
BioMedInformatics 2025, 5(3), 55; https://doi.org/10.3390/biomedinformatics5030055 - 17 Sep 2025
Abstract
►
Show Figures
Background: The selection of machine learning (ML) models in the biomedical sciences often relies on global performance metrics. When these metrics are closely clustered among candidate models, identifying the most suitable model for real-world deployment becomes challenging. Methods: We developed a novel composite
[...] Read more.
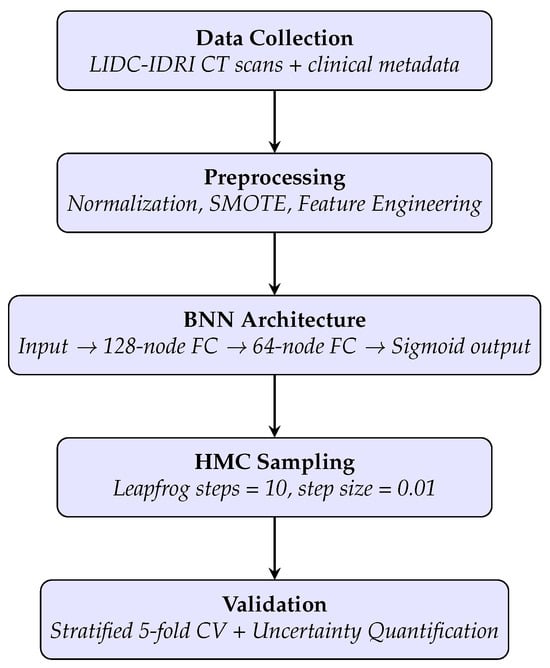
Background: The selection of machine learning (ML) models in the biomedical sciences often relies on global performance metrics. When these metrics are closely clustered among candidate models, identifying the most suitable model for real-world deployment becomes challenging. Methods: We developed a novel composite framework that integrates visual inspection of Model Scoring Distribution Analysis (MSDA) with a new scoring metric (MSDscore). The methodology was implemented within the Digital Phenomics platform as the MSDanalyser tool and tested by generating and evaluating 27 predictive models developed for breast, lung, and renal cancer prognosis. Results: Our approach enabled a detailed inspection of true-positive, false-positive, true-negative, and false-negative distributions across the scoring space, capturing local performance patterns overlooked by conventional metrics. In contrast with the minimal variation between models obtained by global metrics, the MSDA methodology revealed substantial differences in score region behaviour, allowing better discrimination between models. Conclusions: Integrating our composite framework alongside traditional performance metrics provides a complementary and more nuanced approach to model selection in clinical and biomedical settings.
Full article

Graphical abstract



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}