
State of the Art on CAR T-Cell Therapies for Onco-Haematological Disorders and Other Conditions

{kind=link}
Abstract
Simple Summary
Abstract
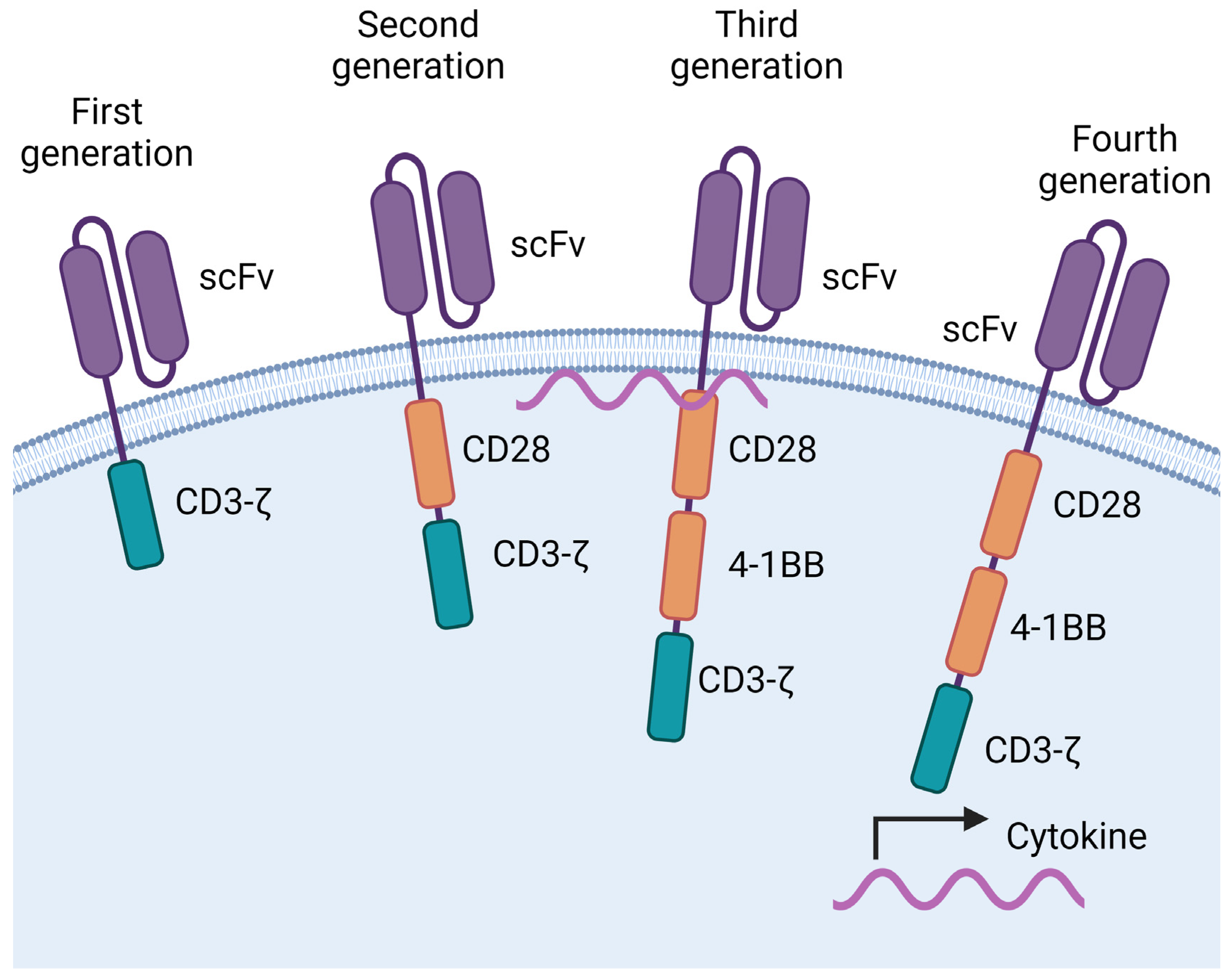
1. Introduction
2. Future Challenges for the Use of CAR T-Cells
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Irvine, D.J.; Maus, M.V.; Mooney, D.J.; Wong, W.W. The future of engineered immune cell therapies. Science 2022, 378, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Passweg, J.R.; Baldomero, H.; Ciceri, F.; de la Cámara, R.; Glass, B.; Greco, R.; Hazenberg, M.D.; Kalwak, K.; McLornan, D.P.; Neven, B.; et al. Hematopoietic cell transplantation and cellular therapies in Europe 2022. CAR-T activity continues to grow; transplant activity has slowed: A report from the EBMT. Bone Marrow Transpl. 2024, 59, 803–812. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Rivière, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef]
- Van Der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef]
- Imai, C.M.I.H.A.R.A.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Sheykhhasan, M.; Manoochehri, H.; Dama, P. Use of CAR T-cell for acute lymphoblastic leukemia (ALL) treatment: A review study. Cancer Gene Ther. 2022, 29, 1080–1096. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson Jr, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Jommi, C.; Bramanti, S.; Pani, M.; Ghirardini, A.; Santoro, A. CAR T-cell therapies in Italy: Patient access barriers and recommendations for health system solutions. Front. Pharmacol. 2022, 13, 915342. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Feng, J.; Gu, T.; Wang, L.; Wang, Y.; Zhou, L.; Hong, R.; Yin, E.T.; Zhang, M.; Lu, P.; et al. CAR T-cell therapies in China: Rapid evolution and a bright future. Lancet Haematol. 2022, 9, e930–e941. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R. and Arslan, Ö. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Ghassemi, S.; Durgin, J.S.; Nunez-Cruz, S.; Patel, J.; Leferovich, J.; Pinzone, M.; Shen, F.; Cummins, K.D.; Plesa, G.; Cantu, V.A.; et al. Rapid manufacturing of non-activated potent CAR T cells. Nat. Biomed. Eng. 2022, 6, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Crompton, J.G.; Sukumar, M.; Roychoudhuri, R.; Clever, D.; Gros, A.; Eil, R.L.; Tran, E.; Hanada, K.I.; Yu, Z.; Palmer, D.C.; et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res. 2015, 75, 296–305. [Google Scholar] [CrossRef]
- Schulthess, D.; Gassull, D.; Makady, A.; Ludlow, A.; Rothman, B.; Ten Have, P.; Wu, Y.; Ekstrom, L.; Minnema, M.; Jagasia, M. Are CAR-T therapies living up to their hype? A study using real-world data in two cohorts to determine how well they are actually working in practice compared with bone marrow transplants. BMJ Evid. Based Med. 2021, 26, 98–102. [Google Scholar] [CrossRef]
- Svoboda, J.; Landsburg, D.J.; Nasta, S.D.; Barta, S.K.; Chong, E.A.; Lariviere, M.J.; Shea, J.; Cervini, A.; Hexner, E.O.; Marshall, A.; et al. Safety and efficacy of armored huCART19-IL18 in patients with relapsed/refractory lymphomas that progressed after anti-CD19 CAR T cells. J. Clin. Oncol. 2024, 42 (Suppl. 16). [Google Scholar] [CrossRef]
- Wutti-In, Y.; Sujjitjoon, J.; Sawasdee, N.; Panya, A.; Kongkla, K.; Yuti, P.; Yongpitakwattana, P.; Thepmalee, C.; Junking, M.; Chieochansin, T.; et al. Development of a novel anti-CD19 CAR containing a fully human scFv and three costimulatory domains. Front. Oncol. 2022, 11, 802876. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.J.; Dong, L.; Liu, Y.C.; Tsao, S.T.; Li, Y.C.; Liu, L.; Gao, Z.; Tan, X.; Lu, D.P.; Zhang, J.P.; et al. Safety and efficacy evaluation of 4SCAR19 chimeric antigen receptor-modified T cells targeting B cell acute lymphoblastic leukemia-three-year follow-up of a multicenter phase I/II study. Blood 2016, 128, 587. [Google Scholar] [CrossRef]
- IQVIA Institute for Human Data Science. Strengthening Pathways for Cell and Gene Therapies: Current State and Future Scenarios. March 2024. Available online: www.iqviainstitute.org (accessed on 2 July 2024).
- Cappell, K.M.; Kochenderfer, J.N. Long-term outcomes following CAR T cell therapy: What we know so far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Li, X.; Chintala, N.K.; Tano, Z.E.; Adusumilli, P.S. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr. Opin. Immunol. 2018, 51, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Del Bufalo, F.; De Angelis, B.; Caruana, I.; Del Baldo, G.; De Ioris, M.A.; Serra, A.; Mastronuzzi, A.; Cefalo, M.G.; Pagliara, D.; Amicucci, M.; et al. GD2-CART01 for relapsed or refractory high-risk neuroblastoma. N. Engl. J. Med. 2023, 388, 1284–1295. [Google Scholar] [CrossRef]
- Yeku, O.O.; Longo, D.L. CAR T cells for neuroblastoma. N. Engl. J. Med. 2023, 388, 1328–1331. [Google Scholar] [CrossRef]
- Schett, G.; Mackensen, A.; Mougiakakos, D. CAR T-cell therapy in autoimmune diseases. Lancet 2023, 402, 2034–2044. [Google Scholar] [CrossRef]
- Mougiakakos, D.; Krönke, G.; Völkl, S.; Kretschmann, S.; Aigner, M.; Kharboutli, S.; Böltz, S.; Manger, B.; Mackensen, A.; Schett, G. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N. Engl. J. Med. 2021, 385, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Krickau, T.; Naumann-Bartsch, N.; Aigner, M.; Kharboutli, S.; Kretschmann, S.; Spoerl, S.; Vasova, I.; Völkl, S.; Woelfle, J.; Mackensen, A.; et al. CAR T-cell therapy rescues adolescent with rapidly progressive lupus nephritis from haemodialysis. Lancet 2024, 403, 1627–1630. [Google Scholar] [CrossRef]
- Müller, F.; Taubmann, J.; Bucci, L.; Wilhelm, A.; Bergmann, C.; Völkl, S.; Aigner, M.; Rothe, T.; Minopoulou, I.; Tur, C.; et al. CD19 CAR T-cell therapy in autoimmune disease—A case series with follow-up. N. Engl. J. Med. 2024, 390, 687–700. [Google Scholar] [CrossRef]
- Müller, F.; Boeltz, S.; Knitza, J.; Aigner, M.; Völkl, S.; Kharboutli, S.; Reimann, H.; Taubmann, J.; Kretschmann, S.; Rösler, W.; et al. CD19-targeted CAR T cells in refractory antisynthetase syndrome. Lancet 2023, 401, 815–818. [Google Scholar] [CrossRef]
- Orvain, C.; Boulch, M.; Bousso, P.; Allanore, Y.; Avouac, J. Is there a place for chimeric antigen receptor–T cells in the treatment of chronic autoimmune rheumatic diseases? Arthritis Rheumatol. 2021, 73, 1954–1965. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Sun, B.; Li, S.; Wei, W.; Liu, X.; Cui, X.; Zhang, X.; Liu, N.; Yan, L.; Deng, Y.; et al. NKG2D-CAR T cells eliminate senescent cells in aged mice and nonhuman primates. Sci. Transl. Med. 2023, 15, eadd1951. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Liu, Y.; Wang, L.; He, Z.; Zhao, X.; Ma, Y.; Jia, Y.; Li, Z.; Yin, N.; Peng, M. A single infusion of engineered long-lived and multifunctional T cells confers durable remission of asthma in mice. Nat. Immunol. 2024, 25, 1059–1072. [Google Scholar] [CrossRef]
- Seif, M.; Kakoschke, T.K.; Ebel, F.; Bellet, M.M.; Trinks, N.; Renga, G.; Pariano, M.; Romani, L.; Tappe, B.; Espie, D.; et al. CAR T cells targeting Aspergillus fumigatus are effective at treating invasive pulmonary aspergillosis in preclinical models. Sci. Transl. Med. 2022, 14, eabh1209. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Chiesa, R.; Georgiadis, C.; Syed, F.; Zhan, H.; Etuk, A.; Gkazi, S.A.; Preece, R.; Ottaviano, G.; Braybrook, T.; Chu, J.; et al. Base-edited CAR7 T cells for relapsed T-cell acute lymphoblastic leukemia. N. Engl. J. Med. 2023, 389, 899–910. [Google Scholar] [CrossRef]
- Hamilton, M.P.; Sugio, T.; Noordenbos, T.; Shi, S.; Bulterys, P.L.; Liu, C.L.; Kang, X.; Olsen, M.N.; Good, Z.; Dahiya, S.; et al. Risk of Second Tumors and T-Cell Lymphoma after CAR T-Cell Therapy. N. Engl. J. Med. 2024, 390, 2047–2060. [Google Scholar] [CrossRef] [PubMed]
- Ozdemirli, M.; Loughney, T.M.; Deniz, E.; Chahine, J.J.; Albitar, M.; Pittaluga, S.; Sadigh, S.; Armand, P.; Uren, A.; Anderson, K.C. Indolent CD4+ CAR T-Cell Lymphoma after Cilta-cel CAR T-Cell Therapy. N. Engl. J. Med. 2024, 390, 2074–2082. [Google Scholar] [CrossRef]
- Mitchell, E.; Vassiliou, G.S. T-Cell Cancer after CAR T-Cell Therapy. N. Engl. J. Med. 2024, 390, 2120–2121. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madrigal, J.A.; Crispín, J.C. State of the Art on CAR T-Cell Therapies for Onco-Haematological Disorders and Other Conditions. Onco 2024, 4, 232-240. https://doi.org/10.3390/onco4030017
Madrigal JA, Crispín JC. State of the Art on CAR T-Cell Therapies for Onco-Haematological Disorders and Other Conditions. Onco. 2024; 4(3):232-240. https://doi.org/10.3390/onco4030017
Chicago/Turabian StyleMadrigal, Jose Alejandro, and José C. Crispín. 2024. "State of the Art on CAR T-Cell Therapies for Onco-Haematological Disorders and Other Conditions" Onco 4, no. 3: 232-240. https://doi.org/10.3390/onco4030017
APA StyleMadrigal, J. A., & Crispín, J. C. (2024). State of the Art on CAR T-Cell Therapies for Onco-Haematological Disorders and Other Conditions. Onco, 4(3), 232-240. https://doi.org/10.3390/onco4030017