
Sparse-Input Neural Networks to Differentiate 32 Primary Cancer Types on the Basis of Somatic Point Mutations


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Theory
2.2. Classifiers
2.3. Sampling Strategies
- Random oversampling, where a subset from minority samples was randomly chosen; these selected samples were replicated and added to the original set.
- Adaptive synthetic (ADASYN) [33], where the distribution of the minority class was used to adaptively generate synthetic samples.
- Random undersampling, where data were randomly removed from the majority class to enforce balancing.
- One-sided selection (OSS) [36], where all majority class examples that were at the boundary or noise were removed from the dataset.
- Edited nearest neighbor (ENN) [37], where samples were removed from the class when the majority of their k nearest neighbors corresponded to a different class.
- Combination of over- and undersampling, which was performed using SMOTE with Tomek links and SMOTE with edited nearest neighbors.
3. Results
3.1. Reported Dataset
3.2. Overall Performance of the Classifiers on the Original Dataset
3.3. Sampling
3.4. Classifier Performance per Primary Cancer Type
4. Discussion
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pavlidis, N.; Pentheroudakis, G. Cancer of unknown primary site. Lancet 2012, 379, 1428–1435. [Google Scholar] [CrossRef]
- Liu, J.; Campen, A.; Huang, S.; Peng, S.; Ye, X.; Palakal, M.; Dunker, A.; Xia, Y.; Li, S. Identification of a gene signature in cell cycle pathway for breast cancer prognosis using gene expression profiling data. BMC Med. Genom. 2008, 1, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golub, T.; Slonim, D.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, J.; Wei, J.; Ringnér, M.; Saal, L.; Ladanyi, M.; Westermann, F.; Berthold, F.; Schwab, M.; Antonescu, C.R.; Peterson, C.; et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat. Med. 2001, 7, 673–679. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Tamayo, P.; Rifkin, R.; Mukherjee, S.; Yeang, C.; Angelo, M.; Ladd, C.; Reich, M.; Latulippe, E.; Mesirov, J.P.; et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc. Natl. Acad. Sci. USA 2001, 98, 15149–15154. [Google Scholar] [CrossRef] [Green Version]
- Tibshirani, R.; Hastie, T.; Narasimhan, B.; Chu, G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6567–6572. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Li, Q.; Chen, Q.; Zhou, Y.; Park, S.; Lee, G.; Grimes, B.; Krysan, K.; Yu, M.; Wang, W.; et al. CancerLocator: Non-invasive cancer diagnosis and tissue-of-origin prediction using methylation profiles of cell-free DNA. Genome Biol. 2017, 18, 53. [Google Scholar] [CrossRef] [Green Version]
- Hao, X.; Luo, H.; Krawczyk, M.; Wei, W.; Wang, W.; Wang, J.; Flagg, K.; Hou, J.; Zhang, H.; Yi, S.; et al. DNA methylation markers for diagnosis and prognosis of common cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 7414–7419. [Google Scholar] [CrossRef] [Green Version]
- Martincorena, I.; Campbell, P. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Amar, D.; Izraeli, S.; Shamir, R. Utilizing somatic mutation data from numerous studies for cancer research: Proof of concept and applications. Oncogene 2017, 36, 33–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Shi, Y.; Li, C.; Kim, J.; Cai, W.; Han, Z.; Feng, D.D. DeepGene: An advanced cancer type classifier based on deep learning and somatic point mutations. BMC Bioinform. 2016, 17, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Bashashati, A.; Roth, A.; Oloumi, A.; Tse, K.; Zeng, T.; Haffari, G.; Hirst, M.; Marra, M.A.; Condon, A.; et al. Feature-based classifiers for somatic mutation detection in tumour-normal paired sequencing data. Bioinformatics 2012, 28, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Xu, L.; Shi, Y.; Salavatipour, M.; Lin, R.G. Using Gene Clustering to Identify Discriminatory Genes with Higher Classification Accuracy. IEEE Symp. Bioinform. BioEng. 2006, 6, 235–242. [Google Scholar]
- Hornik, K.; Stinchcombe, M.; White, H. Multilayer feedforward networks are universal approximators. Neural Netw. 1989, 2, 359–366. [Google Scholar] [CrossRef]
- Cho, J.H.; Lee, D.; Park, J.H.; Lee, I.B. New gene selection method for classification of cancer subtypes considering within-class variation. FEBS Lett. 2003, 551, 3–7. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, J.; Huang, L.-C.; Xu, H.; Zhao, Z. Classification of Cancer Primary Sites Using Machine Learning and Somatic Mutations. BioMed Res. Int. 2015, 2015, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Marquard, A.M.; Birkbak, N.J.; Thomas, C.E.; Favero, F.; Krzystanek, M.; Lefebvre, C.; Ferté, C.; Jamal-Hanjani, M.; Wilson, G.A.; Shafi, S.; et al. TumorTracer: A method to identify the tissue of origin from the somatic mutations of a tumor specimen. BMC Med. Genom. 2015, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. arXiv 2016, arXiv:1603.02754. [Google Scholar]
- Katarzyna, T.; Czerwiska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, 68–83. [Google Scholar]
- International Cancer Genome Consortium. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Sun, X. The Lasso and Its Implementation for Neural Networks. Ph.D. Thesis, National Library of Canada—Bibliotheque Nationale du Canada, Ottawa, ON, Canada, 1999. [Google Scholar]
- Zou, H.; Hastie, T. Regularization and Variable Selection via the Elastic Net. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Lin, Y. Model Selection and Estimation in Regression with Grouped Variables. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 2006, 68, 49–67. [Google Scholar] [CrossRef]
- Simon, N.; Friedman, J.; Hastie, T.; Tibshirani, R. A sparse-group lasso. J. Comput. Graph. Stat. 2013, 22, 231–245. [Google Scholar] [CrossRef]
- Feng, J.; Noah, S. Sparse-input neural networks for high-dimensional nonparametric regression and classification. arXiv 2017, arXiv:1711.07592. [Google Scholar]
- Chollet, F. Keras, Online. 2015. Available online: https://github.com/fchollet/keras (accessed on 1 April 2019).
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Greg, S.; Corrado; Davis, A.; Dean, J.; et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems, Online. 2015. Available online: http://tensorflow.org (accessed on 1 April 2019).
- Diederik, P.K.; Jimmy, B. ADAM: A method for stochastic optimization. ICLR. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Han, H.; Wang, W.Y.; Mao, B.H. Borderline-SMOTE: A new over-sampling method in imbalanced data sets learning. In ICIC Advances in Intelligent Computing; Springer: Berlin, Germany, 2005; pp. 878–887. [Google Scholar]
- He, H.; Bai, Y.; Garcia, E.A.; Li, S. ADASYN: Adaptive synthetic sampling approach for imbalanced learning. In Proceedings of the IEEE International Joint Conference on Neural Networks, Hong Kong, China, 1–8 June 2008; pp. 1322–1328. [Google Scholar]
- Tomek, I. Two Modifications of CNN. IEEE Trans. Syst. Man Commun. 1976, 6, 769–772. [Google Scholar]
- Hart, P. The condensed nearest neighbor rule. IEEE Trans. Inf. Theory 1968, 14, 515–516. [Google Scholar] [CrossRef]
- Kubat, M.; Matwin, S. Addressing the curse of imbalanced training sets: One-sided selection. In Proceedings of the 14th International Conference on Machine Learning, ICML, Nashville, TN, USA, 8–12 July 1997; pp. 179–186. [Google Scholar]
- Wilson, D.L. Asymptotic Properties of Nearest Neighbor Rules Using Edited Data. IEEE Trans. Syst. Man Cybern. 1972, 3, 408–421. [Google Scholar] [CrossRef] [Green Version]
- Levina, E.; Bickel, P.J. Maximum likelihood estimation of intrinsic dimension. Proc. NIPS 2004, 1, 777–784. [Google Scholar]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Hofree, M.; Shen, J.P.; Carter, H.; Gross, A.; Ideker, T. Network-based stratification of tumor mutations. Nat. Methods 2013, 10, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Poline, J.B.; Dumas, G. Experimenting with reproducibility: A case study of robustness in bioinformatics. Gigascience 2018, 7, giy077. [Google Scholar] [CrossRef] [Green Version]
- Le Morvan, M.; Zinovyev, A.; Vert, J.P. NetNorM: Capturing cancer-relevant information in somatic exome mutation data with gene networks for cancer stratification and prognosis. PLoS Comput. Biol. 2017, 13, e1005573. [Google Scholar] [CrossRef] [Green Version]
- Auslander, N.; Wolf, Y.I.; Koonin, E.V. In silico learning of tumor evolution through mutational time series. Proc. Natl. Acad. Sci. USA 2019, 116, 9501–9510. [Google Scholar] [CrossRef] [Green Version]
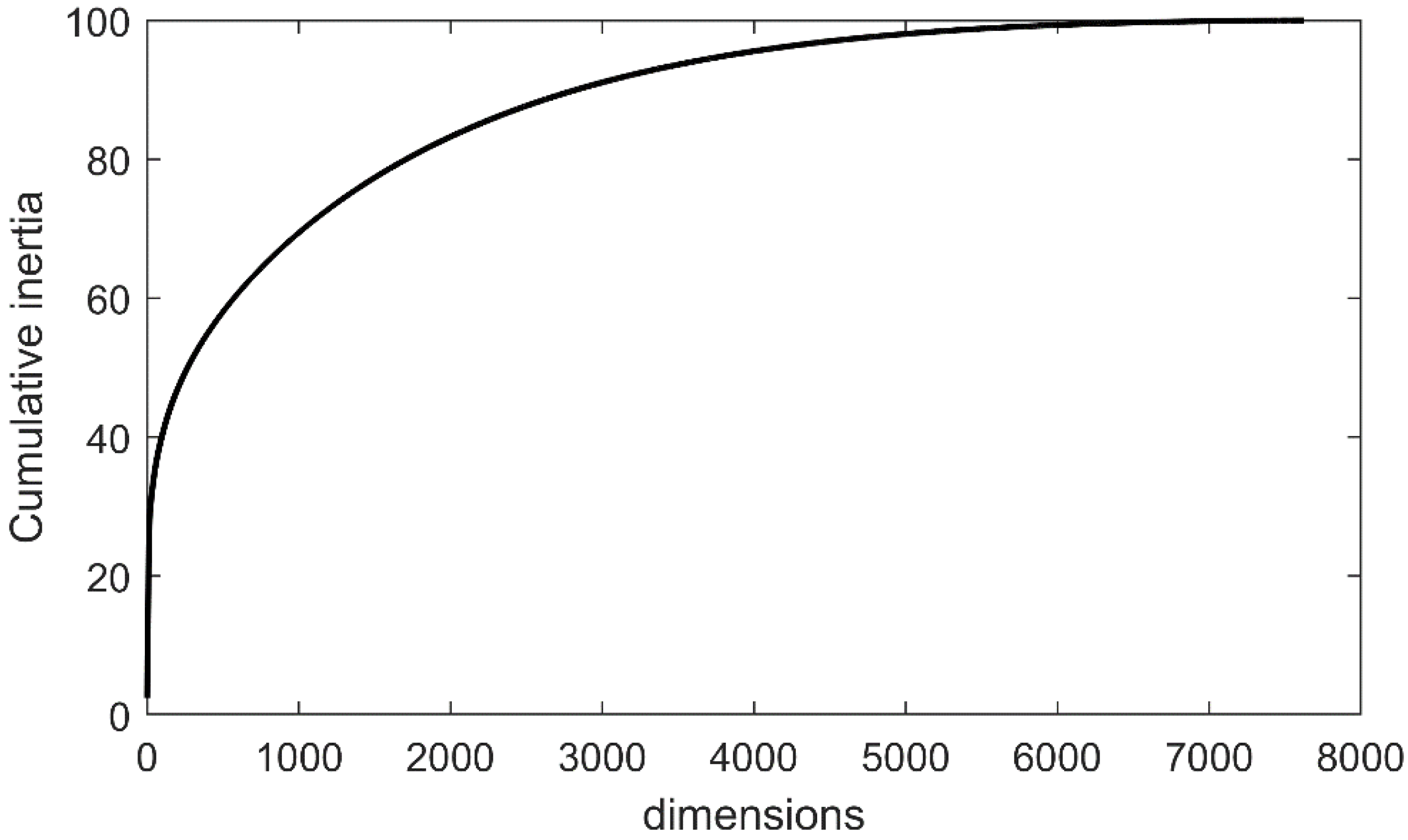



{kind=link}
{kind=link}
{kind=link}
| Ca | Sample Number | Missense Mutations | Nonsense Mutations | Nonstop Mutations | Slice Sites | Total Mutations |
|---|---|---|---|---|---|---|
| ACC | 91 | 7258 | 524 | 15 | 415 | 12,082 |
| BLCA | 130 | 25,108 | 2215 | 47 | 605 | 38,174 |
| BRCA | 993 | 55,063 | 4841 | 133 | 1561 | 83,973 |
| CESC | 194 | 26,606 | 2716 | 84 | 574 | 45,936 |
| CHOL | 36 | 3307 | 316 | 0 | 90 | 5222 |
| COAD | 423 | 163,454 | 12,146 | 184 | 9128 | 324,120 |
| DLBC | 48 | 9623 | 353 | 0 | 188 | 16,403 |
| ESCA | 183 | 33,179 | 2829 | 0 | 851 | 52,576 |
| GBM | 316 | 26,462 | 2230 | 15 | 813 | 46,899 |
| HNSC | 279 | 33,260 | 2686 | 44 | 864 | 49,776 |
| KICH | 66 | 1498 | 103 | 0 | 54 | 2341 |
| KIRP | 171 | 9442 | 515 | 18 | 697 | 15,007 |
| LAML | 197 | 1529 | 117 | 0 | 54 | 2320 |
| LGG | 286 | 6098 | 397 | 7 | 331 | 9256 |
| LIHC | 373 | 32,553 | 1853 | 0 | 1238 | 47,895 |
| LUAD | 230 | 47,700 | 3692 | 56 | 1558 | 69,546 |
| LUSC | 548 | 162,388 | 12,268 | 301 | 11,708 | 263,481 |
| MESO | 83 | 2780 | 177 | 8 | 196 | 5840 |
| OV | 142 | 7106 | 420 | 9 | 244 | 10,926 |
| PAAD | 150 | 19,899 | 1307 | 6 | 974 | 30,138 |
| PCPG | 184 | 3271 | 83 | 0 | 61 | 4212 |
| PRAD | 332 | 7816 | 433 | 12 | 496 | 11,846 |
| READ | 81 | 15,899 | 1724 | 0 | 306 | 23,862 |
| SARC | 259 | 15,457 | 785 | 19 | 332 | 22,185 |
| SKCM | 472 | 243,677 | 15,231 | 111 | 10,522 | 423,963 |
| STAD | 289 | 87,092 | 4423 | 93 | 3852 | 132,196 |
| TGCT | 156 | 2121 | 123 | 0 | 52 | 3428 |
| THCA | 406 | 3000 | 150 | 0 | 77 | 4489 |
| THYM | 121 | 15,938 | 1639 | 0 | 342 | 23,939 |
| UCEC | 248 | 121,440 | 13,472 | 158 | 1942 | 181,159 |
| UCS | 57 | 6003 | 612 | 16 | 275 | 8713 |
| UVM | 80 | 1665 | 73 | 0 | 36 | 2856 |
| Total | 7624 | 1,197,692 | 90,453 | 1336 | 50,436 | 1,974,759 |
| ACC | BLCA | BRCA | CESC | CHOL | COAD | DLBC | ESCA |
| 0.526 | 0.733 | 0.888 | 0.672 | 0.854 | 0.599 | 0.914 | 0.889 |
| GBM | HNSC | KICH | KIRP | LAML | LGG | LIHC | LUAD |
| 0.761 | 0.578 | 0.899 | 0.516 | 0.328 | 0.938 | 0.513 | 0.815 |
| LUSC | MESO | OV | PAAD | PCPG | PRAD | READ | SARC |
| 0.869 | 0.321 | 0.836 | 0.816 | 0.958 | 0.526 | 0.68 | 0.808 |
| SKCM | STAD | TGCT | THCA | THYM | UCEC | UCS | UVM |
| 0.689 | 0.648 | 0.968 | 0.548 | 0.928 | 0.623 | 0.87 | 0.769 |
| Ca. | Sample Number | Missense Mutations | Nonsense Mutations | Nonstop Mutations | Slice Sites | Total Mutations |
|---|---|---|---|---|---|---|
| BRCA | 60 | 522 | 78 | 0 | 68 | 668 |
| LAML | 103 | 752 | 72 | 0 | 22 | 846 |
| PAAD | 98 | 154 | 16 | 6 | 7 | 196 |
| PRAD | 65 | 328 | 28 | 15 | 1 | 427 |
| Learners/Classifiers | Acc | Precision | Recall | F-Score |
|---|---|---|---|---|
| Trained on the 90% of the samples (i.e., 6861) and tested on the 10% of the samples (i.e., 763) | ||||
| Decision Tree | 0.46 (0.40 to 0.51) | 0.48 (0.42 to 0.51) | 0.38 (0.31 to 0.43) | 0.40 (0.34 to 0.44) |
| KNN | 0.44 (0.38 to 0.49) | 0.44 (0.38 to 0.47) | 0.35 (0.30 to 0.39) | 0.33 (0.26 to 0.39) |
| SVM | 0.60 (0.55 to 0.64) | 0.64 (0.60 to 0.68) | 0.47 (0.41 to 0.51) | 0.50 (0.44 to 0.53) |
| XGBoost | 0.66 (0.42 to 0.48) | 0.64 (0.59 to 0.68) | 0.56 (0.51 to 0.60) | 0.58 (0.53 to 0.63) |
| Neural Networks | 0.69 (0.64 to 0.73) | 0.66 (0.61 to 0.70) | 0.57 (0.51 to 0.61) | 0.59 (0.54 to 0.63) |
| SPINN | 0.71 (0.67 to 0.74) | 0.74 (0.70 to 0.77) | 0.62 (0.57 to 0.66) | 0.65 (0.61 to 0.69) |
| Trained on the 80% of the samples (i.e., 6099) and tested on the 20% of the samples (i.e., 1525) | ||||
| Decision Tree | 0.45 (0.38 to 0.51) | 0.45 (0.39 to 0.51) | 0.36 (0.29 to 0.41) | 0.38 (0.32 to 0.43) |
| KNN | 0.43 (0.35 to 0.49) | 0.45 (0.36 to 0.48) | 0.33 (0.26 to 0.38) | 0.32 (0.25 to 0.38) |
| SVM | 0.60 (0.52 to 0.65) | 0.63 (0.56 to 0.68) | 0.47 (0.39 to 0.52) | 0.50 (0.43 to 0.55) |
| XGBoost | 0.65 (0.59 to 0.70) | 0.63 (0.56 to 0.68) | 0.54 (0.49 to 0.58) | 0.56 (0.50 to 0.60) |
| Neural Networks | 0.67 (0.60 to 0.72) | 0.63 (0.55 to 0.68) | 0.55 (0.49 to 0.60) | 0.57 (0.50 to 0.61) |
| SPINN | 0.69 (0.63 to 0.73) | 0.66 (0.61 to 0.71) | 0.59 (0.54 to 0.66) | 0.61 (0.56 to 0.66) |
| Learners/Classifiers | Acc | Precision | Recall | F-Score |
|---|---|---|---|---|
| Trained on the 90% of the samples (i.e., 6173) and tested on the 10% of the samples (i.e., 686) | ||||
| Decision Tree | 0.46 (0.40 to 0.48) | 0.48 (0.42 to 0.50) | 0.38 (0.33 to 0.40) | 0.40 (0.36 to 0.42) |
| KNN | 0.44 (0.39 to 0.46) | 0.44 (0.40 to 0.46) | 0.35 (0.30 to 0.37) | 0.33 (0.29 to 0.36) |
| SVM | 0.61 (0.57 to 0.64) | 0.64 (0.60 to 0.67) | 0.47 (0.41 to 0.49) | 0.51 (0.47 to 0.53) |
| XGBoost | 0.68 (0.63 to 0.71) | 0.65 (0.61 to 0.67) | 0.57 (0.53 to 0.60) | 0.59 (0.56 to 0.61) |
| Neural Networks | 0.70 (0.65 to 0.73) | 0.65 (0.61 to 0.67) | 0.59 (0.55 to 0.63) | 0.60 (0.55 to 0.62) |
| SPINN | 0.73 (0.70 to 0.76) | 0.75 (0.72 to 0.78) | 0.64 (0.60 to 0.67) | 0.67 (0.64 to 0.71) |
| Trained on the 80% of the samples (i.e., 5487) and tested on the 20% of the samples (i.e., 1372) | ||||
| Decision Tree | 0.45 (0.39 to 0.50) | 0.45 (0.40 to 0.50) | 0.36 (0.30 to 0.41) | 0.38 (0.33 to 0.42) |
| KNN | 0.43 (0.39 to 0.46) | 0.45 (0.40 to 0.49) | 0.33 (0.27 to 0.36) | 0.32 (0.27 to 0.35) |
| SVM | 0.60 (0.55 to 0.63) | 0.63 (0.59 to 0.66) | 0.47 (0.42 to 0.50) | 0.50 (0.45 to 0.53) |
| XGBoost | 0.66 (0.62 to 0.69) | 0.64 (0.60 to 0.67) | 0.55 (0.50 to 0.59) | 0.57 (0.51 to 0.60) |
| Neural Networks | 0.68 (0.63 to 0.72) | 0.66 (0.61 to 0.70) | 0.57 (0.52 to 0.61) | 0.58 (0.53 to 0.62) |
| SPINN | 0.71 (0.66 to 0.73) | 0.73 (0.69 to 0.76) | 0.64 (0.60 to 0.67) | 0.66 (0.61 to 0.70) |
| ACC | BLCA | BRCA | CESC | HNSC | KIRP | LGG | LUAD | PAAD | PRAD | STAD | UCS | CHOL | COAD | DLBC | ESCA | GBM | KICH | LAML | LIHC | LUSC | MESO | OV | PCPG | READ | SARC | SKCM | TGCT | THCA | THYM | UCEC | UVM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | 7 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BLCA | 0 | 5 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BRCA | 0 | 0 | 77 | 0 | 1 | 0 | 0 | 0 | 1 | 8 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 6 | 0 | 0 | 0 |
| CESC | 0 | 0 | 1 | 13 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| HNSC | 0 | 2 | 2 | 0 | 16 | 0 | 0 | 0 | 0 | 3 | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| KIRP | 1 | 1 | 0 | 1 | 1 | 12 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| LGG | 0 | 0 | 1 | 0 | 0 | 0 | 25 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| LUAD | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 17 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| PAAD | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 14 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PRAD | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 25 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 2 | 0 | 0 | 0 |
| STAD | 0 | 0 | 1 | 0 | 5 | 0 | 0 | 0 | 1 | 1 | 15 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| UCS | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 |
| CHOL | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| COAD | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 38 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| DLBC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 |
| ESCA | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 16 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| GBM | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 1 | 4 | 1 | 0 | 0 | 0 | 0 | 0 | 21 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| KICH | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| LAML | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 15 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| LIHC | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 1 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 25 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 |
| LUSC | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 53 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| MESO | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| OV | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PCPG | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 12 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| READ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| SARC | 0 | 0 | 4 | 0 | 0 | 0 | 1 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 11 | 0 | 0 | 0 | 0 | 0 | 0 |
| SKCM | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 42 | 0 | 1 | 0 | 0 | 0 |
| TGCT | 0 | 0 | 2 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 8 | 0 | 0 | 0 | 0 |
| THCA | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 35 | 0 | 1 | 0 |
| THYM | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 1 |
| UCEC | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 18 | 0 |
| UVM | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 |
| ACC | BLCA | BRCA | CESC | HNSC | KIRP | LGG | LUAD | PAAD | PRAD | STAD | UCS | CHOL | COAD | DLBC | ESCA | GBM | KICH | LAML | LIHC | LUSC | MESO | OV | PCPG | READ | SARC | SKCM | TGCT | THCA | THYM | UCEC | UVM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | 7 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BLCA | 0 | 4 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| BRCA | 0 | 0 | 76 | 0 | 1 | 0 | 0 | 0 | 1 | 8 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 6 | 0 | 0 | 0 |
| CESC | 0 | 0 | 1 | 13 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| HNSC | 0 | 2 | 2 | 0 | 16 | 0 | 0 | 0 | 0 | 3 | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| KIRP | 1 | 1 | 0 | 1 | 1 | 12 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| LGG | 0 | 0 | 1 | 0 | 0 | 0 | 25 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| LUAD | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 16 | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| PAAD | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 14 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PRAD | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 25 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 2 | 0 | 0 | 0 |
| STAD | 0 | 0 | 1 | 0 | 5 | 0 | 0 | 0 | 1 | 1 | 14 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| UCS | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 |
| CHOL | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| COAD | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 37 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| DLBC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 |
| ESCA | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 16 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| GBM | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 1 | 4 | 1 | 0 | 0 | 0 | 0 | 0 | 21 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| KICH | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| LAML | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 15 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| LIHC | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 1 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 25 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 |
| LUSC | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 53 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| MESO | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| OV | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PCPG | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 12 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| READ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| SARC | 0 | 0 | 4 | 0 | 0 | 0 | 1 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 11 | 0 | 0 | 0 | 0 | 0 | 0 |
| SKCM | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 42 | 0 | 1 | 0 | 0 | 0 |
| TGCT | 0 | 0 | 2 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 8 | 0 | 0 | 0 | 0 |
| THCA | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 35 | 0 | 1 | 0 |
| THYM | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 1 |
| UCEC | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 17 | 0 |
| UVM | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 |
| BRCA | LAML | PAAD | PRAD | |
|---|---|---|---|---|
| Decision tree | 0.40 | 0.45 | 0.41 | 0.19 |
| KNN | 0.39 | 0.44 | 0.39 | 0.18 |
| SVM | 0.53 | 0.55 | 0.54 | 0.25 |
| XGBoost | 0.62 | 0.69 | 0.61 | 0.30 |
| Neural Networks | 0.61 | 0.68 | 0.62 | 0.29 |
| SPINN | 0.64 | 0.72 | 0.65 | 0.30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dikaios, N. Sparse-Input Neural Networks to Differentiate 32 Primary Cancer Types on the Basis of Somatic Point Mutations. Onco 2022, 2, 56-68. https://doi.org/10.3390/onco2020005
Dikaios N. Sparse-Input Neural Networks to Differentiate 32 Primary Cancer Types on the Basis of Somatic Point Mutations. Onco. 2022; 2(2):56-68. https://doi.org/10.3390/onco2020005
Chicago/Turabian StyleDikaios, Nikolaos. 2022. "Sparse-Input Neural Networks to Differentiate 32 Primary Cancer Types on the Basis of Somatic Point Mutations" Onco 2, no. 2: 56-68. https://doi.org/10.3390/onco2020005
APA StyleDikaios, N. (2022). Sparse-Input Neural Networks to Differentiate 32 Primary Cancer Types on the Basis of Somatic Point Mutations. Onco, 2(2), 56-68. https://doi.org/10.3390/onco2020005