1 | 1 | 50 |
| Max no. of SP | 43 | - | 2 | 1456 | 5 |
| Mean no. of SP | 43 | - | 2 | 729 ± 420.5 | 3 ± 1.4 |
| Max no. of Conditions for a SP | 8 | - | 80 | 10 | 6 |
| Mean no. of Conditions for a SP | 8 | - | 80 | 10 | 4 ± 1.1 |
| Accuracy Performance | 96 ± 0.06 | - | 80.8 ± 8.3 | 70 | 96.8 ± 0.9 |
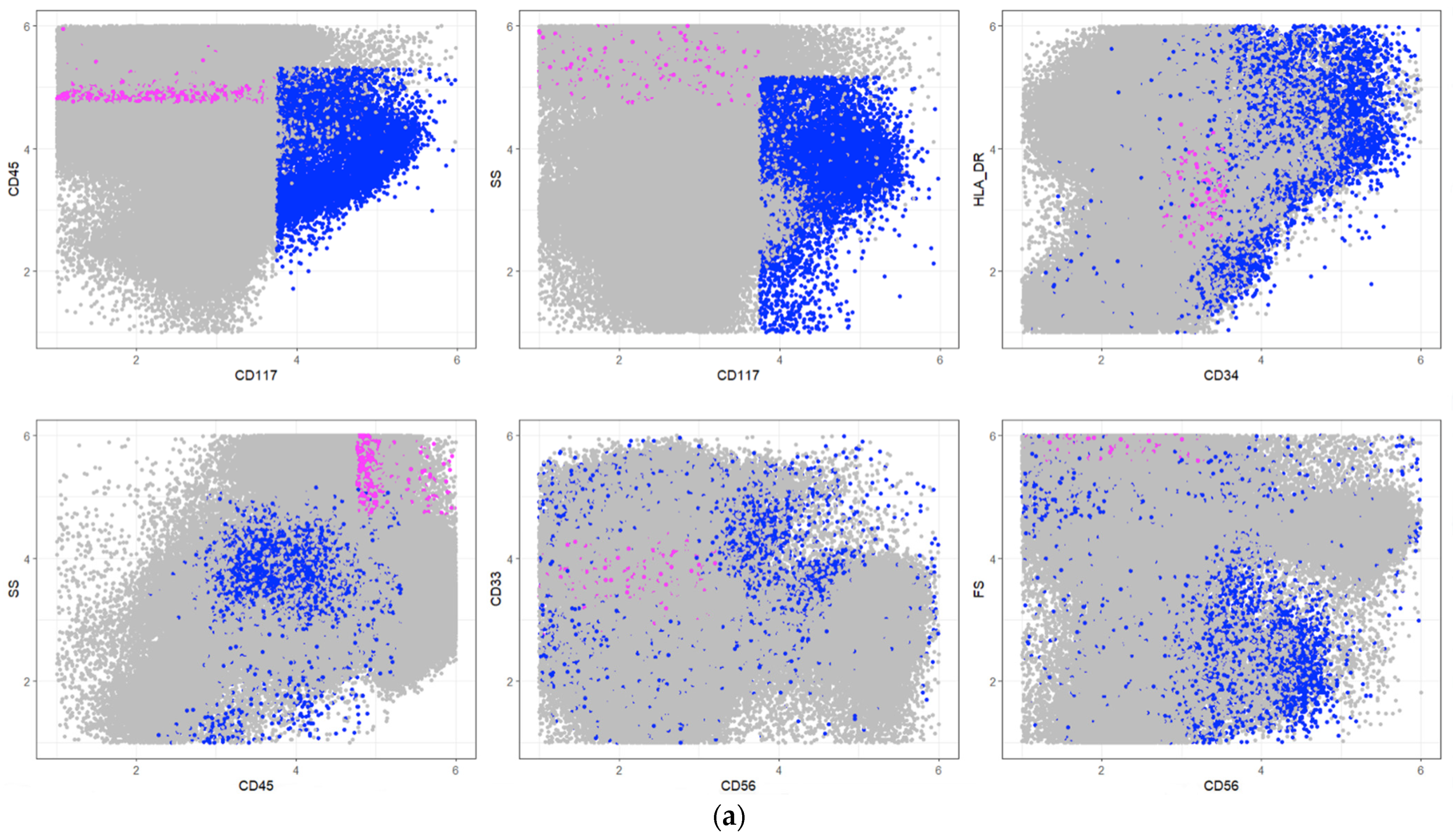
| Pop No. | Cell Types | Description Rule | Frequencies in [%] | |
|---|---|---|---|---|
| PB | BM | |||
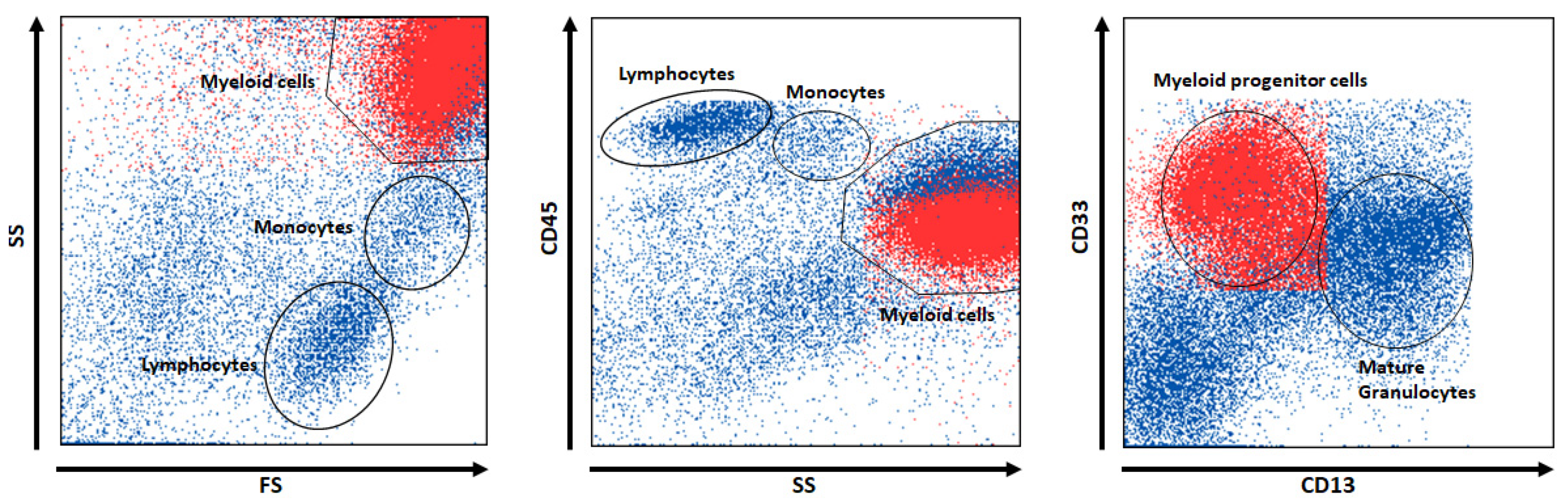
| 1 | Myeloid progenitor cells | SS+, CD33+, CD13- | 5.0 | 43.0 |
| 2 | Subcellular events and aggregates | SS-, HLA_DR-, CD45-, CD117not(+), CD33- | 19.0 | 1.0 |
| 3 | Progenitor B cells | SS-, HLA_DR+, CD4not(+) | 0.3 | 2.0 |
| 4 | Thrombocyte aggregations | SS-, HLA_DR-, CD13-, CD117not(−), CD34not(−), CD117not(+) | 4.2 | 0.5 |
| 5 | CD34+ early progenitor cells | SS (0), CD45-, CD34+ | 1.0 | 8.0 |
| Pop No. | Cell Types | Description Rule | Frequencies in [%] | |
|---|---|---|---|---|
| PB | BM | |||
| 1 | Myeloid progenitor cells | FS+, CD45-, CD13- | 2.0 | 26.9 |
| 2 | Mature granulocyte subset | SS+, CD7not(−), CD117+, CD13+ | 13.0 | 2.9 |
| 3 | T cells | SSnot(−), below average CD33, CD45+, CD13- | 12.2 | 2.1 |
| 4 | Granulocytes subset | Below average HLA_DR, below average CD33, above average CD7, CD117-, SS+, above average CD13 | 9.1 | 1.9 |
| 5 | Hematogones with lymphocyte subset | CD33-, FS-, CD45not(+), CD13- | 2.6 | 8.6 |
| Method/Description | Synthetic Data | Marburg | Dresden | Leukemia |
|---|---|---|---|---|
| Number of Cases per class | (50, 50, 50) | (7, 7) | (22, 22) | (25, 25) |
| Average number of events per case | 1500 | 330,000 | 579,000 | 406,000 |
| Accuracy of Baseline (CART) | 98 | 50 | 46 | 90 |
| Accuracy of RF-FAUST | 64 * | 92 | 96 ± 0.06 | 98 ± 0.5 |
| No. Explanations of RF-Faust | 4 × 4 | 8 × 79 | 8 × 43 | 8 × 212 |
| Accuracy of RF-LIME | 96.3 ± 1.4 | 80.0 ± 0.0 | Not computable | Not computable |
| No. Explanations of RF-LIME | 6 × 2 | 21 × 2 | Not computable | Not computable |
| Accuracy of SuperFlowType | 80 ± 10 | 71.6 ± 15.9 | 70 | Not computable |
| No. Explanations of SuperFlowType | 7 × 3 | 2744 × 10 | 729 × 10 | Not computable |
| Accuracy of ALPODS | 96 | 96.9 ± 0.9 | 96.8 ± 0.9 | 98 ± 2 |
| No. Explanations of ALPODS | 2 × 2 | 3 × 5 | 3 × 4 | 2 × 7 |
| Accuracy of Xgboost-LIME | 99 ± 0.7 | 80.8 ± 13.7 | 80.8 ± 8.3 | Not computable |
| No. Explanations of Xgboost-LIME | 1.5 × 3 | 80 × 2 | 80 × 2 | Not computable |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ultsch, A.; Hoffmann, J.; Röhnert, M.A.; von Bonin, M.; Oelschlägel, U.; Brendel, C.; Thrun, M.C. An Explainable AI System for the Diagnosis of High-Dimensional Biomedical Data. BioMedInformatics 2024, 4, 197-218. https://doi.org/10.3390/biomedinformatics4010013
Ultsch A, Hoffmann J, Röhnert MA, von Bonin M, Oelschlägel U, Brendel C, Thrun MC. An Explainable AI System for the Diagnosis of High-Dimensional Biomedical Data. BioMedInformatics. 2024; 4(1):197-218. https://doi.org/10.3390/biomedinformatics4010013
Chicago/Turabian StyleUltsch, Alfred, Jörg Hoffmann, Maximilian A. Röhnert, Malte von Bonin, Uta Oelschlägel, Cornelia Brendel, and Michael C. Thrun. 2024. "An Explainable AI System for the Diagnosis of High-Dimensional Biomedical Data" BioMedInformatics 4, no. 1: 197-218. https://doi.org/10.3390/biomedinformatics4010013
APA StyleUltsch, A., Hoffmann, J., Röhnert, M. A., von Bonin, M., Oelschlägel, U., Brendel, C., & Thrun, M. C. (2024). An Explainable AI System for the Diagnosis of High-Dimensional Biomedical Data. BioMedInformatics, 4(1), 197-218. https://doi.org/10.3390/biomedinformatics4010013