

IMPI: An Interface for Low-Frequency Point Mutation Identification Exemplified on Resistance Mutations in Chronic Myeloid Leukemia

 , , , ,
, , , ,
Abstract
1. Background
2. Implementation
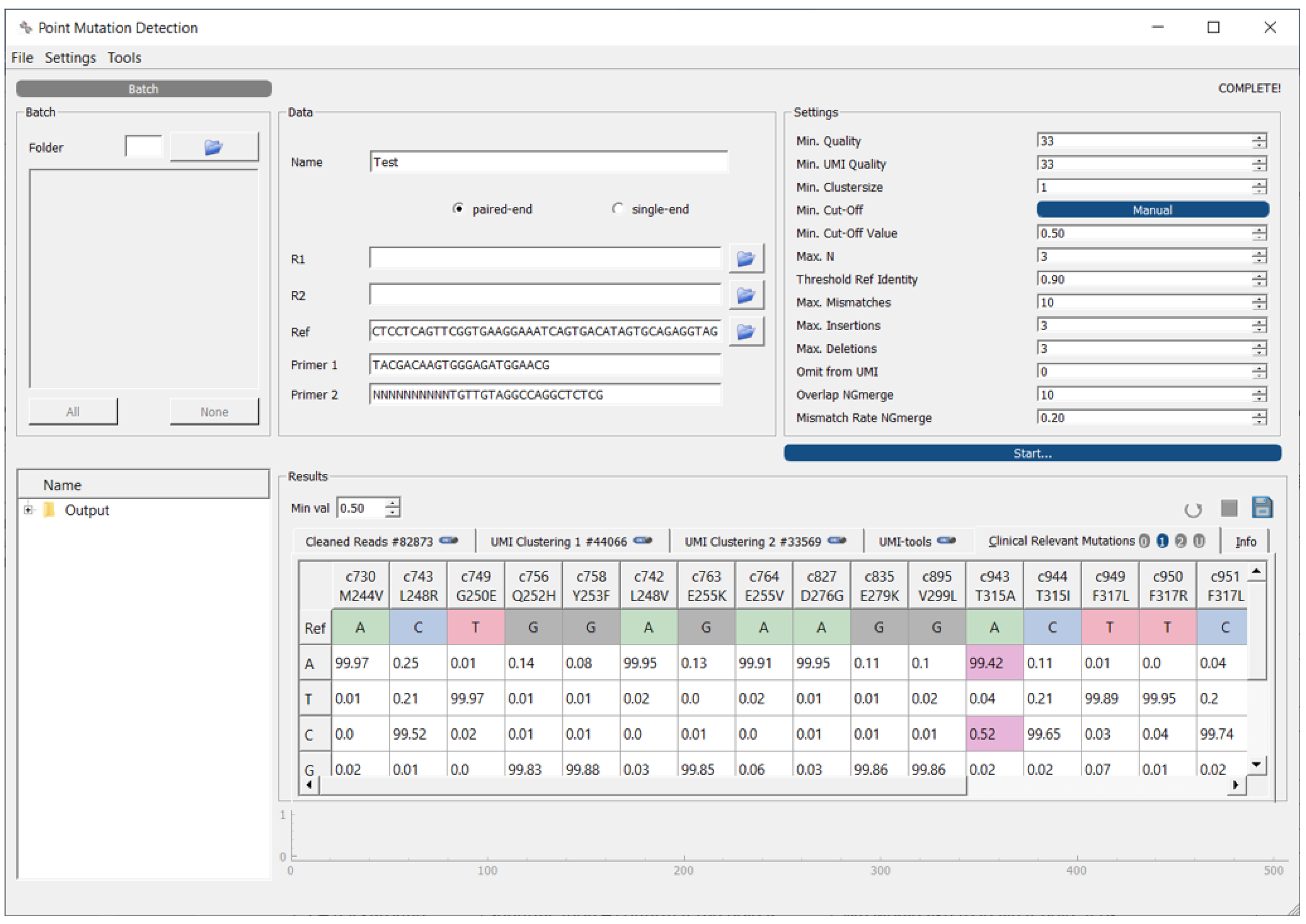
2.1. Software Overview
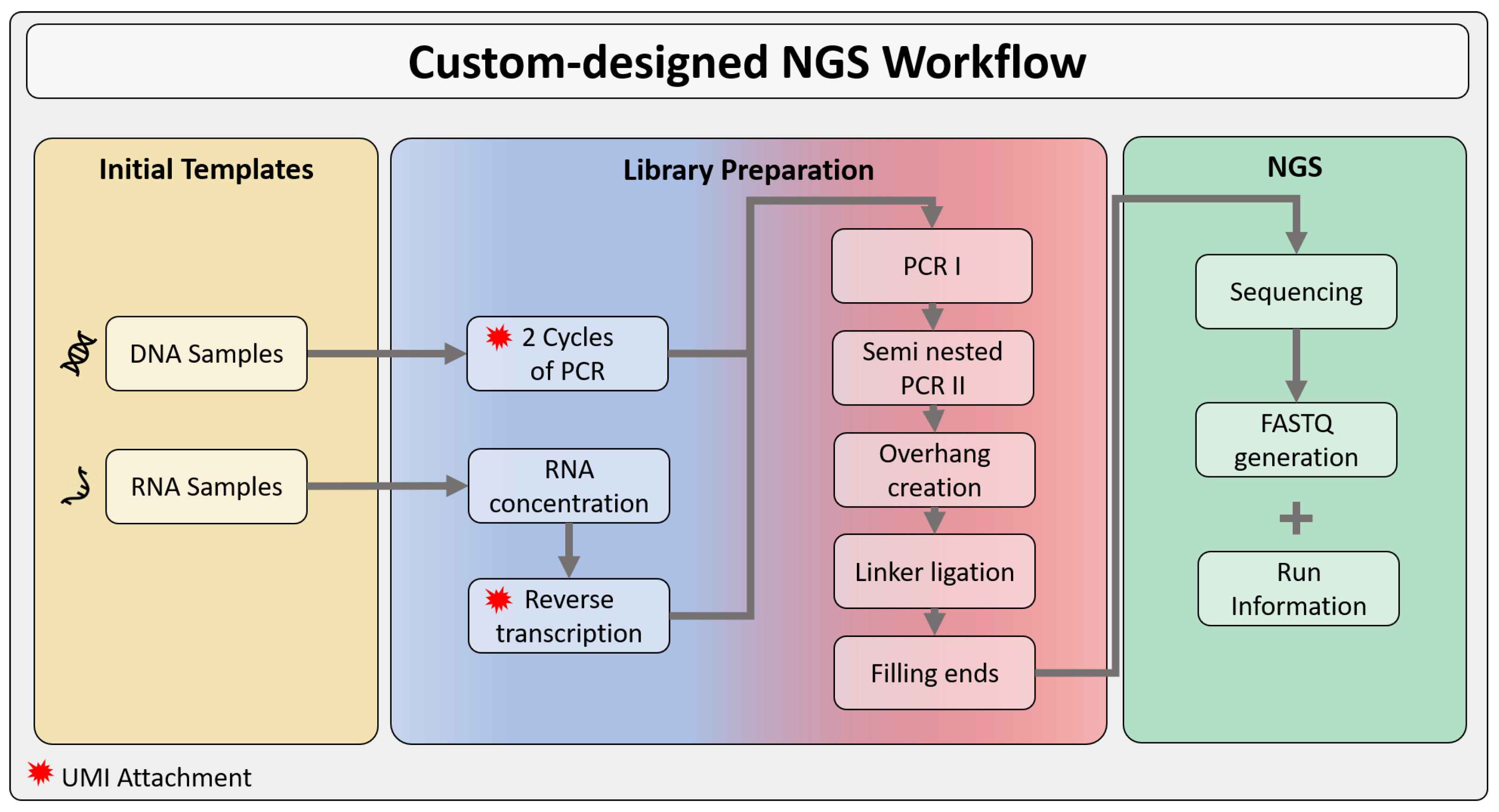
2.2. Input Data
2.3. Data Pre-Processing
2.4. Data Analysis
2.5. Parameter Settings
2.6. Clustering
2.7. Workflow Output
2.8. Parameter Optimization and Data Revision
2.8.1. Parameter Optimization Using an Evolution Strategy (ES)
2.8.2. Wild-Type Correction
2.8.3. Cross-Sample Contamination Analysis
2.9. Output
3. Results
3.1. Dataset
3.1.1. Synthesized Control DNA Samples
3.1.2. Next-Generation Sequencing
3.1.3. Evaluation
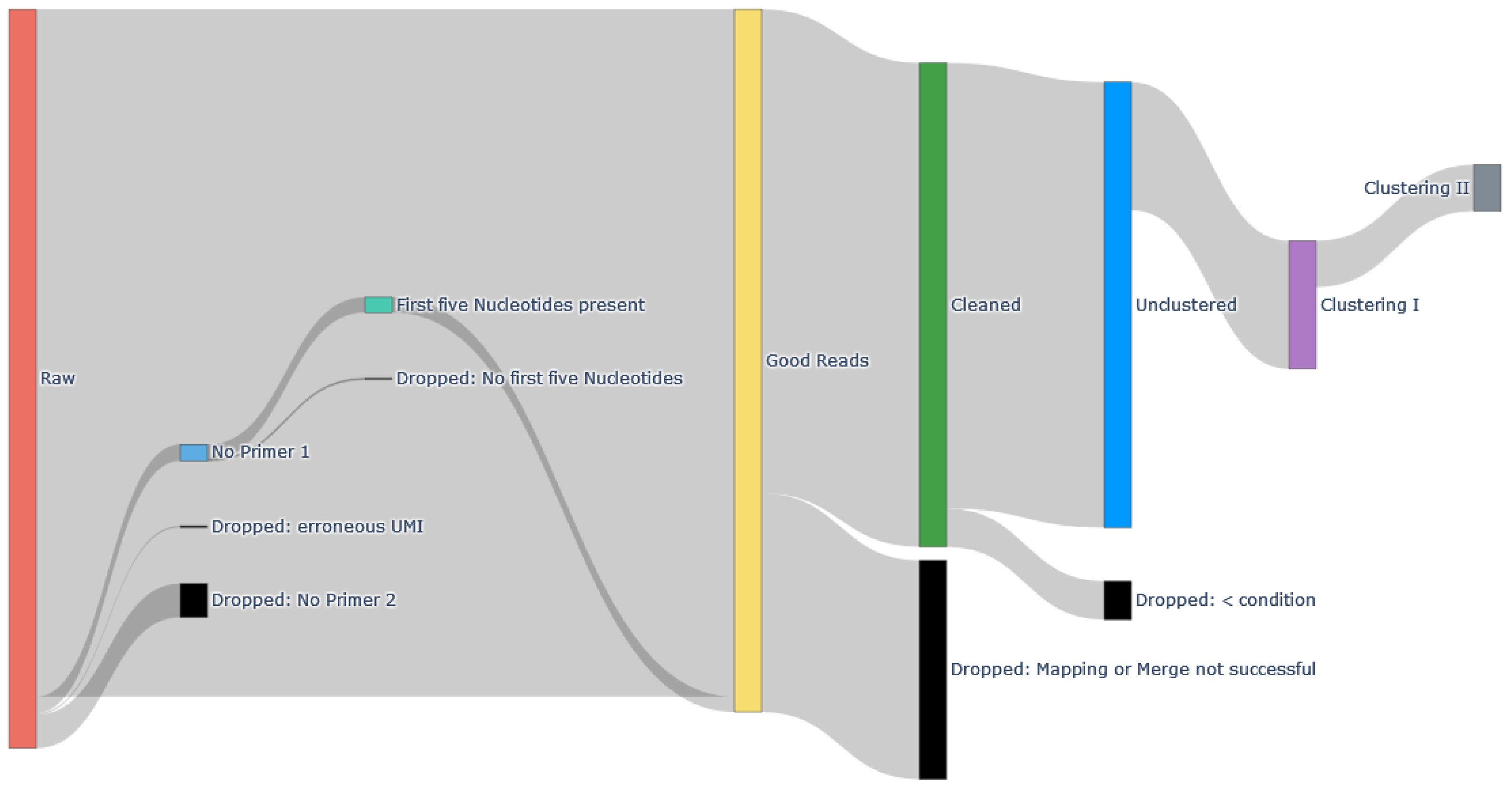
3.2. Sample Pre-Processing
3.3. Cross-Sample Contamination
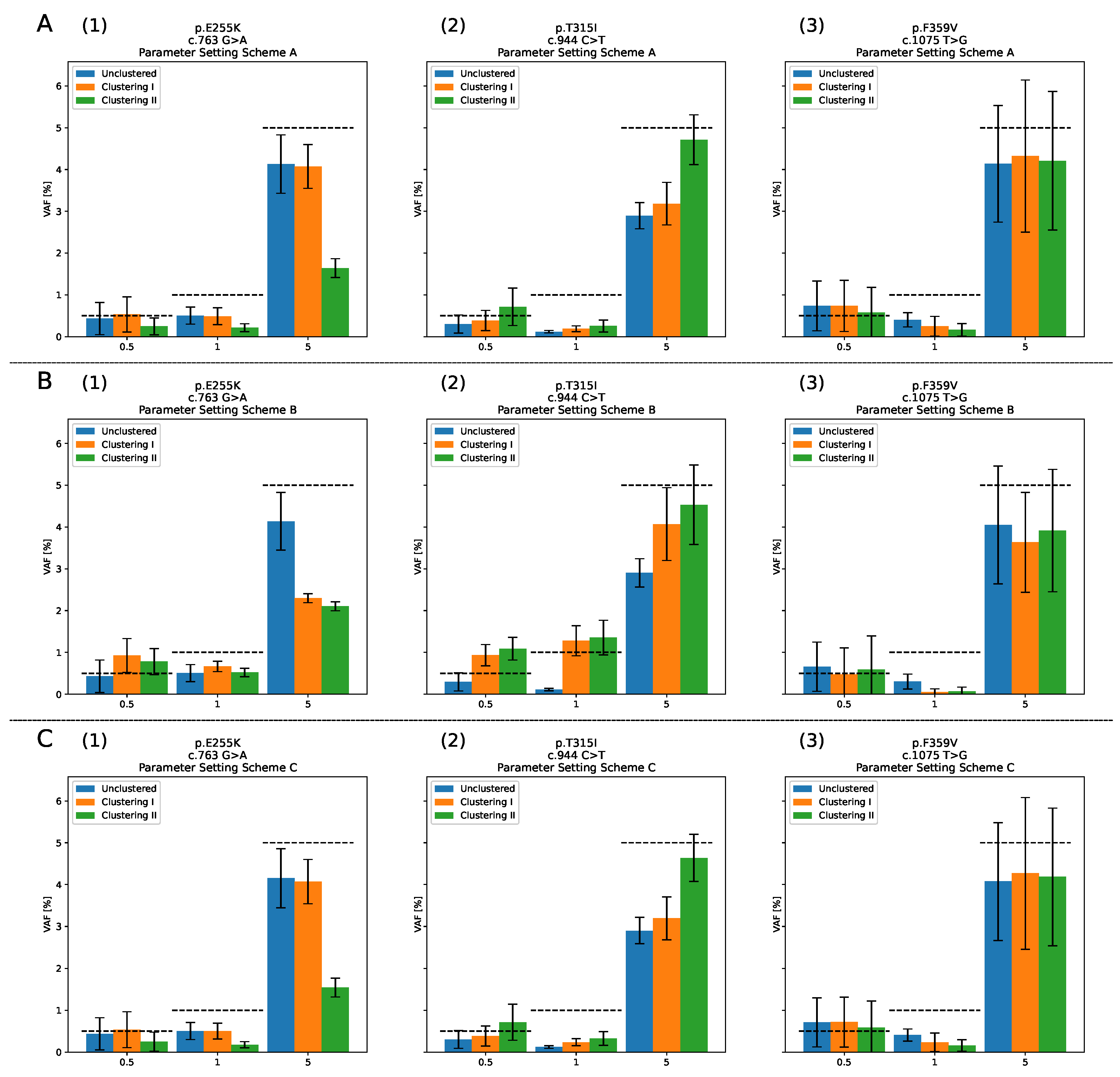
3.4. Raw Data, Clustering I, Clustering II
4. Discussion
4.1. Data Cleaning
4.2. Comparison: Raw Data, Clustering I and Clustering II
4.3. LOD Evaluation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABL1 | Abelson murine leukemia viral oncogene homolog 1 |
| AF | Allele frequency |
| ATP | Adenosine triphosphate |
| BCR | Breakpoint cluster region |
| BWA | Burrows-Wheeler Aligner |
| CIGAR | Concise Idiosyncratic Gapped Alignment Report |
| CML | Chronic myeloid leukemia |
| ES | Evolution strategy |
| GATK | Genome Analysis Toolkit |
| GUI | Graphical user interface |
| IMPI | Interface for Point Mutation Identification |
| LOD | Limit of detection |
| MSE | Mean squared error |
| NGS | Next-generation sequencing |
| OS | Operating system |
| PCR | Polymerase chain reaction |
| PWM | Position weight matrix |
| SAM | Sequence alignment map |
| TKI | Tyrosine kinase inhibitors |
| UMI | Unique molecular identifier |
| VAF | Variant allele frequency |
| wt | Wild-type |
References
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Lin, M.T.; Mosier, S.L.; Thiess, M.; Beierl, K.F.; Debeljak, M.; Tseng, L.H.; Chen, G.; Yegnasubramanian, S.; Ho, H.; Cope, L.; et al. Clinical validation of KRAS, BRAF, and EGFR mutation detection using next-generation sequencing. Am. J. Clin. Pathol. 2014, 141, 856–866. [Google Scholar] [CrossRef]
- Tsiatis, A.C.; Norris-Kirby, A.; Rich, R.G.; Hafez, M.J.; Gocke, C.D.; Eshleman, J.R.; Murphy, K.M. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: Diagnostic and clinical implications. J. Mol. Diagn. 2010, 12, 425–432. [Google Scholar] [CrossRef]
- Schmitt, M.W.; Pritchard, J.R.; Leighow, S.M.; Aminov, B.I.; Beppu, L.; Kim, D.S.; Hodgson, J.G.; Rivera, V.M.; Loeb, L.A.; Radich, J.P. Single-molecule sequencing reveals patterns of preexisting drug resistance that suggest treatment strategies in Philadelphia-positive leukemias. Clin. Cancer Res. 2018, 24, 5321–5334. [Google Scholar] [CrossRef]
- Alikian, M.; Gerrard, G.; Subramanian, P.G.; Mudge, K.; Foskett, P.; Khorashad, J.S.; Lim, A.C.; Marin, D.; Milojkovic, D.; Reid, A.; et al. BCR-ABL1 kinase domain mutations: Methodology and clinical evaluation. Am. J. Hematol. 2012, 87, 298–304. [Google Scholar] [CrossRef]
- Potapov, V.; Ong, J.L. Examining Sources of Error in PCR by Single-Molecule Sequencing. PLoS ONE 2017, 12, e0169774. [Google Scholar] [CrossRef]
- Smith, T.; Heger, A.; Sudbery, I. UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017, 27, 491–499. [Google Scholar] [CrossRef]
- Mansukhani, S.; Barber, L.J.; Kleftogiannis, D.; Moorcraft, S.Y.; Davidson, M.; Woolston, A.; Proszek, P.Z.; Griffiths, B.; Fenwick, K.; Herman, B.; et al. Ultra-sensitive mutation detection and genome-wide DNA copy number reconstruction by error-corrected circulating tumor DNA sequencing. Clin. Chem. 2018, 64, 1626–1635. [Google Scholar] [CrossRef]
- Boltz, V.F.; Rausch, J.; Shao, W.; Hattori, J.; Luke, B.; Maldarelli, F.; Mellors, J.W.; Kearney, M.F.; Coffin, J.M. Ultrasensitive single-genome sequencing: Accurate, targeted, next generation sequencing of HIV-1 RNA. Retrovirology 2016, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.T.; Phillis, S.R.; Yeung, D.T.; Lawrence, D.; Schreiber, A.; Wang, P.; Geoghegan, J.; Lustgarten, S.; Hodgson, G.; Rivera, V.M.; et al. Detection of BCR-ABL1 Compound and Polyclonal Mutants in Chronic Myeloid Leukemia Patients Using a Novel Next Generation Sequencing Approach That Minimises PCR and Sequencing Errors. Blood 2014, 124, 399. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Svensson, V.; Natarajan, K.N.; Ly, L.H.; Miragaia, R.J.; Labalette, C.; Macaulay, I.C.; Cvejic, A.; Teichmann, S.A. Power analysis of single-cell RNA-sequencing experiments. Nat. Methods 2017, 14, 381–387. [Google Scholar] [CrossRef]
- Liu, D. Algorithms for efficiently collapsing reads with Unique Molecular Identifiers. PeerJ 2019, 7, e8275. [Google Scholar] [CrossRef]
- Parekh, S.; Ziegenhain, C.; Vieth, B.; Enard, W.; Hellmann, I. zUMIs-a fast and flexible pipeline to process RNA sequencing data with UMIs. Gigascience 2018, 7, giy059. [Google Scholar] [CrossRef]
- Xia, X. Position weight matrix, gibbs sampler, and the associated significance tests in motif characterization and prediction. Scientifica 2012, 2012, 917540. [Google Scholar] [CrossRef]
- Beyer, H.G.; Schwefel, H.P. Evolution strategies: A comprehensive introduction. Nat. Comput. 2002, 1, 3–52. [Google Scholar] [CrossRef]
- Gaspar, J.M. NGmerge: Merging paired-end reads via novel empirically-derived models of sequencing errors. BMC Bioinform. 2018, 19, 536. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media: Sebastopol, CA, USA, 2020. [Google Scholar]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv 2017, 201178. [Google Scholar]
- Xia, L.; Li, Z.; Zhou, B.; Tian, G.; Zeng, L.; Dai, H.; Li, X.; Liu, C.; Lu, S.; Xu, F.; et al. Statistical analysis of mutant allele frequency level of circulating cell-free DNA and blood cells in healthy individuals. Sci. Rep. 2017, 7, 7526. [Google Scholar] [CrossRef] [PubMed]
- Singh, P. Learn Windows Subsystem for Linux; Apress: Berkley, CA, USA, 2020; pp. 1–17. [Google Scholar]
- Simon, J.S.; Botero, S.; Simon, S.M. Sequencing the peripheral blood B and T cell repertoire—Quantifying robustness and limitations. J. Immunol. Methods 2018, 463, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Tretyakov, K. Matplotlib-Venn: Functions for Plotting Area-Proportional Two-and Three-Way Venn Diagrams in Matplotlib. 2020. Available online: https://pypi.org/project/matplotlib-venn/ (accessed on 28 March 2024).
- Kang, Z.J.; Liu, Y.F.; Xu, L.Z.; Long, Z.J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.X.; Pan, Y.J.; Yan, J.S.; et al. The Philadelphia chromosome in leukemogenesis. Chin. J. Cancer 2016, 35, 48. [Google Scholar] [CrossRef] [PubMed]
- Rumpold, H.; Webersinke, G. Molecular pathogenesis of Philadelphia-positive chronic myeloid leukemia—Is it all BCR-ABL? Curr. Cancer Drug Targets 2011, 11, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Reddy, E.P.; Aggarwal, A.K. The ins and outs of bcr-abl inhibition. Genes Cancer 2012, 3, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J. Translation of the Philadelphia chromosome into therapy for CML. Blood 2008, 112, 4808–4817. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2020, 95, 691–709. [Google Scholar] [CrossRef]
- Ramirez, P.; DiPersio, J.F. Therapy options in imatinib failures. Oncologist 2008, 13, 424–434. [Google Scholar] [CrossRef]
- Chopade, P.; Akard, L.P. Improving Outcomes in Chronic Myeloid Leukemia Over Time in the Era of Tyrosine Kinase Inhibitors. Clin. Lymphoma Myeloma Leuk. 2018, 18, 710–723. [Google Scholar] [CrossRef]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and resistance to BCR-ABL1-targeted therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 28 March 2024).
- Shao, W.; Boltz, V.F.; Spindler, J.E.; Kearney, M.F.; Maldarelli, F.; Mellors, J.W.; Stewart, C.; Volfovsky, N.; Levitsky, A.; Stephens, R.M.; et al. Analysis of 454 sequencing error rate, error sources, and artifact recombination for detection of Low-frequency drug resistance mutations in HIV-1 DNA. Retrovirology 2013, 10, 18. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature Name | Description |
|---|---|
| ID | Read identifier extracted from the FASTQ file source |
| UMI | Extracted unique molecular identifier sequence which was part of the primer |
| UMI_Phred_Quality | Phred quality scores of the UMI encoded in ASCII characters |
| UMI_Avg_Score | Average Phred quality score of the UMI |
| Seq | Nucleotide sequence of the read with its deletions and insertions which are defined by the Concise Idiosyncratic Gapped Alignment Report (CIGAR) [20] information |
| Phred_Quality | Phred quality scores of the whole nucleotide sequence encoded in ASCII characters |
| Avg_Score | Average Phred quality score of the sequence |
| Start | Start position of the sequence within the reference gene sequence |
| Length | Length of the sequence |
| Insertions | Number of insertions |
| Deletions | Number of deletions |
| RefIdentical | Identity of the sequence with the reference gene sequence |
| RefMismatches | Number of mismatches in comparison with the reference gene sequence |
| aaSeq | Translated nucleotide sequence to get the amino acid sequence |
| aaRefIdentical | Identity of the amino acid sequence with the amino acid reference sequence |
| aaRefMismatches | Number of mismatches in comparison with the amino acid reference sequence |
| Parameter Name | Description |
|---|---|
| Min. Quality | All sequences with a lower average Phred quality score are excluded for calculating the AF matrices |
| Min. UMI Quality | All sequences with a lower average Phred quality score of the UMI are excluded |
| Min. Clustersize | When clustering algorithms are applied, all clusters with a size lower than requested are discarded |
| Min. Cut-Off and Min. Cut-Off Value | The minimal cut-off value defines the rate of identical nucleotides of the reads within one cluster to be identical and confirm this nucleotide—if Min. Cut-Off is set Dynamic, the cut-off value depends on the cluster size—the larger the cluster, the higher the need of identical bases for defining a nucleotide at a specific gene location |
| Max. N | When a nucleotide at a specific position is not confirmed and is defined as N—this value defines the maximum N per sequence |
| Threshold Ref Identity | Only sequences with a reference gene correspondence are taken for AF matrix calculation |
| Max. Mismatches, Max. Insertions and Max. Deletions | Definition of the maximal number of mismatches, insertions or deletions |
| Omit from UMI | Since the first few nucleotides within a read show rather low quality IMPI allows for omitting nucleotides from the UMI |
| Overlap NGmerge | Defines the number of nucleotides that are allowed to overlap when using NGmerge (only relevant in pre-processing step) |
| Mismatch Rate NGmerge | Defines the maximum rate of mismatches when in overlapping regions |
| Primer Sequence | |
|---|---|
| Forward | TACGACAAGTGGGAGATGGAACG |
| Reverse | NNNNNNNNNNTGTTGTAGGCCAGGCTCTCG |
| A | B | C | |
|---|---|---|---|
| Min. Quality | 33 | 35 | 34 |
| Min. UMI Quality | 33 | 34 | 31 |
| Min. Clustersize | 1 | 2 | 1 |
| Min. Cut-Off and Min. Cut-Off Value | 0.50 | 0.50 | 0.50 |
| Max. N | 3 | 10 | 11 |
| Threshold Ref Identity | 0.90 | 0.95 | 0.94 |
| Max. Mismatches | 10 | 5 | 11 |
| Max. Insertions | 3 | 1 | 20 |
| Max. Deletions | 3 | 1 | 20 |
| Omit from UMI | 0 | 0 | 0 |
| Mapping | Operating System | GUI/Command-Line-Based | Implementation | |
|---|---|---|---|---|
| UMI-tools [7] | Bowtie2, e.g., | Linux | cmd | Python |
| zUMIs [16] | STAR | Linux | cmd | R |
| UMICollapse [15] | Bowtie2 *, e.g., | Linux | cmd | Java |
| umis [14] | Kallisto/RapMap ** | Windows, Linux | cmd | Python |
| IMPI | Bowtie2 | Windows, Linux | GUI + cmd | Python |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vetter, J.; Burghofer, J.; Malli, T.; Lin, A.M.; Webersinke, G.; Wiederstein, M.; Winkler, S.M.; Schaller, S. IMPI: An Interface for Low-Frequency Point Mutation Identification Exemplified on Resistance Mutations in Chronic Myeloid Leukemia. BioMedInformatics 2024, 4, 1289-1307. https://doi.org/10.3390/biomedinformatics4020071
Vetter J, Burghofer J, Malli T, Lin AM, Webersinke G, Wiederstein M, Winkler SM, Schaller S. IMPI: An Interface for Low-Frequency Point Mutation Identification Exemplified on Resistance Mutations in Chronic Myeloid Leukemia. BioMedInformatics. 2024; 4(2):1289-1307. https://doi.org/10.3390/biomedinformatics4020071
Chicago/Turabian StyleVetter, Julia, Jonathan Burghofer, Theodora Malli, Anna M. Lin, Gerald Webersinke, Markus Wiederstein, Stephan M. Winkler, and Susanne Schaller. 2024. "IMPI: An Interface for Low-Frequency Point Mutation Identification Exemplified on Resistance Mutations in Chronic Myeloid Leukemia" BioMedInformatics 4, no. 2: 1289-1307. https://doi.org/10.3390/biomedinformatics4020071
APA StyleVetter, J., Burghofer, J., Malli, T., Lin, A. M., Webersinke, G., Wiederstein, M., Winkler, S. M., & Schaller, S. (2024). IMPI: An Interface for Low-Frequency Point Mutation Identification Exemplified on Resistance Mutations in Chronic Myeloid Leukemia. BioMedInformatics, 4(2), 1289-1307. https://doi.org/10.3390/biomedinformatics4020071