
Qualification of Human Liver Microsomes for Antibacterial Activity Screening of Drug Metabolites


Abstract
:1. Introduction
2. Materials and Methods
2.1. General
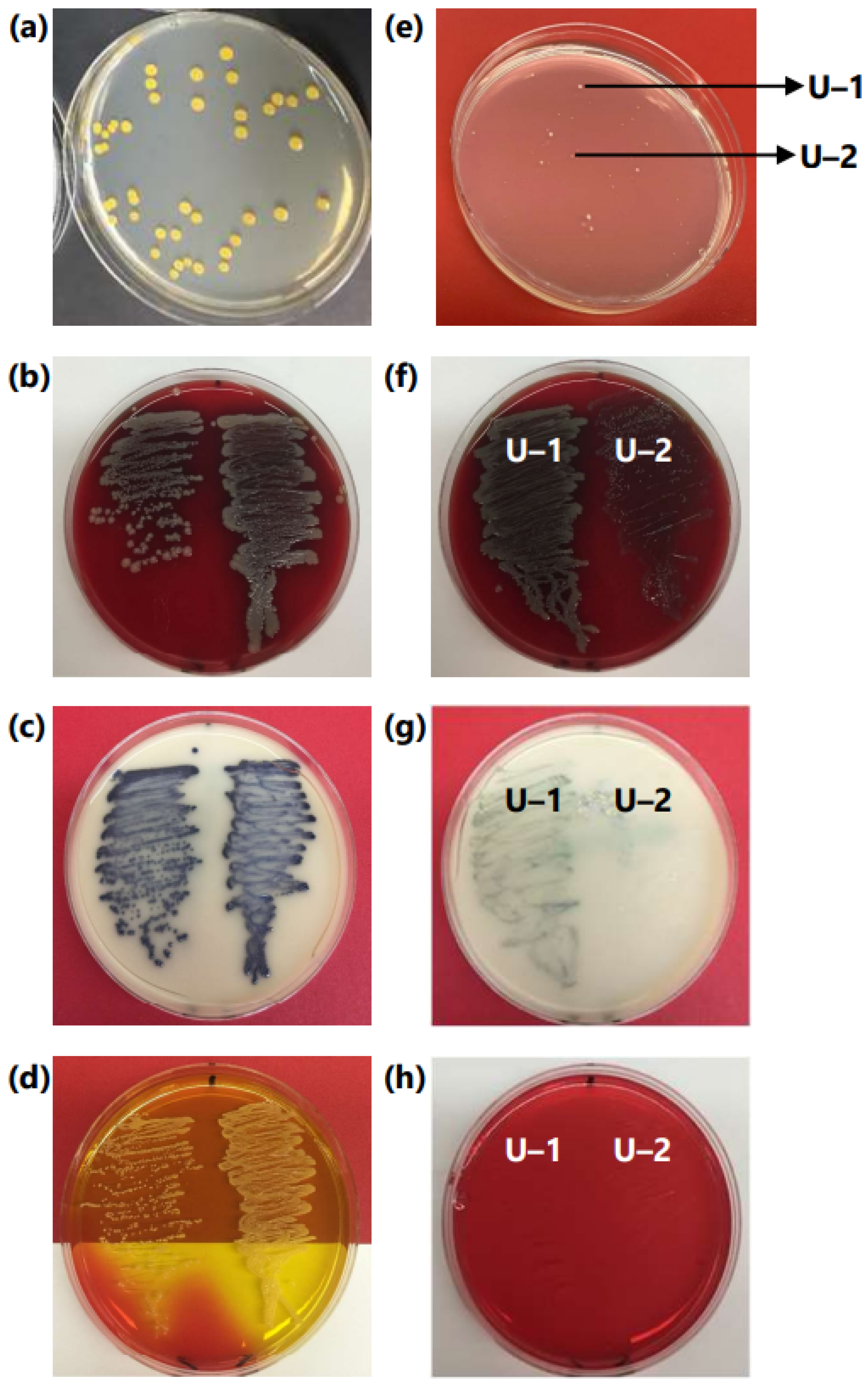
2.2. Initial Observation and Diagnosis of Microbial Content
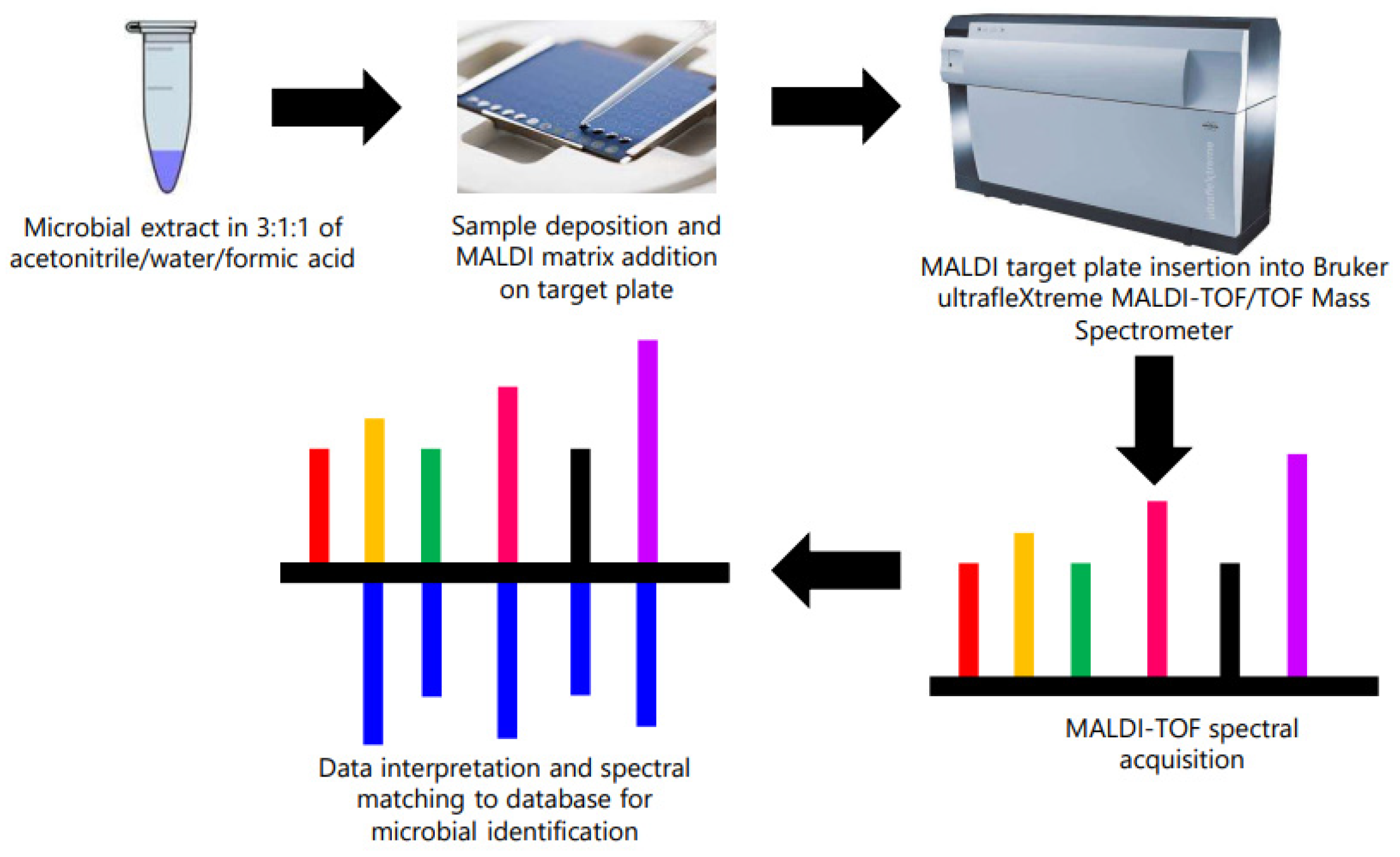
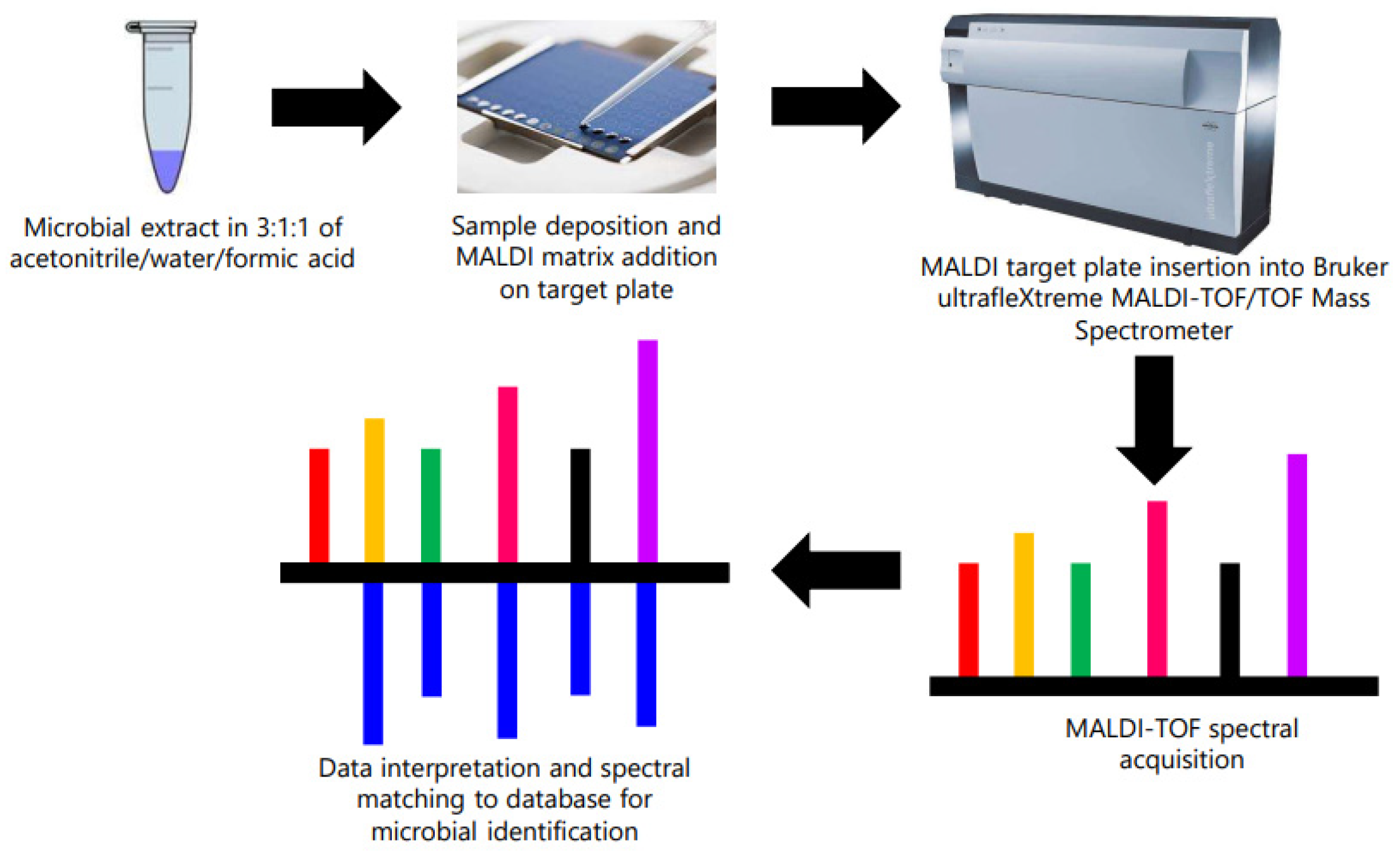
2.3. Identification of Contaminant Bacteria from Microsome Using MALDI-TOF/TOF
2.4. Approaches to Decontaminate Microsomes
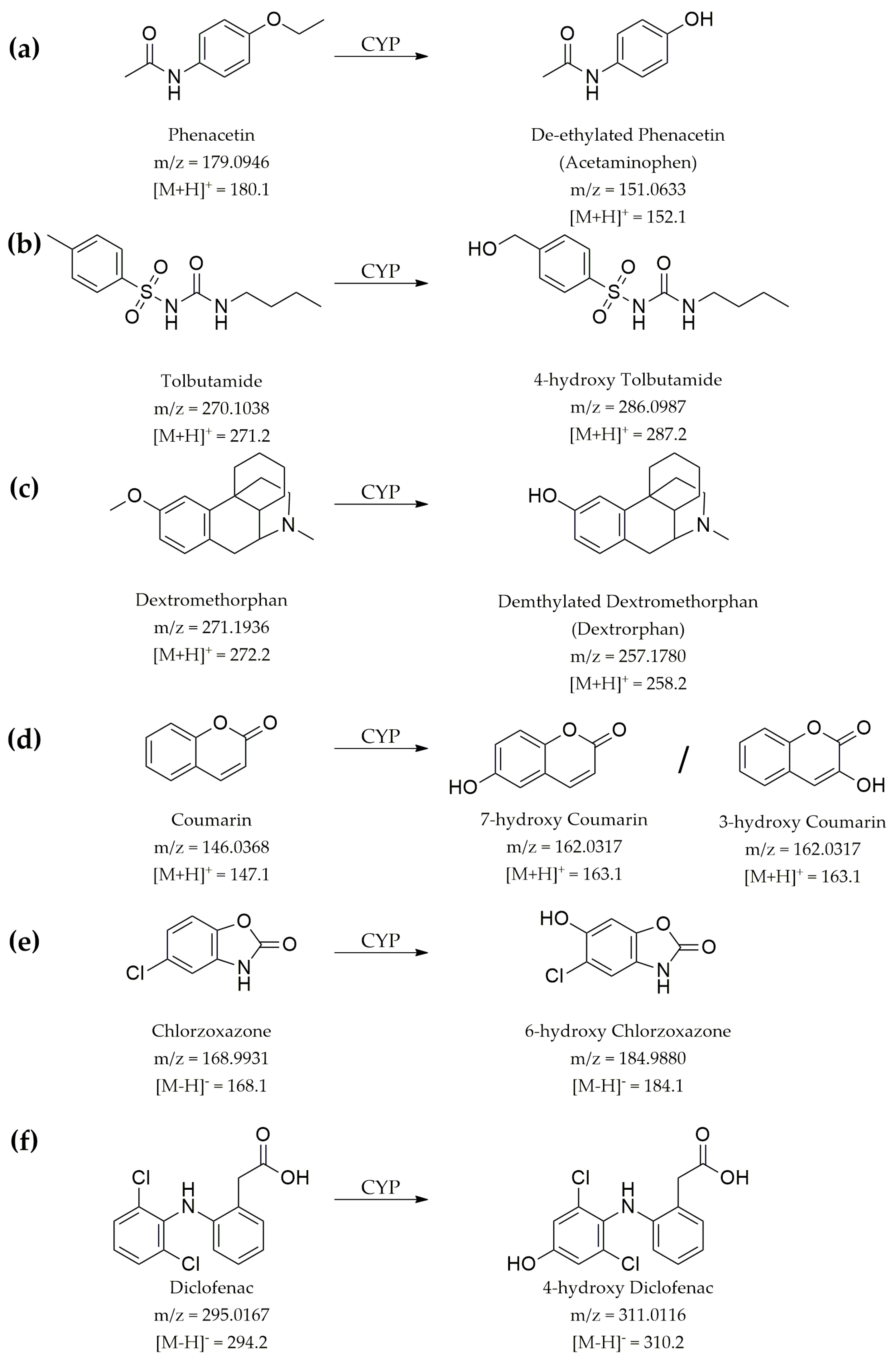
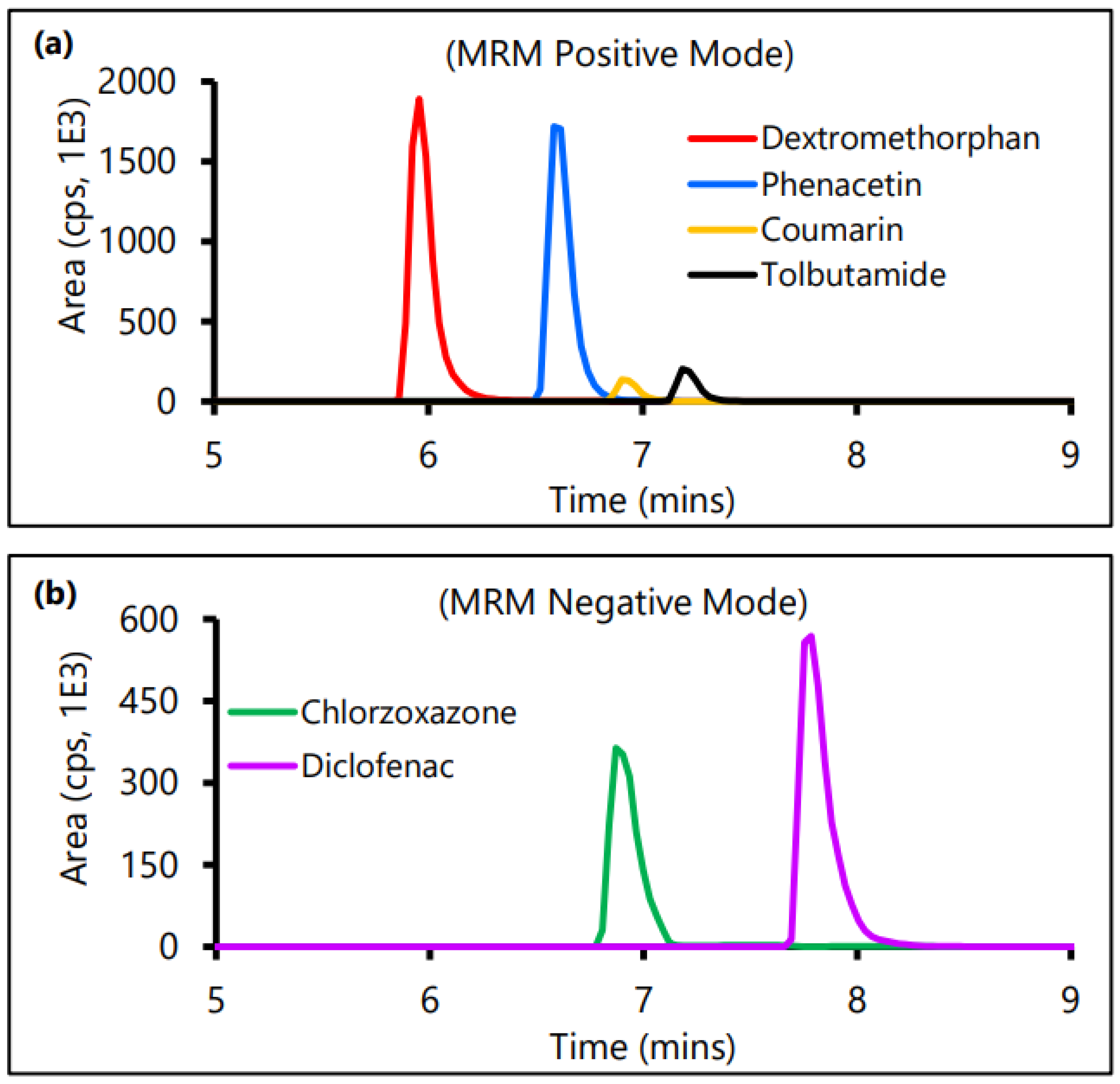
2.5. LC-MS/MS-Based Analytical Method Development for Control CYP Substrates
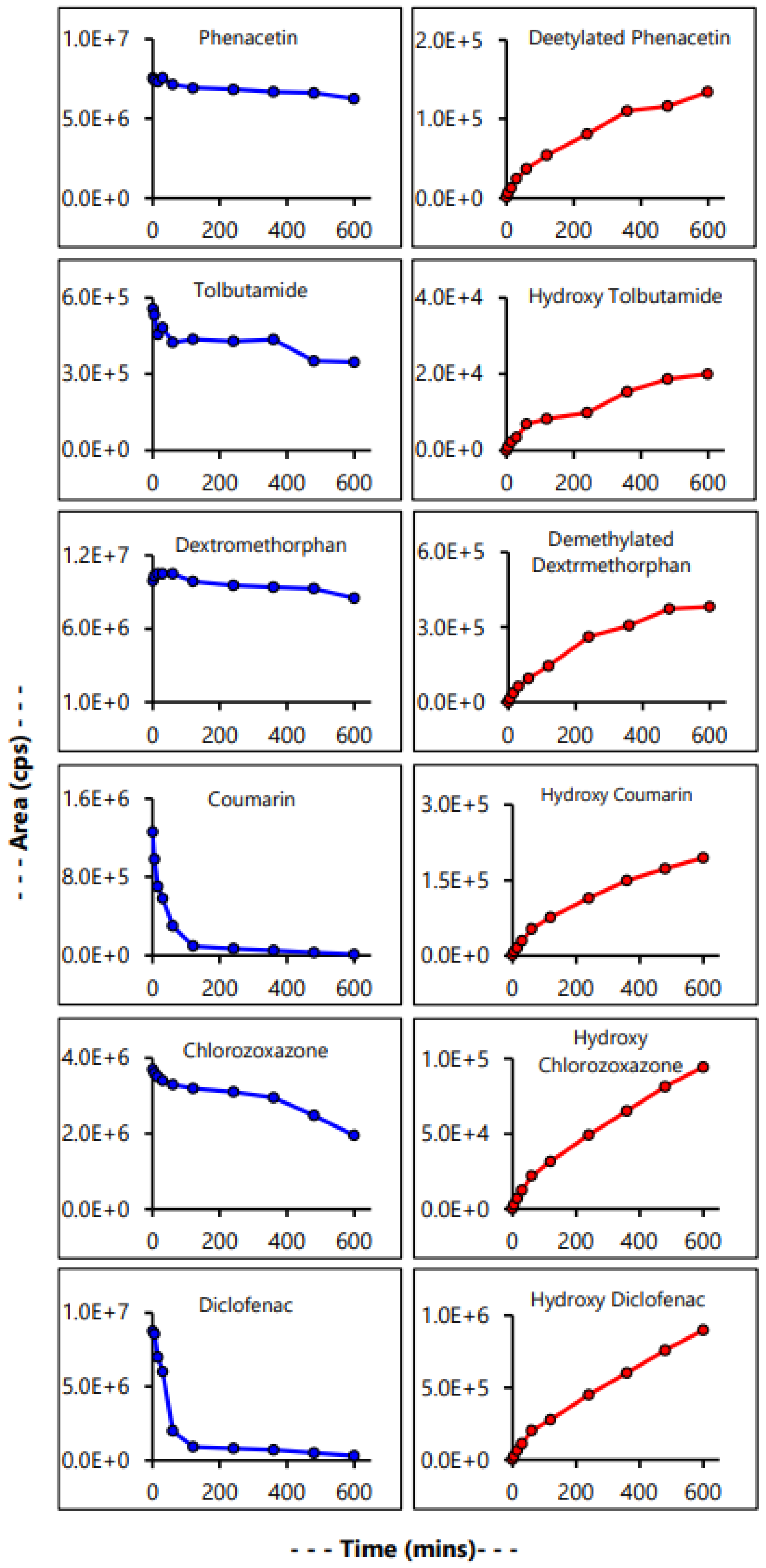
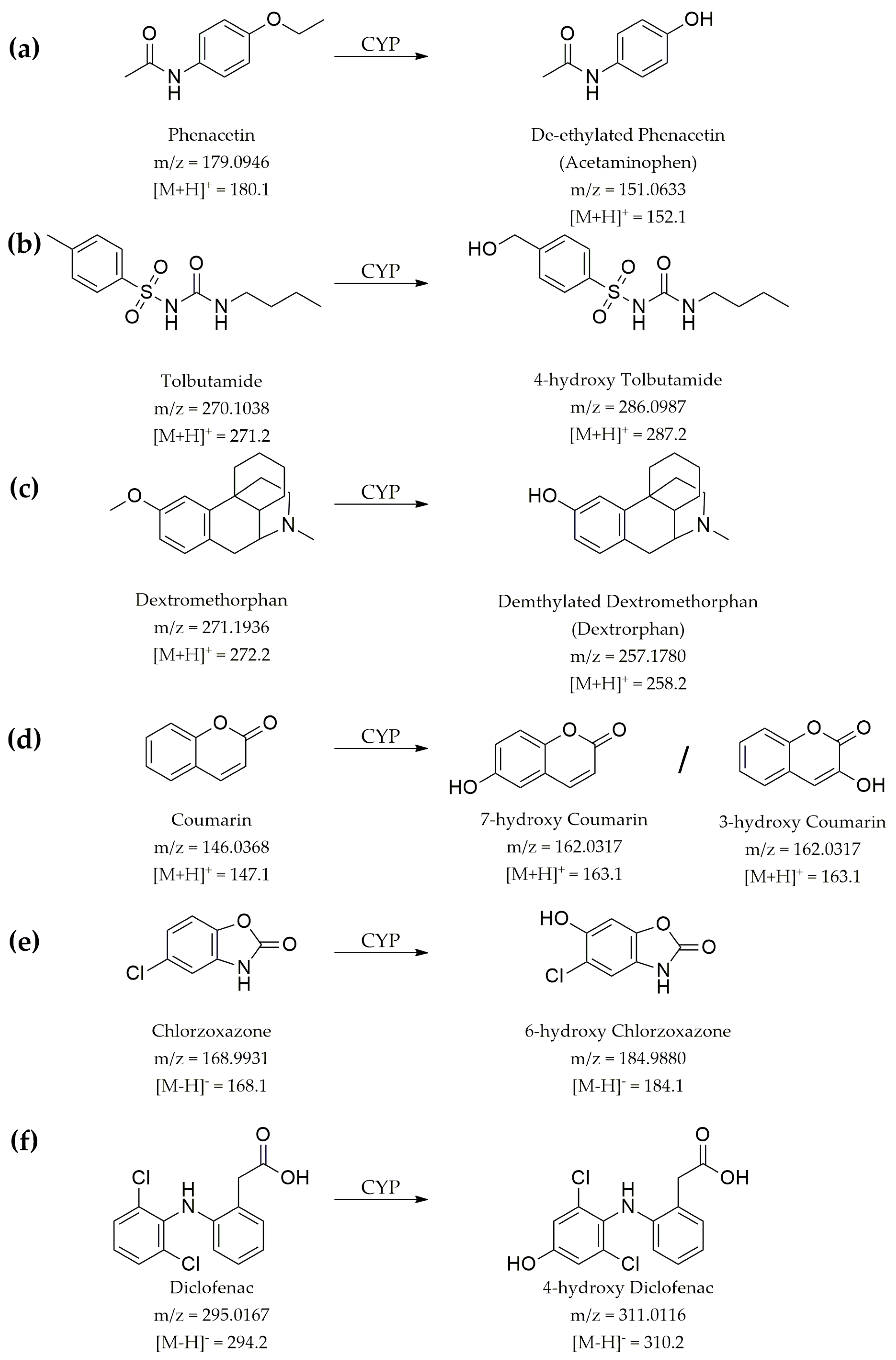
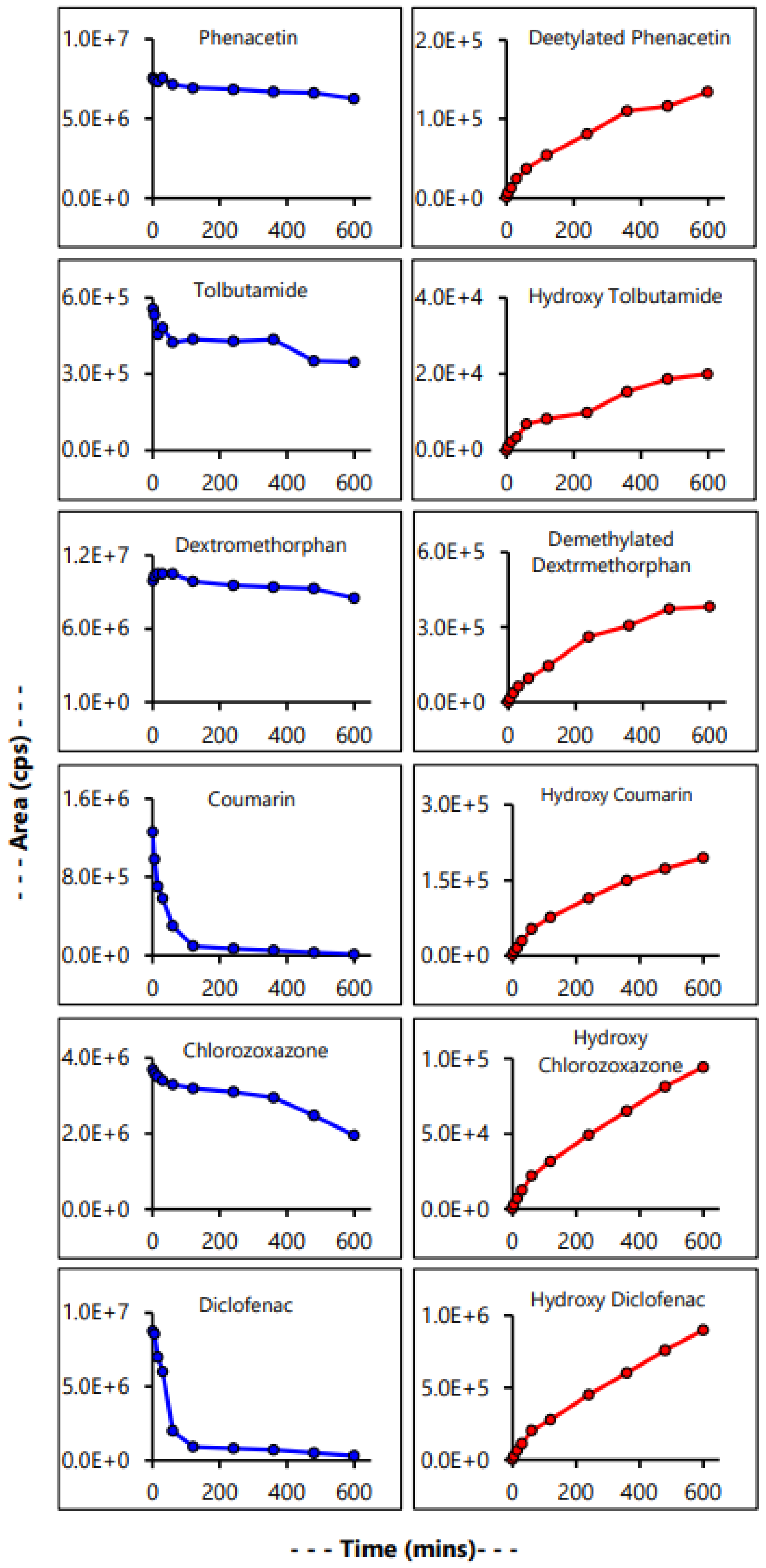
2.6. Time Kinetics Analysis of Drug Metabolism
3. Results and Discussion
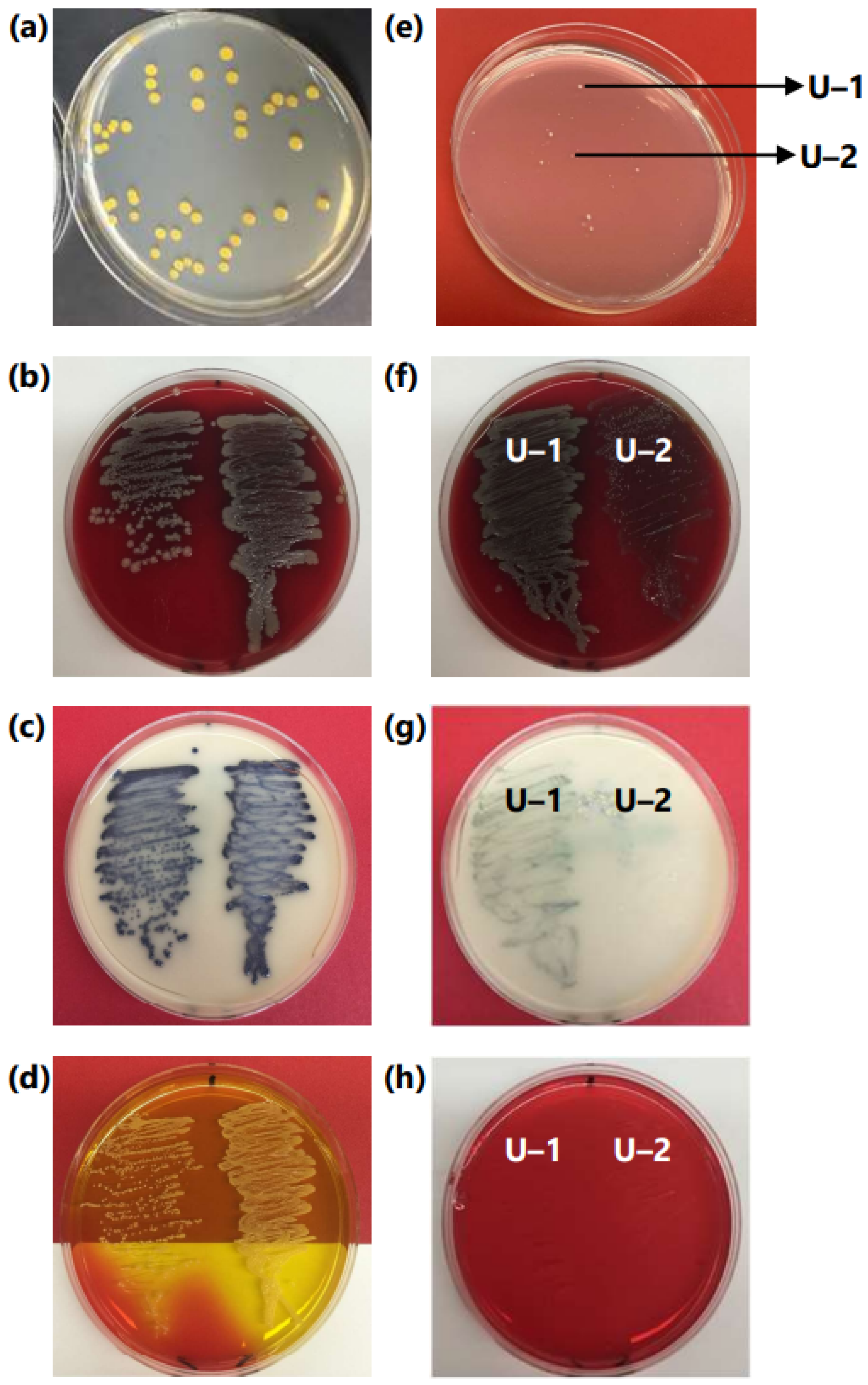
3.1. Initial Observation of Contamination in PM Library Plates
3.2. Assessment of MRSA and Microsome Stock
3.3. MALDI-TOF/TOF Identification of Contaminant Bacteria from Microsomes
3.4. Comparison and Correlation of MALDI-TOF/TOF Data with Biochemical Testing
3.5. Decontamination of Microsome
3.6. Evaluation of Microsomal Drug Metabolism
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stanley, L.A. Chapter 27—Drug Metabolism. In Pharmacognosy; Badal, S., Delgoda, R., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 527–545. [Google Scholar]
- Tillement, J.P.; Tremblay, D. 5.02—Clinical Pharmacokinetic Criteria for Drug Research. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Oxford, UK, 2007; pp. 11–30. [Google Scholar]
- Pinto, N.; Dolan, M.E. Clinically relevant genetic variations in drug metabolizing enzymes. Curr. Drug Metab. 2011, 12, 487–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prescott, L.F.; Forrest, J.A.; Adjepon-Yamoah, K.K.; Finlayson, N.D. Drug metabolism in liver disease. J. Clin. Pathol. Suppl. (R. Coll. Pathol.) 1975, 9, 62–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paintaud, G.; Bechtel, Y.; Brientini, M.P.; Miguet, J.P.; Bechtel, P.R. Effects of liver diseases on drug metabolism. Therapie 1996, 51, 384–389. [Google Scholar] [PubMed]
- Rautio, J.; Meanwell, N.A.; Di, L.; Hageman, M.J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 2018, 17, 559–587. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, H.; Liang, X.; Roberts, M.S. Chapter 30—Hepatic Metabolism in Liver Health and Disease. In Liver Pathophysiology; Muriel, P., Ed.; Academic Press: Boston, MA, USA, 2017; pp. 391–400. [Google Scholar]
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug Metabolism in the Liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef]
- Ilowite, N.T.; Laxer, R.M. Chapter 6—Pharmacology and Drug Therapy. In Textbook of Pediatric Rheumatology, 6th ed.; Cassidy, J.T., Laxer, R.M., Petty, R.E., Lindsley, C.B., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2011; pp. 71–126. [Google Scholar]
- Jia, L.; Liu, X. The conduct of drug metabolism studies considered good practice (II): In vitro experiments. Curr. Drug Metab. 2007, 8, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Lechón, M.J.; Castell, J.V.; Donato, M.T. Hepatocytes—The choice to investigate drug metabolism and toxicity in man: In vitro variability as a reflection of in vivo. Chem. Biol. Interact. 2007, 168, 30–50. [Google Scholar] [CrossRef]
- Gasser, R. Importance of Drug Metabolism in Drug Discovery and Development. In Molecular and Applied Aspects of Oxidative Drug Metabolizing Enzymes; Arinç, E., Schenkman, J.B., Hodgson, E., Eds.; Springer: Boston, MA, USA, 1999; pp. 183–193. [Google Scholar]
- Gibson, G.G.; Skett, P. Pathways of drug metabolism. In Introduction to Drug Metabolism; Gibson, G.G., Skett, P., Eds.; Springer: Boston, MA, USA, 1994; pp. 1–34. [Google Scholar]
- Zhang, Z.; Tang, W. Drug metabolism in drug discovery and development. Acta Pharm. Sin. B 2018, 8, 721–732. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Yoo, J.; Dueñas-González, A. Chapter 13—DNA Methyltransferase Inhibitors for Cancer Therapy. In Epigenetic Technological Applications; Zheng, Y.G., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 265–290. [Google Scholar]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Kumar, A.P.; Lukman, S.; Nguyen, M.N. Drug Repurposing and Multi-Target Therapies. In Encyclopedia of Bioinformatics and Computational Biology; Ranganathan, S., Gribskov, M., Nakai, K., Schönbach, C., Eds.; Academic Press: Oxford, UK, 2019; pp. 780–791. [Google Scholar]
- Roy, A.; Chaguturu, R. Chapter 3—Holistic Drug Targeting. In Innovative Approaches in Drug Discovery; Patwardhan, B., Chaguturu, R., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 65–88. [Google Scholar]
- Demkow, U. Chapter 11—Next Generation Sequencing in Pharmacogenomics. In Clinical Applications for Next-Generation Sequencing; Demkow, U., Płoski, R., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 217–240. [Google Scholar]
- Osakwe, O.; Rizvi, S.A.A. Introduction. In Social Aspects of Drug Discovery, Development and Commercialization; Osakwe, O., Rizvi, S.A.A., Eds.; Academic Press: Boston, MA, USA, 2016; pp. xvii–xxx. [Google Scholar]
- Ayon, N.J.; Gutheil, W.G. Dimensionally Enhanced Antibacterial Library Screening. ACS Chem. Biol. 2019, 14, 2887–2894. [Google Scholar] [CrossRef]
- Ayon, N.J.; Gutheil, W.G. Correction to “Dimensionally Enhanced Antibacterial Library Screening”. ACS Chem. Biol. 2021, 16, 1610–1611. [Google Scholar] [CrossRef]
- Knights, K.M.; Stresser, D.M.; Miners, J.O.; Crespi, C.L. In Vitro Drug Metabolism Using Liver Microsomes. Curr. Protoc. Pharmacol. 2016, 74, 7.8.1–7.8.24. [Google Scholar] [CrossRef] [PubMed]
- Plongla, R.; Miller, M.B. Chapter 12—Molecular Testing for Diseases Associated with Bacterial Infections. In Diagnostic Molecular Pathology; Coleman, W.B., Tsongalis, G.J., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 139–150. [Google Scholar]
- Patel, R. MALDI-TOF MS for the Diagnosis of Infectious Diseases. Clin. Chem. 2015, 61, 100–111. [Google Scholar] [CrossRef] [Green Version]
- McElvania Tekippe, E.; Shuey, S.; Winkler, D.W.; Butler, M.A.; Burnham, C.A. Optimizing identification of clinically relevant Gram-positive organisms by use of the Bruker Biotyper matrix-assisted laser desorption ionization-time of flight mass spectrometry system. J. Clin. Microbiol. 2013, 51, 1421–1427. [Google Scholar] [CrossRef] [Green Version]
- Westblade, L.F.; Jennemann, R.; Branda, J.A.; Bythrow, M.; Ferraro, M.J.; Garner, O.B.; Ginocchio, C.C.; Lewinski, M.A.; Manji, R.; Mochon, A.B.; et al. Multicenter study evaluating the Vitek MS system for identification of medically important yeasts. J. Clin. Microbiol. 2013, 51, 2267–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF mass spectrometry: An emerging technology for microbial identification and diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, S.; Weinert, K.; Wagner, C.; Gunzl, B.; Wieser, A.; Maier, T.; Kostrzewa, M. Novel, improved sample preparation for rapid, direct identification from positive blood cultures using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. J. Mol. Diagn. 2011, 13, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Ayon, N.J. Metabolomics and Chemical Library Screening for Antibacterial Drug Discovery. Ph.D. Thesis, University of Missouri, Kansas City, MA, USA, 2020. [Google Scholar]
- Smith, A.C.; Hussey, M.A. Gram Stain Protocol. Am. Soc. Microbiol. 2005, 1, 1–9. [Google Scholar]
- McClelland, R. Gram’s stain: The key to microbiology. MLO Med. Lab Obs. 2001, 33, 20–22, 25–28, quiz 30-21. [Google Scholar]
- Reiner, K. Catalase Test Protocol. Am. Soc. Microbiol. 2010. [Google Scholar]
- Schmitt, B.H.; Cunningham, S.A.; Dailey, A.L.; Gustafson, D.R.; Patel, R. Identification of anaerobic bacteria by Bruker Biotyper matrix-assisted laser desorption ionization-time of flight mass spectrometry with on-plate formic acid preparation. J. Clin. Microbiol. 2013, 51, 782–786. [Google Scholar] [CrossRef] [Green Version]
- Schulthess, B.; Bloemberg, G.V.; Zbinden, R.; Bottger, E.C.; Hombach, M. Evaluation of the Bruker MALDI Biotyper for identification of Gram-positive rods: Development of a diagnostic algorithm for the clinical laboratory. J. Clin. Microbiol. 2014, 52, 1089–1097. [Google Scholar] [CrossRef] [Green Version]
- Schulthess, B.; Bloemberg, G.V.; Zbinden, A.; Mouttet, F.; Zbinden, R.; Böttger, E.C.; Hombach, M. Evaluation of the Bruker MALDI Biotyper for Identification of Fastidious Gram-Negative Rods. J. Clin. Microbiol. 2016, 54, 543. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, P.R.; Lee, T.F.; Du, S.H.; Teng, S.H.; Liao, C.H.; Sheng, W.H.; Teng, L.J. Bruker biotyper matrix-assisted laser desorption ionization-time of flight mass spectrometry system for identification of Nocardia, Rhodococcus, Kocuria, Gordonia, Tsukamurella, and Listeria species. J. Clin. Microbiol. 2014, 52, 2371–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katara, G.; Hemvani, N.; Chitnis, S.; Chitnis, V.; Chitnis, D.S. Surface disinfection by exposure to germicidal UV light. Indian J. Med. Microbiol. 2008, 26, 241–242. [Google Scholar] [CrossRef] [PubMed]
- Fellows, P.J. 11—Pasteurisation. In Food Processing Technology, 4th ed.; Fellows, P.J., Ed.; Woodhead Publishing: Soston, UK, 2017; pp. 563–580. [Google Scholar]
- Chapman, G.H. The Significance of Sodium Chloride in Studies of Staphylococci. J. Bacteriol. 1945, 50, 201–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.; Hedin, G. Use of Cefoxitin-Based Selective Broth for Improved Detection of Methicillin-Resistant Staphylococcus aureus. J. Clin. Microbiol. 2006, 44, 592. [Google Scholar] [CrossRef] [Green Version]
- Hermsen, E.D.; Sullivan, C.J.; Rotschafer, J.C. Polymyxins: Pharmacology, pharmacokinetics, pharmacodynamics, and clinical applications. Infect. Dis. Clin. N. Am. 2003, 17, 545–562. [Google Scholar] [CrossRef]
- Sud, I.J.; Feingold, D.S. Action of antifungal imidazoles on Staphylococcus aureus. Antimicrob. Agents Chemother. 1982, 22, 470–474. [Google Scholar] [CrossRef] [Green Version]
- Ayon, N.J.; Sharma, A.D.; Gutheil, W.G. LC-MS/MS-Based Separation and Quantification of Marfey’s Reagent Derivatized Proteinogenic Amino Acid DL-Stereoisomers. J. Am. Soc. Mass. Spectrom. 2019, 30, 448–458. [Google Scholar] [CrossRef]
- Walsky, R.L.; Obach, R.S. Validated assays for human cytochrome P450 activities. Drug Metab. Dispos. 2004, 32, 647–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Roy, A. Early Probe and Drugg Discovery in Academia: A Minireview. High-Throughput 2018, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Buxton, R. Blood Agar Plates and Hemolysis Protocols. Am. Soc. Microbiol. 2005. [Google Scholar]
- Peterson, J.F.; Riebe, K.M.; Hall, G.S.; Wilson, D.; Whittier, S.; Palavecino, E.; Ledeboer, N.A. Spectra MRSA, a new chromogenic agar medium to screen for methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 2010, 48, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Sharp, S.E.; Searcy, C. Comparison of mannitol salt agar and blood agar plates for identification and susceptibility testing of Staphylococcus aureus in specimens from cystic fibrosis patients. J. Clin. Microbiol. 2006, 44, 4545–4546. [Google Scholar] [CrossRef] [Green Version]
- Bartholomew, J.W.; Mittwer, T. The Gram stain. Bacteriol. Rev. 1952, 16, 1–29. [Google Scholar] [CrossRef]
- Denton, M.; Kerr, K.G. Microbiological and clinical aspects of infection associated with Stenotrophomonas maltophilia. Clin. Microbiol. Rev. 1998, 11, 57–80. [Google Scholar] [CrossRef] [Green Version]
- Crossman, L.C.; Gould, V.C.; Dow, J.M.; Vernikos, G.S.; Okazaki, A.; Sebaihia, M.; Saunders, D.; Arrowsmith, C.; Carver, T.; Peters, N.; et al. The complete genome, comparative and functional analysis of Stenotrophomonas maltophilia reveals an organism heavily shielded by drug resistance determinants. Genome Biol. 2008, 9, R74. [Google Scholar] [CrossRef] [Green Version]
- Mahdi, O.; Eklund, B.; Fisher, N. Laboratory culture and maintenance of Stenotrophomonas maltophilia. Curr. Protoc. Microbiol. 2014, 32, Unit-6F.1. [Google Scholar] [CrossRef] [Green Version]
- Hsueh, P.R.; Teng, L.J.; Ho, S.W.; Hsieh, W.C.; Luh, K.T. Clinical and microbiological characteristics of Flavobacterium indologenes infections associated with indwelling devices. J. Clin. Microbiol. 1996, 34, 1908–1913. [Google Scholar] [CrossRef] [Green Version]
- Mukerji, R.; Kakarala, R.; Smith, S.J.; Kusz, H.G. Chryseobacterium indologenes: An emerging infection in the USA. BMJ Case Rep. 2016, 2016, bcr2016214486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, G.; Muthusamy, S.; Raveendran, V.; Joseph, N.M.; Easow, J.M. Unforeseeable presentation of Chryseobacterium indologenes infection in a paediatric patient. BMC Res. Notes 2016, 9, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, G.; Roskot, N.; Smalla, K. Genotypic and phenotypic relationships between clinical and environmental isolates of Stenotrophomonas maltophilia. J. Clin. Microbiol. 1999, 37, 3594–3600. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-T.; Lin, C.-Y.; Chen, Y.-H.; Hsueh, P.-R. Update on infections caused by Stenotrophomonas maltophilia with particular attention to resistance mechanisms and therapeutic options. Front. Microbiol. 2015, 6, 893. [Google Scholar] [CrossRef] [PubMed]
- Pompilio, A.; Piccolomini, R.; Picciani, C.; D’Antonio, D.; Savini, V.; Di Bonaventura, G. Factors associated with adherence to and biofilm formation on polystyrene by Stenotrophomonas maltophilia: The role of cell surface hydrophobicity and motility. FEMS Microbiol. Lett. 2008, 287, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Christakis, G.B.; Perlorentzou, S.P.; Chalkiopoulou, I.; Athanasiou, A.; Legakis, N.J. Chryseobacterium indologenes Non-Catheter-Related Bacteremia in a Patient with a Solid Tumor. J. Clin. Microbiol. 2005, 43, 2021. [Google Scholar] [CrossRef] [Green Version]
- Schoch, P.E.; Cunha, B.A. Pseudomonas maltophilia. Infect. Control. 1987, 8, 169–173. [Google Scholar] [CrossRef]
- Kirby, J.T.; Sader, H.S.; Walsh, T.R.; Jones, R.N. Antimicrobial susceptibility and epidemiology of a worldwide collection of Chryseobacterium spp.: Report from the SENTRY Antimicrobial Surveillance Program (1997–2001). J. Clin. Microbiol. 2004, 42, 445–448. [Google Scholar] [CrossRef] [Green Version]
- Ansel, H.C.; Norred, W.P.; Roth, I.L. Antimicrobial activity of dimethyl sulfoxide against Escherichia coli, Pseudomonas aeruginosa, and Bacillus megaterium. J. Pharm. Sci. 1969, 58, 836–839. [Google Scholar] [CrossRef]
- Fedorka-Cray, P.J.; Cray, W.C., Jr.; Anderson, G.A.; Nickerson, K.W. Bacterial tolerance of 100% dimethyl sulfoxide. Can. J. Microbiol. 1988, 34, 688–689. [Google Scholar] [CrossRef] [PubMed]
- Dyrda, G.; Boniewska-Bernacka, E.; Man, D.; Barchiewicz, K.; Słota, R. The effect of organic solvents on selected microorganisms and model liposome membrane. Mol. Biol. Rep. 2019, 46, 3225–3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Hochlowski, J.; Tang, H.; Hepp, D.; Beckner, C.; Kantor, S.; Schmitt, R. Studies on Repository Compound Stability in DMSO under Various Conditions. J. Biomol. Screen. 2003, 8, 292–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauret, N.; Gauthier, A.; Nicoll-Griffith, D.A. Effect of common organic solvents on in vitro cytochrome P450-mediated metabolic activities in human liver microsomes. Drug Metab. Dispos. 1998, 26, 1–4. [Google Scholar] [PubMed]
- Nirogi, R.; Kandikere, V.; Bhyrapuneni, G.; Ponnamaneni, R.K.; Palacharla, R.; Manoharan, A. Effect of dimethyl sulfoxide on in vitro cytochrome P4501A2 mediated phenacetin O-deethylation in human liver microsomes. Drug Metab. Dispos. 2011, 39, 2162–2164. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control CYP Substrate | Q1 | Q3 | DP (V) | EP (V) | CEP (V) | CE (V) | Retention Time (min) |
|---|---|---|---|---|---|---|---|
| Phenacetin | 180.1 | 110.1 | 46 | 9.5 | 12 | 27 | 6.59 |
| Tolbutamide | 271.2 | 172.1 | 41 | 6.5 | 16 | 18 | 7.19 |
| Dextromethorphan | 272.2 | 213.1 | 61 | 7.5 | 16 | 35 | 5.96 |
| Coumarin | 147.1 | 103.1 | 46 | 3.0 | 13 | 23 | 6.90 |
| Chlorzoxazone | 168.1 | 132.1 | −50 | −11.5 | −10 | −28 | 6.87 |
| Diclofenac | 294.2 | 250.1 | −25 | −7.0 | −14 | −16 | 7.75 |
| Sample | Identified Microorganism | BioTyper ID Score |
|---|---|---|
| U–1 | Stenotrophomonas maltophilia | 2.395 |
| U–2 | Chryseobacterium inodologenes | 2.556 |
| Control (Bacterial Test Standard) | Escherichia coli | 2.303 |
| Control CYP Substrate | % Metabolized | Rate (pmol/mg/min) per µM of Substrate |
|---|---|---|
| Phenacetin | 65.98% | 6.4 |
| Tolbutamide | 51.55% | 4.2 |
| Dextromethorphan | 54.59% | 2.6 |
| Coumarin | 100% | 6.7 |
| Chlorzoxazone | 60.05% | 2.9 |
| Diclofenac | 95.42% | 25.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jubaer, N. Qualification of Human Liver Microsomes for Antibacterial Activity Screening of Drug Metabolites. Appl. Microbiol. 2023, 3, 104-118. https://doi.org/10.3390/applmicrobiol3010009
Jubaer N. Qualification of Human Liver Microsomes for Antibacterial Activity Screening of Drug Metabolites. Applied Microbiology. 2023; 3(1):104-118. https://doi.org/10.3390/applmicrobiol3010009
Chicago/Turabian StyleJubaer, Navid. 2023. "Qualification of Human Liver Microsomes for Antibacterial Activity Screening of Drug Metabolites" Applied Microbiology 3, no. 1: 104-118. https://doi.org/10.3390/applmicrobiol3010009
APA StyleJubaer, N. (2023). Qualification of Human Liver Microsomes for Antibacterial Activity Screening of Drug Metabolites. Applied Microbiology, 3(1), 104-118. https://doi.org/10.3390/applmicrobiol3010009