
A Comparison of the Microbial Populations in a Culture-Dependent and a Culture-Independent Analysis of Industrial Water Samples


Abstract
1. Introduction
2. Materials and Methods
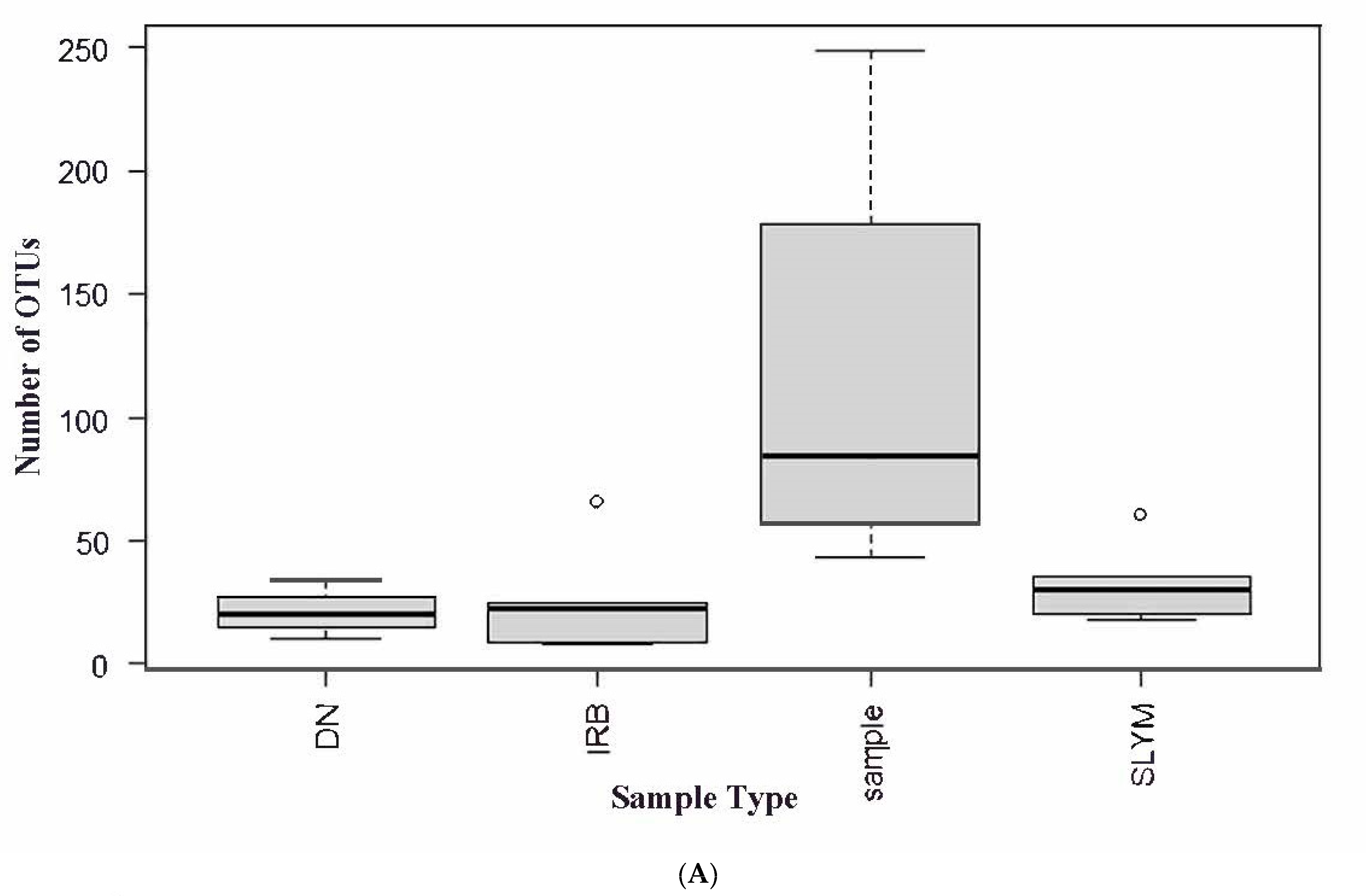
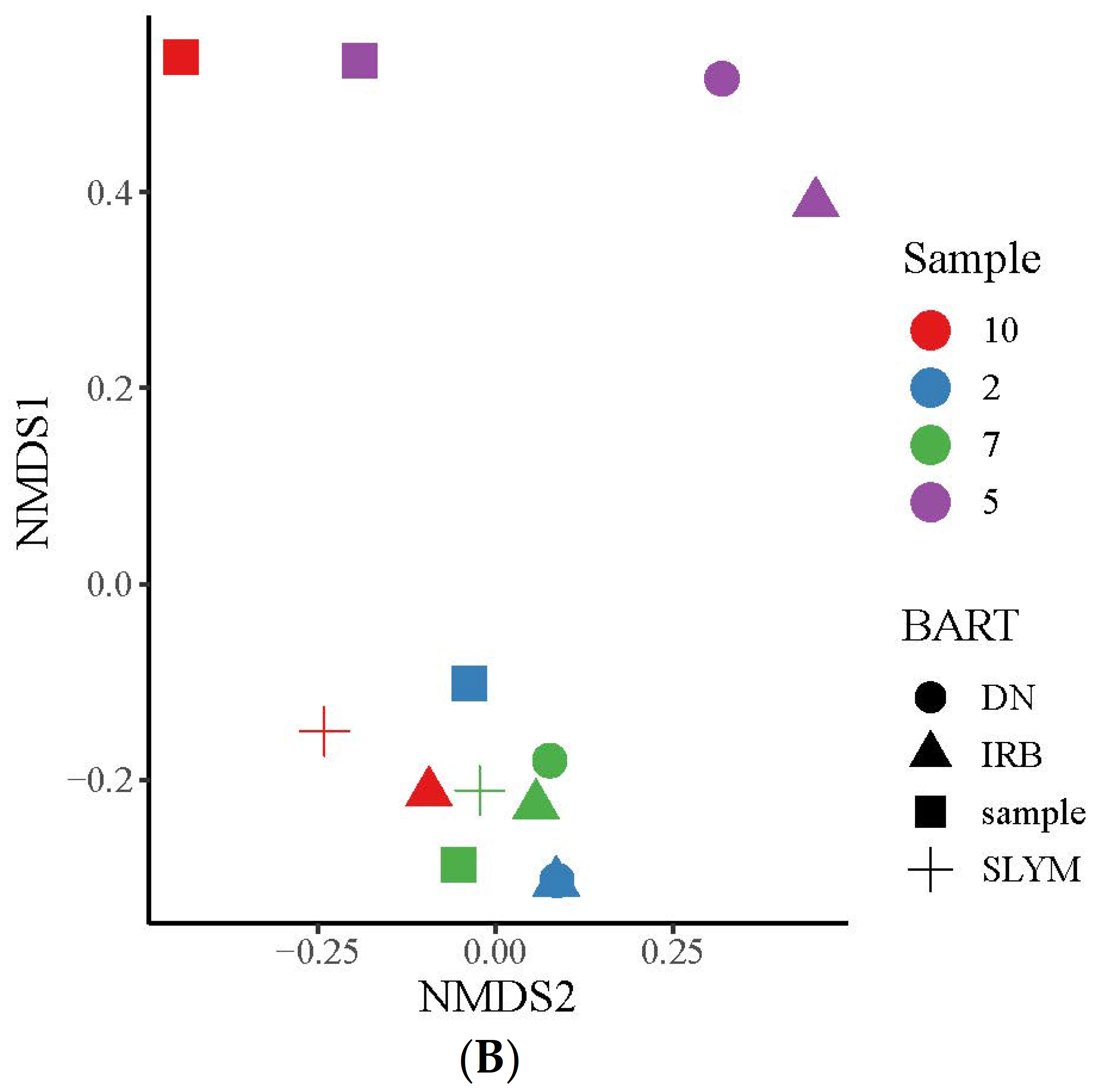
3. Results
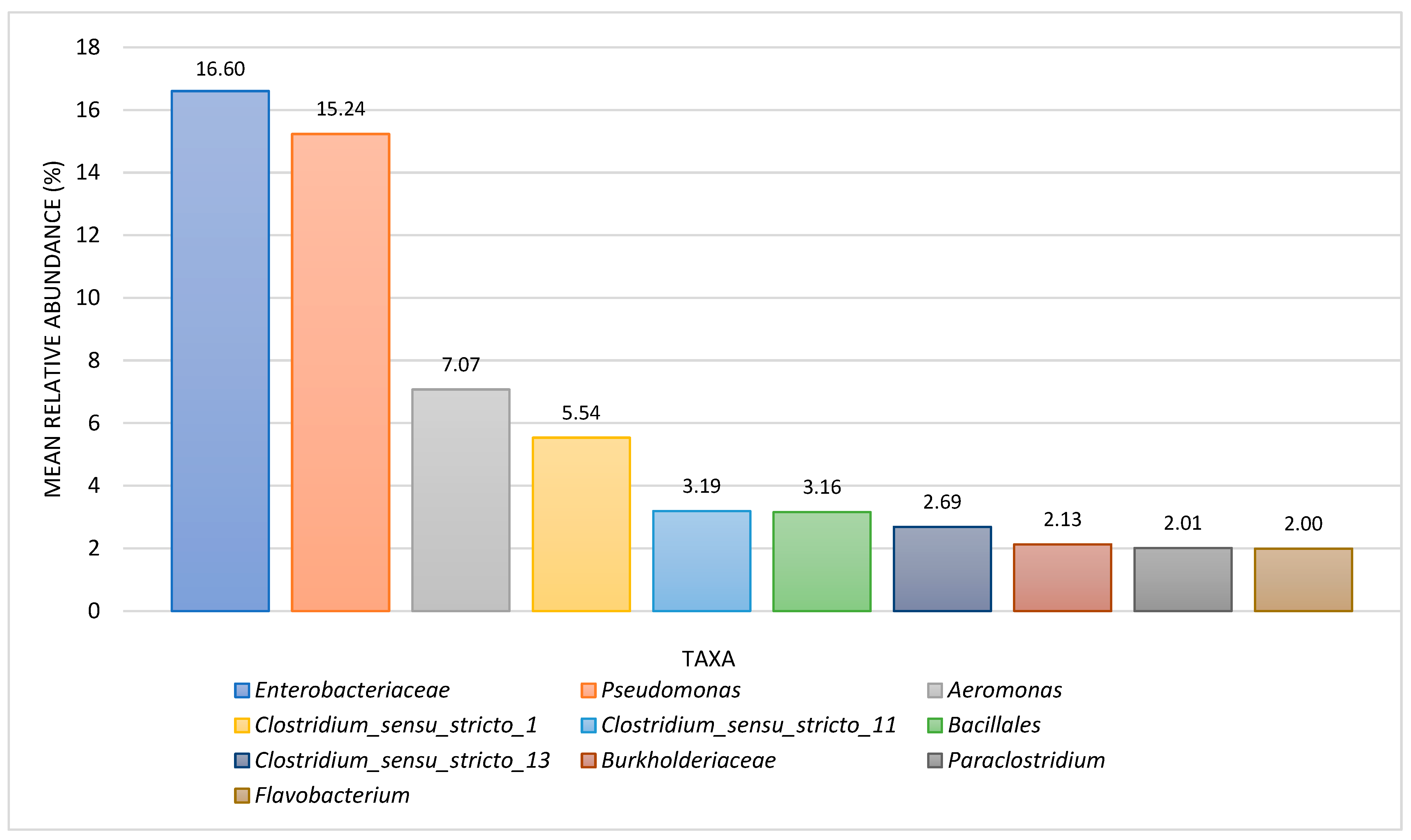
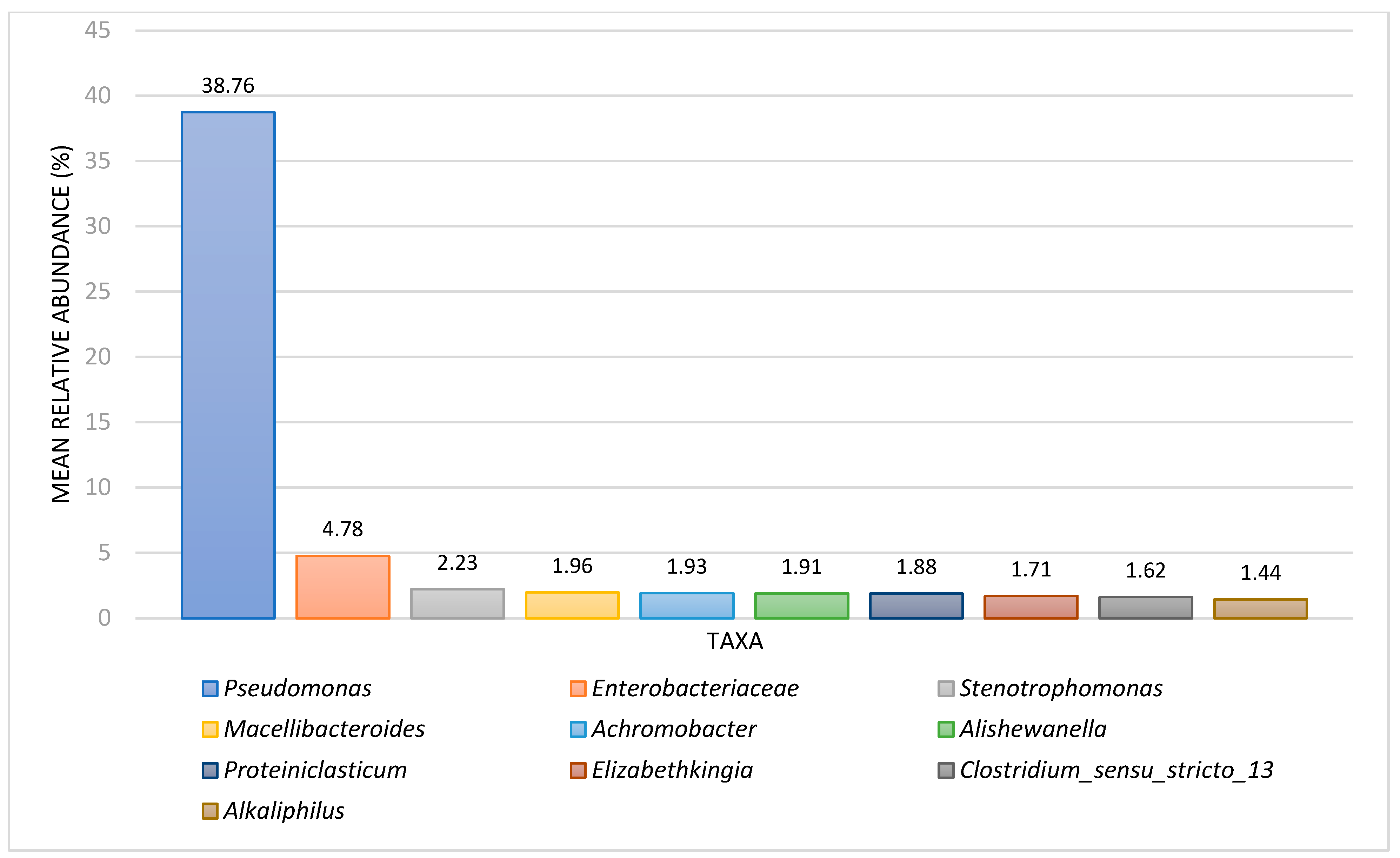
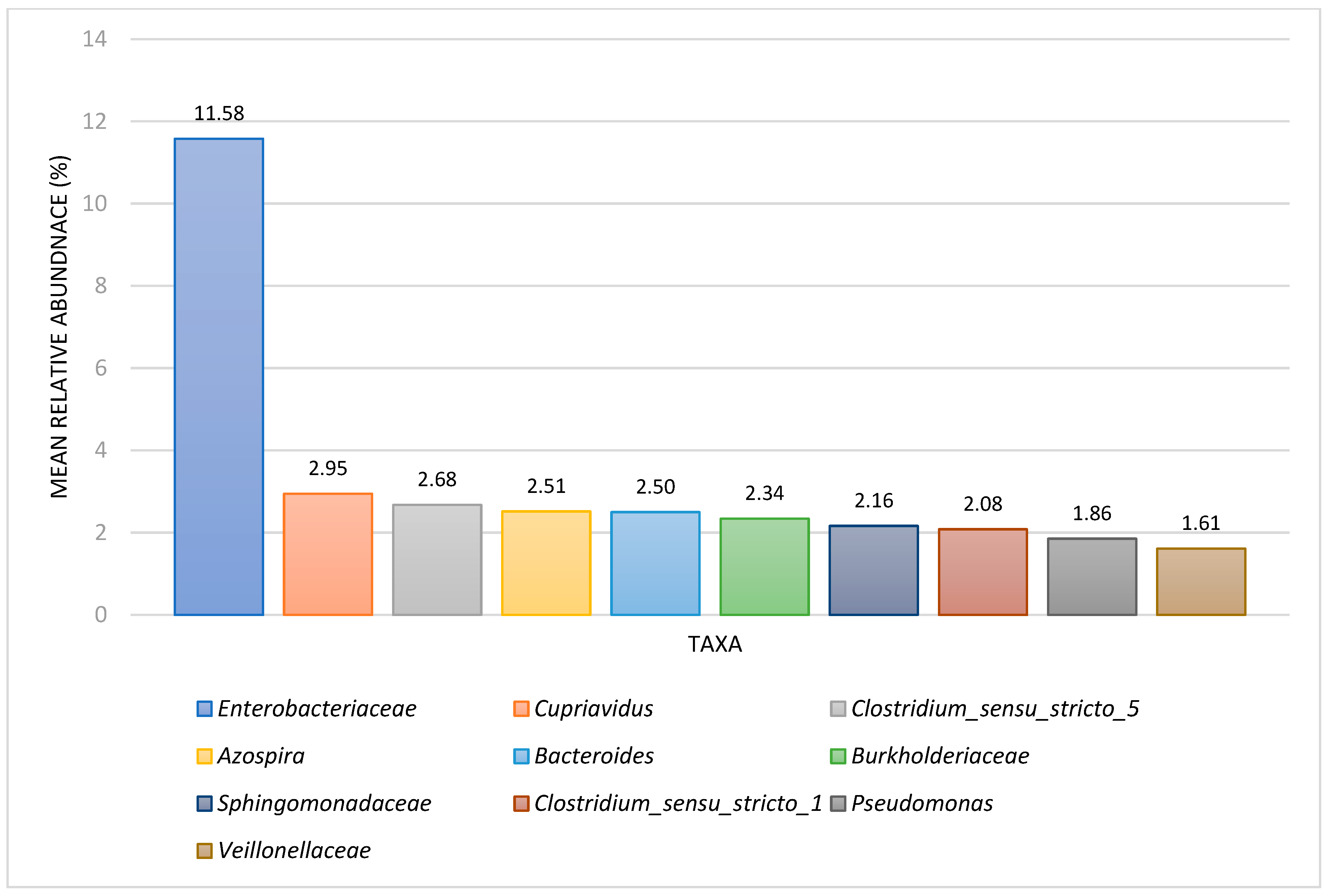
3.1. Comparison between Slime-Forming Bacteria (SLYM) BART and NGS Results
3.2. Comparison between Iron-Related Bacteria (IRB) BART and NGS Results
3.3. Comparison between Denitrifying (DN) BART and NGS Results
3.4. Comparison between Acid-Producing Bacteria (APB) BART and NGS Results
3.5. NGS and BART Analysis of Water Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gracias, K.S.; McKillip, J.L. A review of conventional detection and enumeration methods for pathogenic bacteria in food. Can. J. Microbiol. 2004, 50, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Reasoner, D.J.; Geldreich, E.E. A new medium for the enumeration and subculture of bacteria from potable water. Appl. Environ. Microbiol. 1985, 49, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Clesceri, L.S.; Greenberg, A.E.; Trussell, R.R. Standard Methods for the Examination of Water and Wastewater, 21st ed.; APHA: Washington, DC, USA, 2005. [Google Scholar]
- Clesceri, L.S.; Greenberg, A.E.; Eaton, A.D. Standard Methods for the Examination of Water and Wastewater, 20th ed.; APHA: Washington, DC, USA, 1998. [Google Scholar]
- Brown, V.I.; Lowbury, E.J. Use of an improved cetrimide agar medium and other culture methods for Pseudomonas aeruginosa. J. Clin. Pathol. 1965, 18, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Elazhary, M.A.; Saheb, S.A.; Roy, R.S.; Lagacé, A. A simple procedure for the preliminary identification of aerobic gram-negative intestinal bacteria with special reference to the Enterobacteriaceae. Can. J. Comp. Med. 1973, 37, 43–46. [Google Scholar] [PubMed]
- Bopp, C.A.; Sumner, J.W.; Morris, G.K.; Wells, J.G. Isolation of Legionella spp. from environmental water samples by low-pH treatment and use of a selective medium. J. Clin. Microbiol. 1981, 13, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, M.; Lagier, J.C.; Raoult, D.; Khelaifia, S. Bacterial culture through selective and non-selective conditions: The evolution of culture media in clinical microbiology. New Microbes New Infect. 2019, 34, 100622. [Google Scholar] [CrossRef] [PubMed]
- Kamuran, A.; Serap, C.; Esin, O.Y.; Gülcihan, G. Advance methods for the qualitative and quantitative determination of microorganisms. Microchem. J. 2021, 166, 106188. [Google Scholar] [CrossRef]
- Razban, B.; Nelson, K.Y.; Cullimore, D.R.; Cullimore, J.; McMartin, D.W. Quantitative bacteriological assessment of aerobic wastewater treatment quality and plant performance. J. Environ. Sci. Health Part A Toxic Hazard. Subst. Environ. Eng. 2012, 47, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Little, B.J.; Wagner, P.A.; Lewandowski, Z. The Role of Biomineralization in Microbiologically Influenced Corrosion. In Proceedings of the CORROSION/98, San Diego, CA, USA, 22–27 March 1998; NACE International: Houston, TX, USA, 1998. Paper No. 294. [Google Scholar]
- Little, B.; Wagner, P.; Hart, K.; Ray, R.; Lavoie, D.; Nealson, K.; Aguilar, C. The role of biomineralization in microbiologically influenced corrosion. Biodegradation 1998, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Lu, Y.; Yang, X.; Shi, X. Carbon and steel surfaces modified by Leptothrix discophora SP-6: Characterization and implications. Environ. Sci. Technol. 2007, 41, 7987–7996. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y.; Chang, W.; Lou, Y.; Qian, H. Microbiologically influenced corrosion of FeCoNiCrMn high-entropy alloys by Pseudomonas aeruginosa biofilm. Front. Microbiol. 2022, 13, 1009310. [Google Scholar] [CrossRef] [PubMed]
- Cullimore, D.R. Standard Methods for BART Testers, Fourth Edition. Available online: https://www.dbi.ca/StandMeth/StandardMethod-4th.pdf (accessed on 19 December 2013).
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. SINTAX, a simple non-Bayesian taxonomy classifier for 16S and ITS 513 sequences. bioRxiv 2016. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl. Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Gieg, L.; Sargent, J.; Bagaria, H.; Place, T.; Sharma, M.; Shen, Y.; Kiesman, D. Synergistic effect of biocide and biodispersant to mitigate microbiologically influenced corrosion in crude oil transmission pipelines. In CORROSION 2020-15090; NACE International: Houston, TX, USA, 2020. [Google Scholar]
- Vaz-Moreira, I.; Egas, C.; Nunes, O.C.; Manaia, C.M. Culture-dependent and culture-independent diversity surveys target different bacteria: A case study in a freshwater sample. Antonie Van Leeuwenhoek 2011, 100, 245–257. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Group/Taxon | Slime-Forming Bacteria BARTs (%), n = 28 | Iron-Related Bacteria BARTs, (%) n = 26 | Denitrifying Bacteria BARTs, (%), n = 21 | Acid-Producing Bacteria BARTs, (%), n = 14 | Interpretation Notes |
|---|---|---|---|---|---|
| Enteric bacteria | 28.6 | 46.2 | ----- | ----- | Enteric bacteria encompass all identified organisms under the family Enterobacteriaceae. |
| Pseudomonas | 21.4 | 15.4 | ----- | ----- | Pseudomonads include all identified organisms under the genus Pseudomonas. |
| Slime-forming bacteria/ prokaryotes | 100 | ----- | ----- | ----- | The slime-forming/prokaryotes and bacteria metabolic groupings included slime formers, both bacterial and prokaryotic, as well as viscous bulking bacteria. |
| Iron-related bacteria | ----- | 84.6 | ----- | ----- | The iron-related bacteria metabolic grouping included both iron-reducing bacteria and iron-oxidizing bacteria. |
| Anaerobic bacteria | ----- | 65.4 | ----- | ----- | The anaerobic metabolic grouping included methane oxidizing, methane- producing—acetotrophic, methane-producing—hydrogenotrophic, methane producing—methylotrophic, methane-oxidizing prokaryotes, syntrophs, sulfate-reducing bacteria, foaming, and fermentative prokaryotes. |
| Acid-producing and/or fermentative metabolism | ----- | ----- | ----- | 100 | The acid-producing and fermentative metabolic grouping included acid-producing prokaryotes, fermentative prokaryotes, fermentative thiosulfate-reducing prokaryotes, and acetogens. |
| Denitrifying bacteria | ----- | ----- | 100 | ----- | The denitrifying grouping included nitrite-reducing and nitrate-reducing organisms. |
| Agreements for the listed microorganisms and metabolic groupings were only considered if the types of BARTs had reaction patterns for them. | |||||
| Sample Top 3 Genera | IRB BART Top 3 Genera | SLYM BART Top 3 Genera | DN BART Top 3 Genera | |
|---|---|---|---|---|
| Sample 10 | 1. Phaeodactylibacter 2. Hydrogenophaga 3. JGI_0001001-H03 | 1. Pseudomonas 2. Shewanella 3. Proteiniclasticum | 1. Clostridium_sensu_stricto_1 2. Shewanella 3. Pseudomonas | |
| Sample 9 | 1. Hydrogenophaga 2. Flavobacterium 3. Simplicispira | 1. Pseudomonas 2. Chryseobacterium 3. Flavobacterium | ||
| Sample 8 | 1. Pseudomonas 2. Rheinheimera 3. Flavobacterium | 1. Pseudomonas 2. Thermovirga 3. Halomonas | ||
| Sample 7 | 1. Pseudomonas 2. Alishewanella 3. Sphingopyxis | 1. Pseudomonas 2. Stenotrophomos 3. Alishewanella | 1. Clostridium_sensu_stricto _1 2. Pseudomonas 3. Stenotrophomonas | 1. Pseudomonas 2. Stenotrophomonas 3. Alishewanella |
| Sample 6 | 1. Nubsella 2. Sphingopyxis 3. Aminobacter | 1. Aeromonas 2. Morganella 3. Hafina-Obesumbacterium | ||
| Sample 5 | 1. Cupriavidus 2. Pedobacter 3. Sphingobacterium | 1. Providencia 2. Pseudomonas 3. Alcaligenes | 1. Sphingobacterium 2. Stenotrophomonas 3. Clostridium_sensu_stricto _18 | |
| Sample 4 | 1. Hyphomicrobium 2. Pir4_lineage 3. Haliscomenobacter | 1. Pseudomonas 2. Morganella 3. Aeromonas | ||
| Sample 3 | 1. Pseudomonas 2. Legionella 3. Porphyrobacter | 1. Clostridium_sensu_stricto _1 2. Lysinibacillus 3. Paraclostridium | ||
| Sample 2 | 1. Pseudomonas 2. Methyloversatilis 3. Cupriavidus | 1. Pseudomonas 2. Morganella 3. Acinetobacter | 1. Pseudomonas 2. Alkaliphilus 3. Cupriavidus | |
| Sample 1 | 1. Pseudomonas 2. Cupriavidus 3. Sphingobacterium | 1. Pseudomonas 2. Sphingobacterium 3. Stenotrophomonas |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McIlwaine, D.B.; Moore, M.; Corrigan, A.; Niemaseck, B.; Nicoletti, D. A Comparison of the Microbial Populations in a Culture-Dependent and a Culture-Independent Analysis of Industrial Water Samples. Appl. Microbiol. 2024, 4, 1079-1090. https://doi.org/10.3390/applmicrobiol4030073
McIlwaine DB, Moore M, Corrigan A, Niemaseck B, Nicoletti D. A Comparison of the Microbial Populations in a Culture-Dependent and a Culture-Independent Analysis of Industrial Water Samples. Applied Microbiology. 2024; 4(3):1079-1090. https://doi.org/10.3390/applmicrobiol4030073
Chicago/Turabian StyleMcIlwaine, Douglas B., Mackenzie Moore, Alexsandra Corrigan, Benjamin Niemaseck, and Danika Nicoletti. 2024. "A Comparison of the Microbial Populations in a Culture-Dependent and a Culture-Independent Analysis of Industrial Water Samples" Applied Microbiology 4, no. 3: 1079-1090. https://doi.org/10.3390/applmicrobiol4030073
APA StyleMcIlwaine, D. B., Moore, M., Corrigan, A., Niemaseck, B., & Nicoletti, D. (2024). A Comparison of the Microbial Populations in a Culture-Dependent and a Culture-Independent Analysis of Industrial Water Samples. Applied Microbiology, 4(3), 1079-1090. https://doi.org/10.3390/applmicrobiol4030073