
Tumor-Agnostic Biomarkers: Heed Caution, and Why Cell of Origin Still Matters


{kind=link}
Abstract
:Simple Summary
Abstract
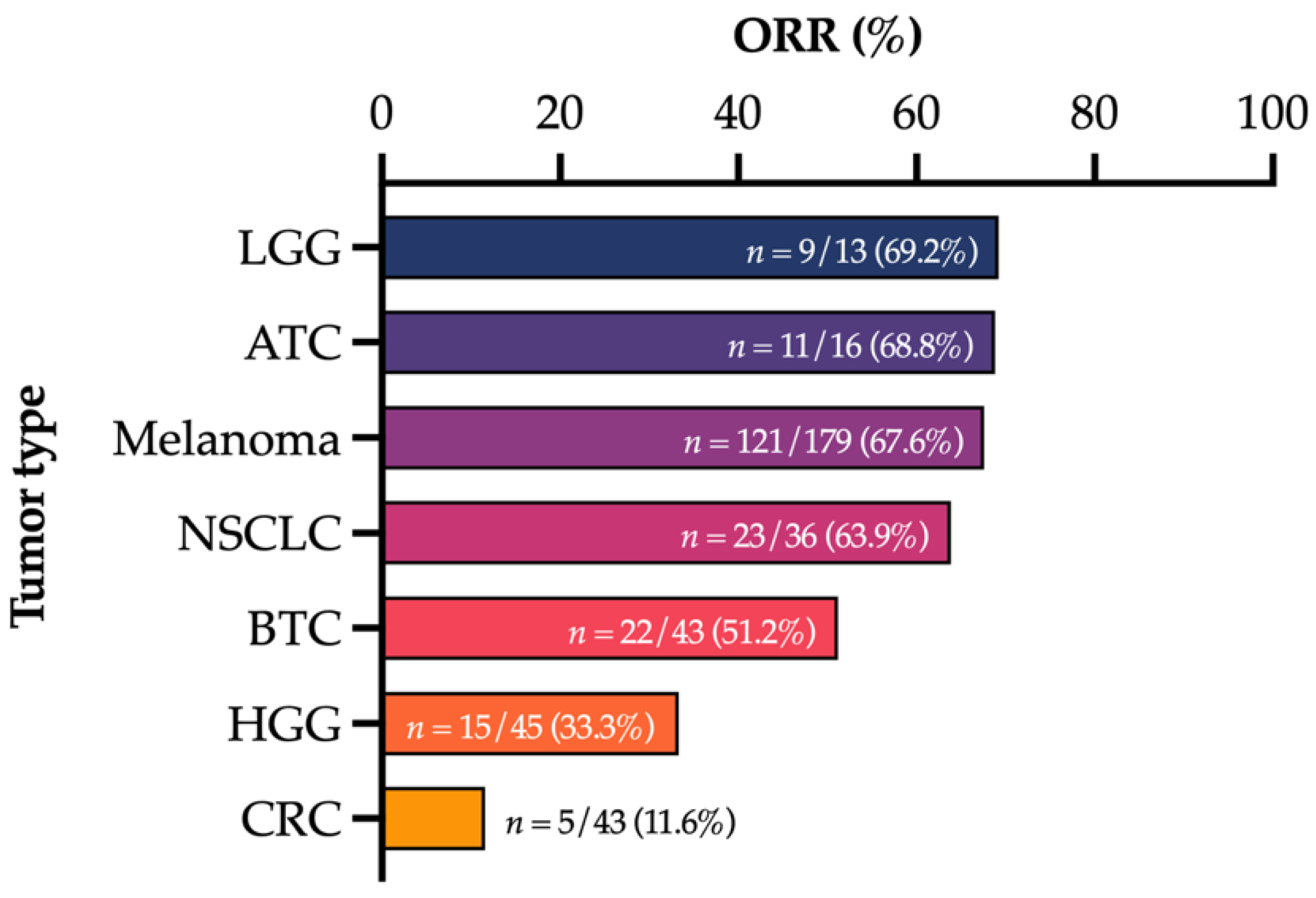
1. Introduction
2. Conclusions
Funding
Conflicts of Interest
References
- DeVita, V.T.; Chu, E. A History of Cancer Chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [Green Version]
- Photopoulos, J. The Future of Tissue-Agnostic Drugs. Nature 2020, 585, S16–S18. [Google Scholar] [CrossRef]
- Offin, M.; Liu, D.; Drilon, A. Tumor-Agnostic Drug Development. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 184–187. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in Patients with Advanced or Metastatic NTRK Fusion-Positive Solid Tumours: Integrated Analysis of Three Phase 1-2 Trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Advanced Solid Tumours Treated with Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Adashek, J.J.; Subbiah, V.; Kurzrock, R. From Tissue-Agnostic to N-of-One Therapies: (R)Evolution of the Precision Paradigm. Trends Cancer 2021, 7, 15–28. [Google Scholar] [CrossRef]
- Drilon, A.E.; Subbiah, V.; Oxnard, G.R.; Bauer, T.M.; Velcheti, V.; Lakhani, N.J.; Besse, B.; Park, K.; Patel, J.D.; Cabanillas, M.E.; et al. A Phase 1 Study of LOXO-292, a Potent and Highly Selective RET Inhibitor, in Patients with RET-Altered Cancers. J. Clin. Oncol. 2018, 36, 102. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-Mediated Re-Activation of MAPK Signaling Contributes to Insensitivity of BRAF Mutant Colorectal Cancers to RAF Inhibition with Vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of Colon Cancer to BRAF(V600E) Inhibition through Feedback Activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative Genomic Analyses Identify MITF as a Lineage Survival Oncogene Amplified in Malignant Melanoma. Nature 2005, 436, 117–122. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK Fusion-Positive Cancers and TRK Inhibitor Therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Lee, J.W.; Soung, Y.H.; Kim, S.Y.; Park, W.S.; Nam, S.W.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Absence of EGFR Mutation in the Kinase Domain in Common Human Cancers besides Non-Small Cell Lung Cancer. Int. J. Cancer 2005, 113, 510–511. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.J.; Pilié, P.G.; Rashid, N.U.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.B.; Lim, B.; et al. High Tumor Mutation Burden Fails to Predict Immune Checkpoint Blockade Response across All Cancer Types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Merino, D.M.; McShane, L.M.; Fabrizio, D.; Funari, V.; Chen, S.-J.; White, J.R.; Wenz, P.; Baden, J.; Barrett, J.C.; Chaudhary, R.; et al. Establishing Guidelines to Harmonize Tumor Mutational Burden (TMB): In Silico Assessment of Variation in TMB Quantification across Diagnostic Platforms: Phase I of the Friends of Cancer Research TMB Harmonization Project. J. Immunother. Cancer 2020, 8, e000147. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Yates, L.R.; Seoane, J.; Le Tourneau, C.; Siu, L.L.; Marais, R.; Michiels, S.; Soria, J.C.; Campbell, P.; Normanno, N.; Scarpa, A.; et al. The European Society for Medical Oncology (ESMO) Precision Medicine Glossary. Ann. Oncol. 2018, 29, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M., Jr.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus Trametinib in Patients with Previously Untreated BRAFV600E-Mutant Metastatic Non-Small-Cell Lung Cancer: An Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Subbiah, V.; Lassen, U.; Élez, E.; Italiano, A.; Curigliano, G.; Javle, M.; de Braud, F.; Prager, G.W.; Greil, R.; Stein, A.; et al. Dabrafenib plus Trametinib in Patients with BRAFV600E-Mutated Biliary Tract Cancer (ROAR): A Phase 2, Open-Label, Single-Arm, Multicentre Basket Trial. Lancet Oncol. 2020, 21, 1234–1243. [Google Scholar] [CrossRef]
- Subbiah, V.; Stein, A.; van den Bent, M.; Wick, A.; de Vos, F.Y.; von Bubnoff, N.; van Linde, M.E.; Lai, A.; Prager, G.W.; Campone, M.; et al. Abstract CT025: Dabrafenib plus Trametinib in BRAF V600E-Mutant High-Grade (HGG) and Low-Grade Glioma (LGG). Cancer Res. 2021, 81, CT025. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4023–4031. [Google Scholar] [CrossRef] [Green Version]
- Ferone, G.; Lee, M.C.; Sage, J.; Berns, A. Cells of Origin of Lung Cancers: Lessons from Mouse Studies. Genes Dev. 2020, 34, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C.; Govindan, R. Clinical Implications of Genomic Discoveries in Lung Cancer. N. Engl. J. Med. 2016, 374, 1864–1873. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, K.D.; Proost, N.; Brouns, I.; Adriaensen, D.; Song, J.-Y.; Berns, A. Cell of Origin of Small Cell Lung Cancer: Inactivation of Trp53 and Rb1 in Distinct Cell Types of Adult Mouse Lung. Cancer Cell 2011, 19, 754–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tata, P.R.; Chow, R.D.; Saladi, S.V.; Tata, A.; Konkimalla, A.; Bara, A.; Montoro, D.; Hariri, L.P.; Shih, A.R.; Mino-Kenudson, M.; et al. Developmental History Provides a Roadmap for the Emergence of Tumor Plasticity. Dev. Cell 2018, 44, 679–693.e5. [Google Scholar] [CrossRef] [Green Version]
- Mollaoglu, G.; Jones, A.; Wait, S.J.; Mukhopadhyay, A.; Jeong, S.; Arya, R.; Camolotto, S.A.; Mosbruger, T.L.; Stubben, C.J.; Conley, C.J.; et al. The Lineage-Defining Transcription Factors SOX2 and NKX2-1 Determine Lung Cancer Cell Fate and Shape the Tumor Immune Microenvironment. Immunity 2018, 49, 764–779.e9. [Google Scholar] [CrossRef] [Green Version]
- Yatabe, Y.; Kosaka, T.; Takahashi, T.; Mitsudomi, T. EGFR Mutation Is Specific for Terminal Respiratory Unit Type Adenocarcinoma. Am. J. Surg. Pathol. 2005, 29, 633–639. [Google Scholar] [CrossRef]
- Yatabe, Y.; Dacic, S.; Borczuk, A.C.; Warth, A.; Russell, P.A.; Lantuejoul, S.; Beasley, M.B.; Thunnissen, E.; Pelosi, G.; Rekhtman, N.; et al. Best Practices Recommendations for Diagnostic Immunohistochemistry in Lung Cancer. J. Thorac. Oncol. 2019, 14, 377–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inamura, K. Clinicopathological Characteristics and Mutations Driving Development of Early Lung Adenocarcinoma: Tumor Initiation and Progression. Int. J. Mol. Sci. 2018, 19, 1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herriges, M.; Morrisey, E.E. Lung Development: Orchestrating the Generation and Regeneration of a Complex Organ. Development 2014, 141, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Cheung, W.K.C.; Nguyen, D.X. Lineage Factors and Differentiation States in Lung Cancer Progression. Oncogene 2015, 34, 5771–5780. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [CrossRef] [PubMed]
- Wilkerson, M.D.; Yin, X.; Walter, V.; Zhao, N.; Cabanski, C.R.; Hayward, M.C.; Miller, C.R.; Socinski, M.A.; Parsons, A.M.; Thorne, L.B.; et al. Differential Pathogenesis of Lung Adenocarcinoma Subtypes Involving Sequence Mutations, Copy Number, Chromosomal Instability, and Methylation. PLoS ONE 2012, 7, e36530. [Google Scholar] [CrossRef] [Green Version]
- Westover, D.; Zugazagoitia, J.; Cho, B.C.; Lovly, C.M.; Paz-Ares, L. Mechanisms of Acquired Resistance to First- and Second-Generation EGFR Tyrosine Kinase Inhibitors. Ann. Oncol. 2018, 29, i10–i19. [Google Scholar] [CrossRef]
- Garraway, L.A.; Sellers, W.R. Lineage Dependency and Lineage-Survival Oncogenes in Human Cancer. Nat. Rev. Cancer 2006, 6, 593–602. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, A.C. Tumor-Agnostic Biomarkers: Heed Caution, and Why Cell of Origin Still Matters. Onco 2021, 1, 95-100. https://doi.org/10.3390/onco1020008
Tan AC. Tumor-Agnostic Biomarkers: Heed Caution, and Why Cell of Origin Still Matters. Onco. 2021; 1(2):95-100. https://doi.org/10.3390/onco1020008
Chicago/Turabian StyleTan, Aaron C. 2021. "Tumor-Agnostic Biomarkers: Heed Caution, and Why Cell of Origin Still Matters" Onco 1, no. 2: 95-100. https://doi.org/10.3390/onco1020008
APA StyleTan, A. C. (2021). Tumor-Agnostic Biomarkers: Heed Caution, and Why Cell of Origin Still Matters. Onco, 1(2), 95-100. https://doi.org/10.3390/onco1020008