
Cancer Immunotherapy: Targeting TREX1 Has the Potential to Unleash the Host Immunity against Cancer Cells


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
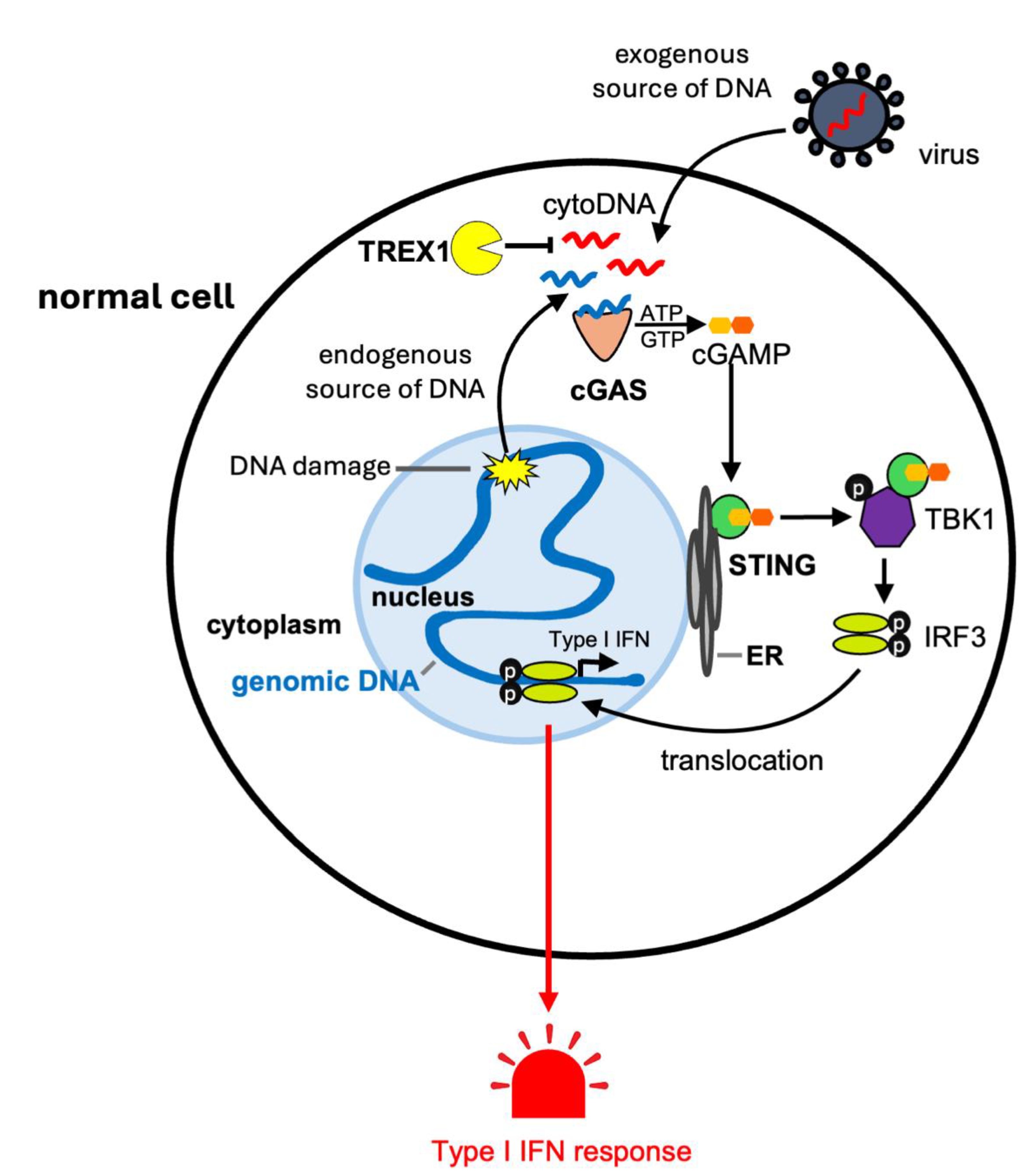
1.1. The cGAS-STING Pathway and Inflammation
1.2. TREX1 Degrades Cytosolic DNA Limiting cGAS-STING Signaling
1.3. Impact of TREX1 on Genomic Stability
1.4. Cancer Immunotherapy
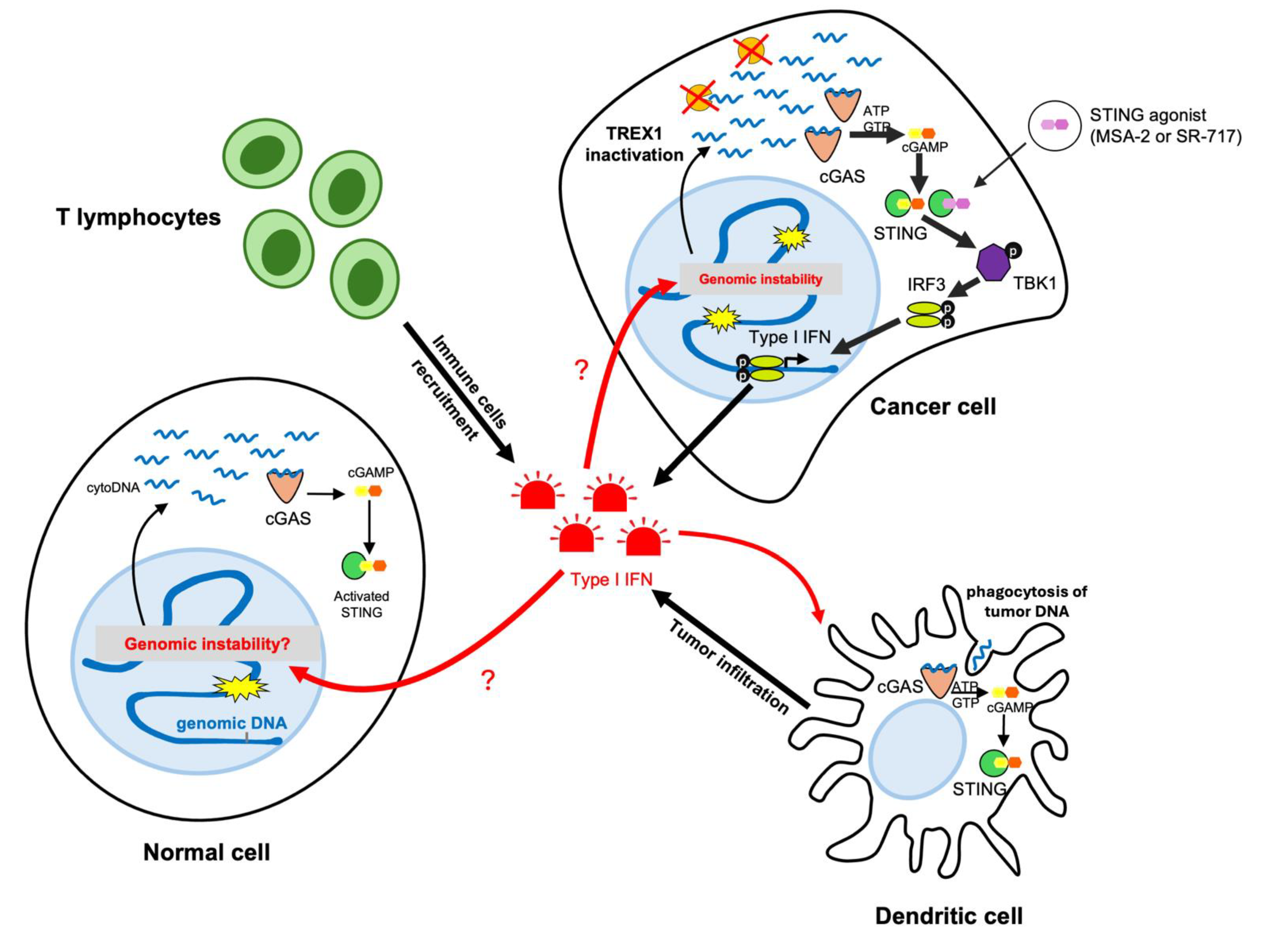
2. Inactivation of TREX1 Promotes Type I Interferon Response Unleashing Tumor Immunogenicity
2.1. Cancer Rejection through Activation of the cGAS-STING Pathway
2.2. TREX1 Induction by Chemo- and Radio-Therapies
2.3. Promising Efficacy of TREX1 Inactivation in Pre-Clinical Cancer Models
2.4. Depletion of TREX1 Remodels the Immune Cell Population in the Tumor Microenvironment
3. Combining TREX1 Inactivation with Immune Checkpoint Blockade Shows Promising Potential for Immune Clearance of Cancer Cells
4. Limitations and Future Perspectives for TREX1 Targeting in Cancer Therapy
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, T.; Chen, Z.J. The CGAS–CGAMP–STING Pathway Connects DNA Damage to Inflammation, Senescence, and Cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Samson, N.; Ablasser, A. The CGAS–STING Pathway and Cancer. Nat. Cancer 2022, 3, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Pathare, G.R.; Decout, A.; Glück, S.; Cavadini, S.; Makasheva, K.; Hovius, R.; Kempf, G.; Weiss, J.; Kozicka, Z.; Guey, B.; et al. Structural Mechanism of CGAS Inhibition by the Nucleosome. Nature 2020, 587, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Volkman, H.E.; Cambier, S.; Gray, E.E.; Stetson, D.B. Tight Nuclear Tethering of CGAS Is Essential for Preventing Autoreactivity. Elife 2019, 8, e47491. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA Damage Response in Immuno-Oncology: Developments and Opportunities. Nat. Rev. Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef]
- Técher, H.; Pasero, P. The Replication Stress Response on a Narrow Path Between Genomic Instability and Inflammation. Front. Cell Dev. Biol. 2021, 9, 702584. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Kirchmair, A.; Sato, A.; Buqué, A.; Rybstein, M.; Petroni, G.; Bloy, N.; Finotello, F.; Stafford, L.; Navarro Manzano, E.; et al. Mitochondrial DNA Drives Abscopal Responses to Radiation That Are Inhibited by Autophagy. Nat. Immunol. 2020, 21, 1160–1171. [Google Scholar] [CrossRef]
- Erdal, E.; Haider, S.; Rehwinkel, J.; Harris, A.L.; McHugh, P.J. A Prosurvival DNA Damage-Induced Cytoplasmic Interferon Response Is Mediated by End Resection Factors and Is Limited by Trex1. Genes Dev. 2017, 31, 353–369. [Google Scholar] [CrossRef]
- Kitai, Y.; Kawasaki, T.; Sueyoshi, T.; Kobiyama, K.; Ishii, K.J.; Zou, J.; Akira, S.; Matsuda, T.; Kawai, T. DNA-Containing Exosomes Derived from Cancer Cells Treated with Topotecan Activate a STING-Dependent Pathway and Reinforce Antitumor Immunity. J. Immunol. 2017, 198, 1649–1659. [Google Scholar] [CrossRef]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Della Corte, C.M.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-Cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.J.; LeBert, N.; Chitre, A.A.; Koo, C.X.E.; Nga, X.H.; Ho, S.S.W.; Khatoo, M.; Tan, N.Y.; Ishii, K.J.; Gasser, S. Genome-Derived Cytosolic DNA Mediates Type I Interferon-Dependent Rejection of B Cell Lymphoma Cells. Cell Rep. 2015, 11, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.S.W.; Zhang, W.Y.L.; Tan, N.Y.J.; Khatoo, M.; Suter, M.A.; Tripathi, S.; Cheung, F.S.G.; Lim, W.K.; Tan, P.H.; Ngeow, J.; et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 2016, 44, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Lu, C.; Jin, Q.; Lu, H.; Chen, X.; Tian, L.; Zhang, Y.; Ortega, J.; Zhang, J.; Siteni, S.; et al. MLH1 Deficiency-Triggered DNA Hyperexcision by Exonuclease 1 Activates the CGAS-STING Pathway. Cancer Cell 2021, 39, 109–121.e5. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal Instability Drives Metastasis through a Cytosolic DNA Response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef]
- Crow, Y.J.; Manel, N. Aicardi–Goutières Syndrome and the Type I Interferonopathies. Nat. Rev. Immunol. 2015, 15, 429–440. [Google Scholar] [CrossRef]
- Yang, Y.G.; Lindahl, T.; Barnes, D.E. Trex1 Exonuclease Degrades SsDNA to Prevent Chronic Checkpoint Activation and Autoimmune Disease. Cell 2007, 131, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Stetson, D.B.; Ko, J.S.; Heidmann, T.; Medzhitov, R. Trex1 Prevents Cell-Intrinsic Initiation of Autoimmunity. Cell 2008, 134, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Fye, J.M.; Orebaugh, C.D.; Coffin, S.R.; Hollis, T.; Perrino, F.W. Dominant Mutation of the TREX1 Exonuclease Gene in Lupus and Aicardi-Goutieres Syndrome. J. Biol. Chem. 2011, 286, 32373–32382. [Google Scholar] [CrossRef]
- Lehtinen, D.A.; Harvey, S.; Mulcahy, M.J.; Hollis, T.; Perrino, F.W. The TREX1 Double-Stranded DNA Degradation Activity Is Defective in Dominant Mutations Associated with Autoimmune Disease. J. Biol. Chem. 2008, 283, 31649–31656. [Google Scholar] [CrossRef]
- Huang, K.W.; Wu, C.Y.; Toh, S.I.; Liu, T.C.; Tu, C.I.; Lin, Y.H.; Cheng, A.J.; Kao, Y.T.; Chu, J.W.; Hsiao, Y.Y. Molecular Insight into the Specific Enzymatic Properties of TREX1 Revealing the Diverse Functions in Processing RNA and DNA/RNA Hybrids. Nucleic Acids Res. 2023, 51, 11927–11940. [Google Scholar] [CrossRef] [PubMed]
- Lee-Kirsch, M.A.; Gong, M.; Chowdhury, D.; Senenko, L.; Engel, K.; Lee, Y.A.; De Silva, U.; Bailey, S.L.; Witte, T.; Vyse, T.J.; et al. Mutations in the Gene Encoding the 3′-5′ DNA Exonuclease TREX1 Are Associated with Systemic Lupus Erythematosus. Nat. Genet. 2007, 39, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Tani, T.; Mathsyaraja, H.; Campisi, M.; Li, Z.H.; Haratani, K.; Fahey, C.G.; Ota, K.; Mahadevan, N.R.; Shi, Y.; Saito, S.; et al. TREX1 Inactivation Unleashes Cancer Cell STING-Interferon Signaling and Promotes Antitumor Immunity. Cancer Discov. 2024, 14, 752–765. [Google Scholar] [CrossRef] [PubMed]
- Toufektchan, E.; Dananberg, A.; Striepen, J.; Hickling, J.H.; Shim, A.; Chen, Y.; Nichols, A.; Paez, M.A.D.; Mohr, L.; Bakhoum, S.F.; et al. Intratumoral TREX1 Induction Promotes Immune Evasion by Limiting Type I IFN. Cancer Immunol. Res. 2024, 12, 673–686. [Google Scholar] [CrossRef]
- Lim, J.; Rodriguez, R.; Williams, K.; Silva, J.; Gutierrez, A.G.; Tyler, P.; Baharom, F.; Sun, T.; Lin, E.; Martin, S.; et al. The Exonuclease TREX1 Constitutes an Innate Immune Checkpoint Limiting CGAS/STING-Mediated Antitumor Immunity. Cancer Immunol. Res. 2024, 12, 663–672. [Google Scholar] [CrossRef]
- Lau, A.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA Tumor Virus Oncogenes Antagonize the CGAS-STING DNA-Sensing Pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef]
- Técher, H. T-Rex Escaped from the Cytosolic Park: Re-Thinking the Impact of TREX1 Exonuclease Deficiencies on Genomic Stability. BioEssays 2024, 46, 2400066. [Google Scholar] [CrossRef] [PubMed]
- Técher, H.; Gopaul, D.; Heuzé, J.; Bouzalmad, N.; Leray, B.; Vernet, A.; Mettling, C.; Moreaux, J.; Pasero, P.; Lin, Y.-L. MRE11 and TREX1 Control Senescence by Coordinating Replication Stress and Interferon Signaling. Nat. Commun. 2024, 15, 5423. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Xiao, N.; Zhang, S.; Zhou, X.; Zhang, Y.; Lu, Z.; Fu, Y.; Huang, M.; Xu, S.; Chen, Q. Suppression of TREX1 Deficiency-Induced Cellular Senescence and Interferonopathies by Inhibition of DNA Damage Response. iScience 2023, 26, 107090. [Google Scholar] [CrossRef]
- Giordano, A.M.S.; Luciani, M.; Gatto, F.; Alezz, M.A.; Beghè, C.; Della Volpe, L.; Migliara, A.; Valsoni, S.; Genua, M.; Dzieciatkowska, M.; et al. DNA Damage Contributes to Neurotoxic Inflammation in Aicardi-Goutières Syndrome Astrocytes. J. Exp. Med. 2022, 219, e20211121. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.; Rapp, A.; Berndt, N.; Staroske, W.; Schuster, M.; Dobrick-Mattheuer, M.; Kretschmer, S.; König, N.; Kurth, T.; Wieczorek, D.; et al. RPA and Rad51 Constitute a Cell Intrinsic Mechanism to Protect the Cytosol from Self DNA. Nat. Commun. 2016, 7, 11752. [Google Scholar] [CrossRef] [PubMed]
- Abt, E.R.; Le, T.M.; Dann, A.M.; Capri, J.R.; Poddar, S.; Lok, V.; Li, L.; Liang, K.; Creech, A.L.; Rashid, K.; et al. Reprogramming of Nucleotide Metabolism by Interferon Confers Dependence on the Replication Stress Response Pathway in Pancreatic Cancer Cells. Cell Rep. 2022, 38, 110236. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, S.D.; Ando, S.; Holley, J.A.; Sugie, A.; Zhao, F.R.; Poddar, S.; Kato, R.; Miner, C.A.; Nitta, Y.; Krishnamurthy, S.R.; et al. Inherited C-Terminal TREX1 Variants Disrupt Homology-Directed Repair to Cause Senescence and DNA Damage Phenotypes in Drosophila, Mice, and Humans. Nat. Commun. 2024, 15, 4696. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.; Maagdenberg, A.M.J.M.v.D.; Jen, J.C.; Kavanagh, D.; Bertram, P.; Spitzer, D.; Liszewski, M.K.; Barilla-Labarca, M.L.; Terwindt, G.M.; Kasai, Y.; et al. C-Terminal Truncations in Human 3′-5′ DNA Exonuclease TREX1 Cause Autosomal Dominant Retinal Vasculopathy with Cerebral Leukodystrophy. Nat. Genet. 2007, 39, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Nader, G.P.d.F.; Agüera-Gonzalez, S.; Routet, F.; Gratia, M.; Maurin, M.; Cancila, V.; Cadart, C.; Palamidessi, A.; Ramos, R.N.; San Roman, M.; et al. Compromised Nuclear Envelope Integrity Drives TREX1-Dependent DNA Damage and Tumor Cell Invasion. Cell 2021, 184, 5230–5246.e22. [Google Scholar] [CrossRef]
- Sharma, P.; Goswami, S.; Raychaudhuri, D.; Siddiqui, B.A.; Singh, P.; Nagarajan, A.; Liu, J.; Subudhi, S.K.; Poon, C.; Gant, K.L.; et al. Immune Checkpoint Therapy—Current Perspectives and Future Directions. Cell 2023, 186, 1652–1669. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Dissecting the Mechanisms of Immune Checkpoint Therapy. Nat. Rev. Immunol. 2020, 20, 75–76. [Google Scholar] [CrossRef]
- Chamoto, K.; Yaguchi, T.; Tajima, M.; Honjo, T. Insights from a 30-Year Journey: Function, Regulation and Therapeutic Modulation of PD1. Nat. Rev. Immunol. 2023, 23, 682–695. [Google Scholar] [CrossRef]
- Pang, K.; Shi, Z.D.; Wei, L.Y.; Dong, Y.; Ma, Y.Y.; Wang, W.; Wang, G.Y.; Cao, M.Y.; Dong, J.J.; Chen, Y.A.; et al. Research Progress of Therapeutic Effects and Drug Resistance of Immunotherapy Based on PD-1/PD-L1 Blockade. Drug Resist. Updates 2023, 66, 100907. [Google Scholar] [CrossRef]
- De Martino, M.; Vanpouille-Box, C.; Galluzzi, L. Immunological Barriers to Immunotherapy in Primary and Metastatic Breast Cancer. EMBO Mol. Med. 2021, 13, e14393. [Google Scholar] [CrossRef]
- de Miguel, M.; Calvo, E. Clinical Challenges of Immune Checkpoint Inhibitors. Cancer Cell 2020, 38, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Hong, Z.; Zhang, C.; Wang, L.; Han, Z.; Ma, D. Immune Checkpoint Therapy for Solid Tumours: Clinical Dilemmas and Future Trends. Signal Transduct. Target. Ther. 2023, 8, 320. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I Interferons in Anticancer Immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Kitajima, S.; Tani, T.; Springer, B.F.; Campisi, M.; Osaki, T.; Haratani, K.; Chen, M.; Knelson, E.H.; Mahadevan, N.R.; Ritter, J.; et al. MPS1 Inhibition Primes Immunogenicity of KRAS-LKB1 Mutant Lung Cancer. Cancer Cell 2022, 40, 1128–1144.e8. [Google Scholar] [CrossRef]
- Pan, B.S.; Perera, S.A.; Piesvaux, J.A.; Presland, J.P.; Schroeder, G.K.; Cumming, J.N.; Wesley Trotter, B.; Altman, M.D.; Buevich, A.V.; Cash, B.; et al. An Orally Available Non-Nucleotide STING Agonist with Antitumor Activity. Science 2020, 369, eaba6098. [Google Scholar] [CrossRef]
- Chin, E.N.; Yu, C.; Vartabedian, V.F.; Jia, Y.; Kumar, M.; Gamo, A.M.; Vernier, W.; Ali, S.H.; Kissai, M.; Lazar, D.C.; et al. Antitumor Activity of a Systemic STING-Activating Non-Nucleotide CGAMP Mimetic. Science 2020, 369, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA Exonuclease Trex1 Regulates Radiotherapy-Induced Tumour Immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.P.; Chapman, J.R.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory DsDNA from Irradiated Cancer Cells to DCs. Cancer Immunol. Res. 2018, 6, 910–920. [Google Scholar] [CrossRef]
- Hemphill, W.O.; Simpson, S.R.; Liu, M.; Salsbury, F.R.; Hollis, T.; Grayson, J.M.; Perrino, F.W. TREX1 as a Novel Immunotherapeutic Target. Front. Immunol. 2021, 12, 660184. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Lira, M.C.; Vanpouille-Box, C.; Galluzzi, L. Adaptive Inhibition of CGAS Signaling by TREX1. Trends Cancer 2024, 10, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Won, J.K.; Bakhoum, S.F. The Cytosolic DNA-Sensing CGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Mohr, L.; Toufektchan, E.; von Morgen, P.; Chu, K.; Kapoor, A.; Maciejowski, J. ER-Directed TREX1 Limits CGAS Activation at Micronuclei. Mol. Cell 2021, 81, 724–738.e9. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.; Partch, C.L. Substituted Pyrrolo [1,2-a]Pyrazines and Pyrrolo [1,2-a][1,4]Diazepines as TREX1 Inhibitors. U.S. Patent 11,583,538, 21 February 2023. [CrossRef]
- Letourneau, J.J.; Elamparuthi, K.S.; Huang, C.-Y.; Venkata, V.B. NOVEL CYCLIC TREX1 INHIBITORS 2020.
- Flowers, S.; Petronella, B.A.; McQueney, M.S.; Fanelli, B.; Eisenberg, W.; Uveges, A.; Roden, A.L.; Salowe, S.; Bommireddy, V.; Letourneau, J.J.; et al. A Novel TREX1 Inhibitor, VB-85680, Upregulates Cellular Interferon Responses. PLoS ONE 2024, 19, e0305962. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Yang, X.; Wang, P.; He, H.; Chen, Y.; Yu, L.; Fang, H.; Wang, F.; Huang, Z. Systematic Pan-Cancer Analysis Identifies CGAS as an Immunological and Prognostic Biomarker. Ann. Transl. Med. 2023, 11, 121. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Q.; Liu, C.; Hu, Y.; Liu, Y.; Lyu, Y.; Liao, Y.; Chen, L.; Yang, H.; Mao, Y. Identification of RP11-770J1.4 as Immune-related LncRNA Regulating the CTXN1–CGAS–STING Axis in Histologically Lower-grade Glioma. MedComm 2023, 4, e458. [Google Scholar] [CrossRef]
- Epstein, R.S.; Roy, U.K.B.; Aapro, M.; Salimi, T.; Moran, D.; Krenitsky, J.; Leone-Perkins, M.L.; Girman, C.; Schlusser, C.; Crawford, J. Cancer Patients’ Perspectives and Experiences of Chemotherapy-Induced Myelosuppression and Its Impact on Daily Life. Patient Prefer. Adherence 2021, 15, 453–465. [Google Scholar] [CrossRef]
- Epstein, R.S.; Aapro, M.S.; Basu Roy, U.K.; Salimi, T.; Krenitsky, J.A.; Leone-Perkins, M.L.; Girman, C.; Schlusser, C.; Crawford, J. Patient Burden and Real-World Management of Chemotherapy-Induced Myelosuppression: Results from an Online Survey of Patients with Solid Tumors. Adv. Ther. 2020, 37, 3606–3618. [Google Scholar] [CrossRef]
- Murayama, T.; Mahadevan, N.R.; Meador, C.B.; Ivanova, E.V.; Pan, Y.; Knelson, E.H.; Tani, T.; Nakayama, J.; Ma, X.; Thai, T.C.; et al. Targeting TREX1 Induces Innate Immune Response in Drug-Resistant Small-Cell Lung Cancer. Cancer Res. Commun. 2024, 4, 2399–2414. [Google Scholar] [CrossRef]
- Raso, M.C.; Djoric, N.; Walser, F.; Hess, S.; Schmid, F.M.; Burger, S.; Knobeloch, K.P.; Penengo, L. Interferon-Stimulated Gene 15 Accelerates Replication Fork Progression Inducing Chromosomal Breakage. J. Cell Biol. 2020, 219, e202002175. [Google Scholar] [CrossRef] [PubMed]
- Kidiyoor, G.R.; Li, Q.; Bastianello, G.; Bruhn, C.; Giovannetti, I.; Mohamood, A.; Beznoussenko, G.V.; Mironov, A.; Raab, M.; Piel, M.; et al. ATR Is Essential for Preservation of Cell Mechanics and Nuclear Integrity during Interstitial Migration. Nat. Commun. 2020, 11, 4828. [Google Scholar] [CrossRef] [PubMed]
- Sladitschek-Martens, H.L.; Guarnieri, A.; Brumana, G.; Zanconato, F.; Battilana, G.; Xiccato, R.L.; Panciera, T.; Forcato, M.; Bicciato, S.; Guzzardo, V.; et al. YAP/TAZ Activity in Stromal Cells Prevents Ageing by Controlling CGAS–STING. Nature 2022, 607, 790–798. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hawillo, K.; Kemiha, S.; Técher, H. Cancer Immunotherapy: Targeting TREX1 Has the Potential to Unleash the Host Immunity against Cancer Cells. Onco 2024, 4, 322-334. https://doi.org/10.3390/onco4040022
Hawillo K, Kemiha S, Técher H. Cancer Immunotherapy: Targeting TREX1 Has the Potential to Unleash the Host Immunity against Cancer Cells. Onco. 2024; 4(4):322-334. https://doi.org/10.3390/onco4040022
Chicago/Turabian StyleHawillo, Karim, Samira Kemiha, and Hervé Técher. 2024. "Cancer Immunotherapy: Targeting TREX1 Has the Potential to Unleash the Host Immunity against Cancer Cells" Onco 4, no. 4: 322-334. https://doi.org/10.3390/onco4040022
APA StyleHawillo, K., Kemiha, S., & Técher, H. (2024). Cancer Immunotherapy: Targeting TREX1 Has the Potential to Unleash the Host Immunity against Cancer Cells. Onco, 4(4), 322-334. https://doi.org/10.3390/onco4040022