
Isolation and Optimization of Phages Infecting Members of the Streptococcus bovis/Streptococcus equinus Complex

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and Storage of Ruminal Contents and Cattle Fecal Matter
2.2. Bacterial Host and Bacteriophage Isolation
2.3. Double Layer Assay and Bacteriophage Propagation
2.4. Phage Genetic and Phenotypic Characterization
2.4.1. Whole Genome Sequencing
2.4.2. Genome Annotation and Phylogenetic Analysis
2.4.3. Transmission Electron Microscopy (TEM)
2.4.4. Assessing Inhibition of Bacterial Growth
2.4.5. Lifestyle Characterization
2.4.6. Stability of Phages in Rumen Fluid
2.5. Phage Training to Increase Killing Efficiency
3. Results
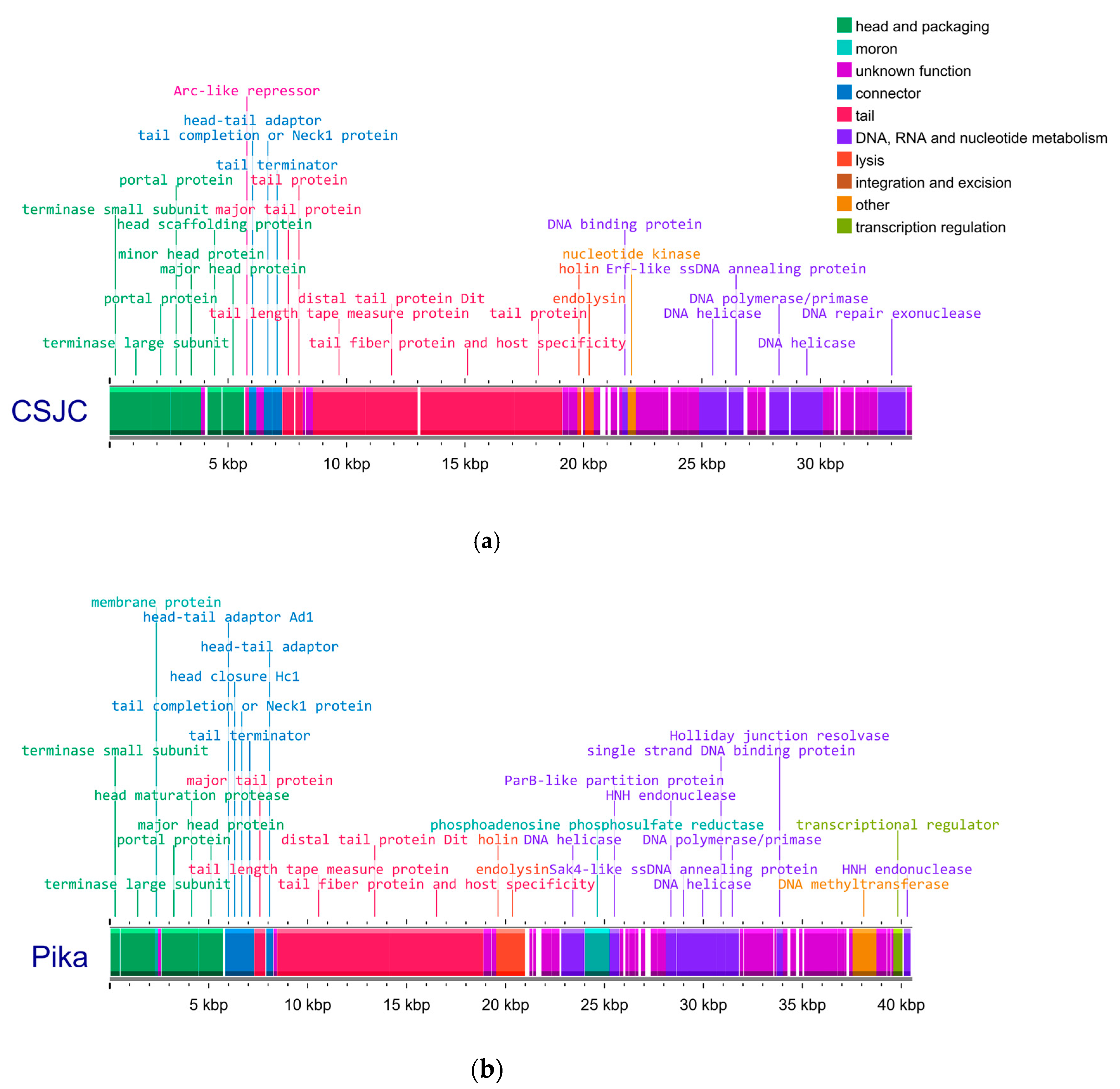
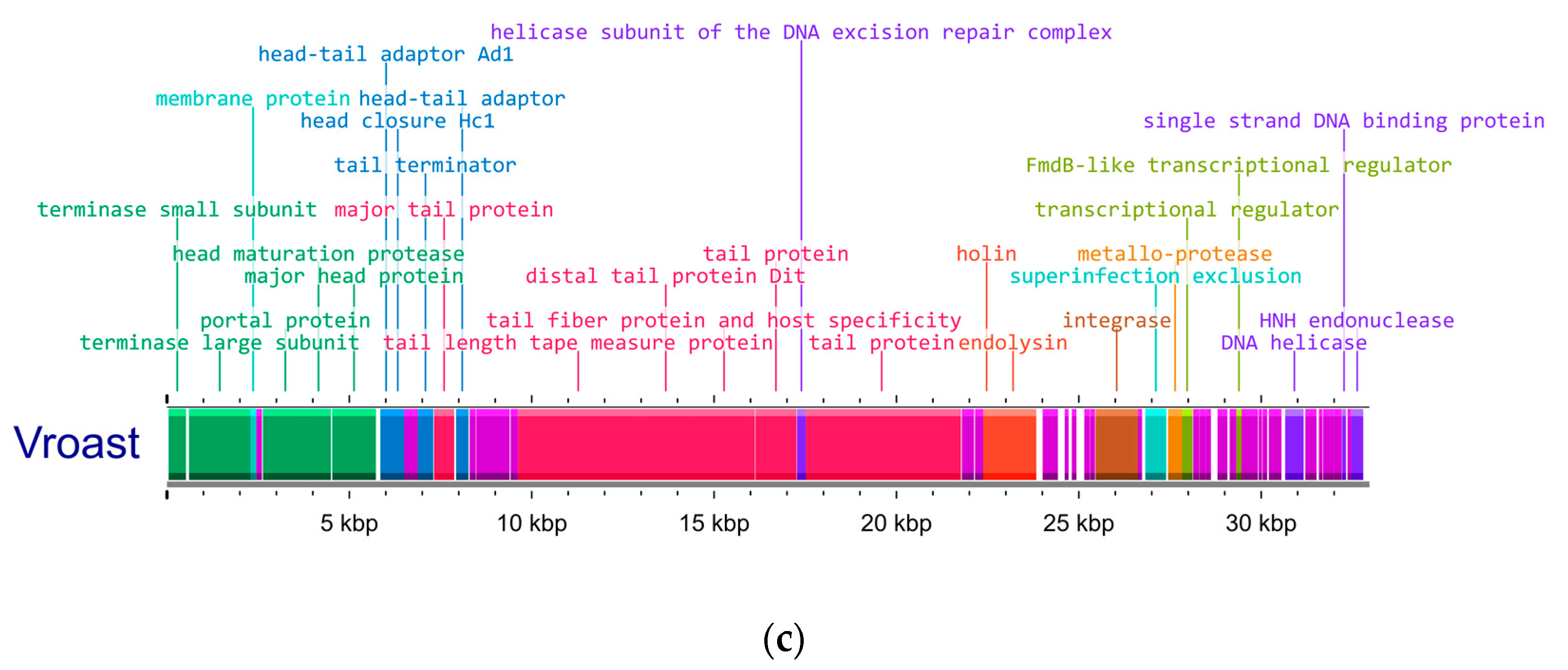
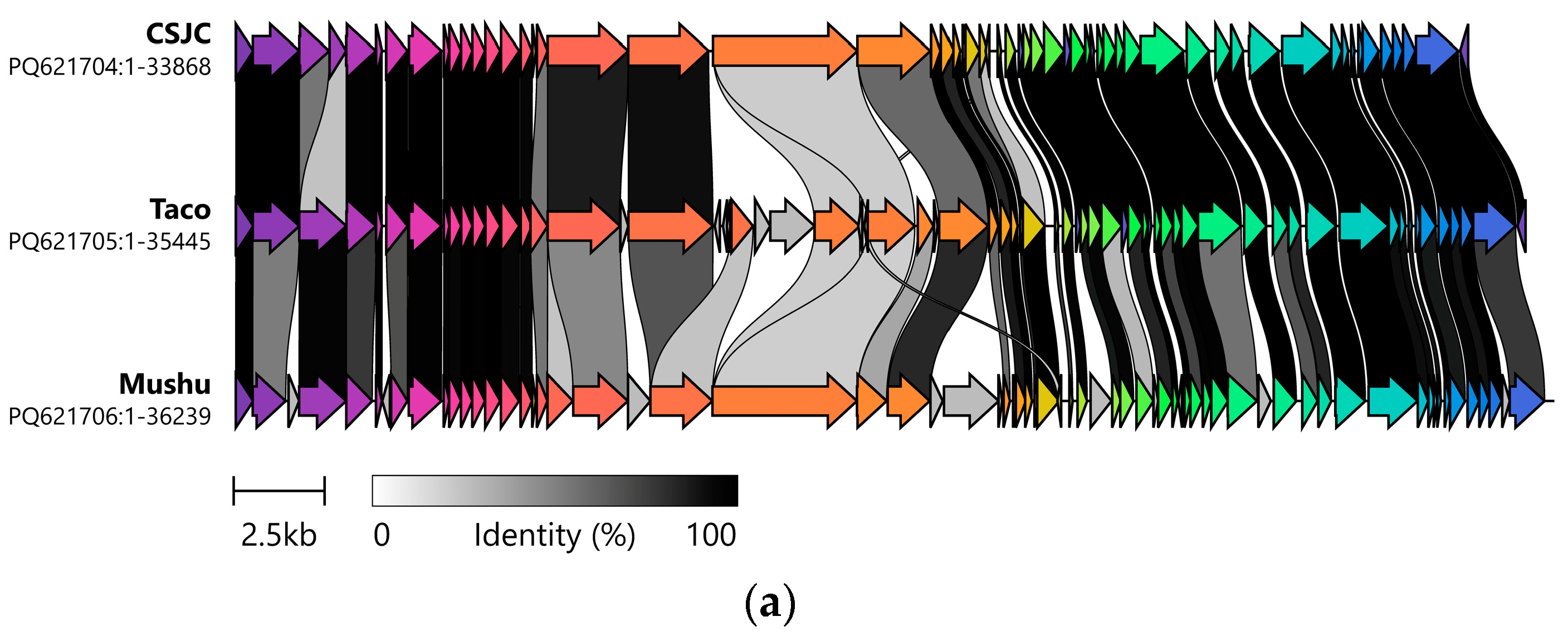
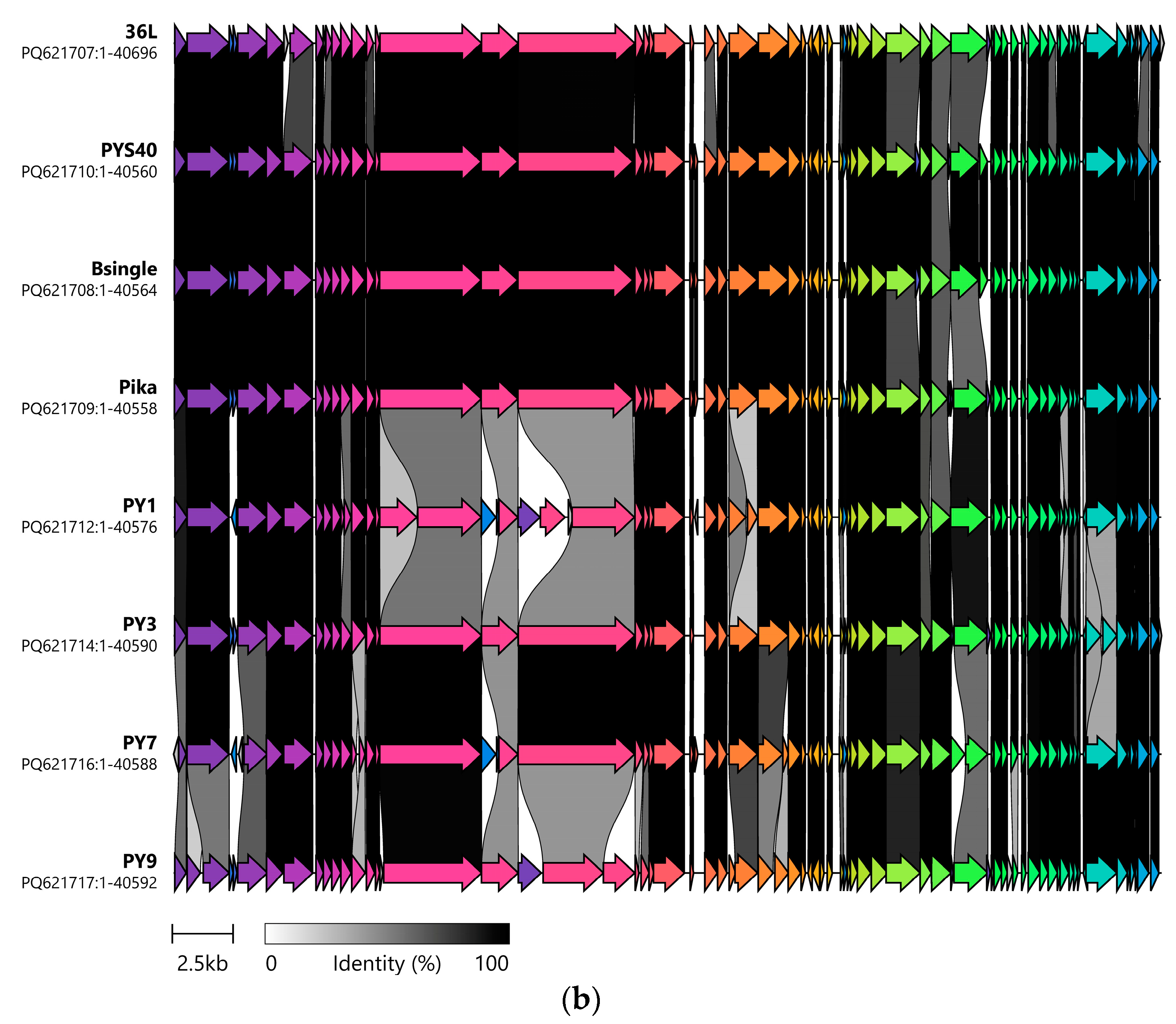
3.1. Genetic Analysis of Isolated Phages Reveals Two Clusters of Unique Phages That Have Strong Intercluster Homology
3.2. The Isolated Streptococcus Bacteriophages Have a Limited Host Range but Are Generally Effective at Inhibiting Bacterial Growth In Vitro, and Retain Activity When Exposed to Ruminal Fluid In Vitro
3.3. All but One of the Sequenced Phages Are Lytic, but Further Analysis Is Required for Cluster 2 Phages
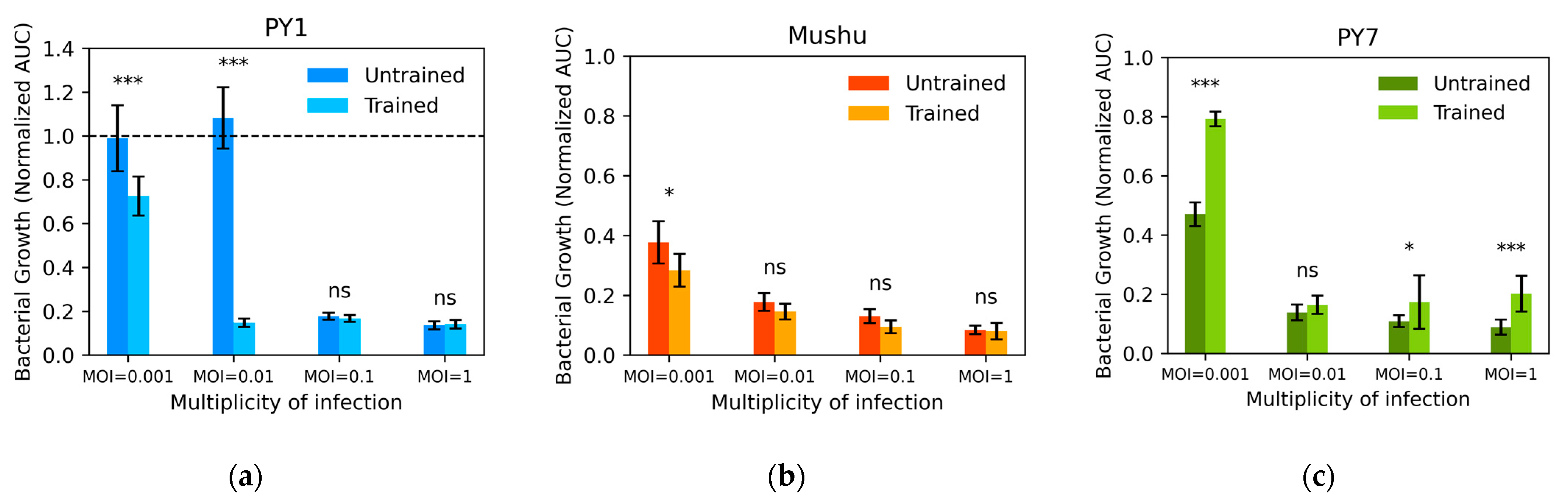
3.4. Phage Training by Serial Passaging Had Inconsistent Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Knight, R. USDA Economic Research Service. Cattle & Beef—Sector at a Glance. 2025. Available online: https://www.ers.usda.gov/topics/animal-products/cattle-beef/sector-at-a-glance (accessed on 30 December 2024).
- Ibidhi, R.; Ben Salem, H. Water footprint of livestock products and production systems: A review. Anim. Prod. Sci. 2020, 60, 1369–1380. [Google Scholar] [CrossRef]
- Godfray, H.C.J.; Aveyard, P.; Garnett, T.; Hall, J.W.; Key, T.J.; Lorimer, J.; Pierrehumbert, R.T.; Scarborough, P.; Springmann, M.; Jebb, S.A. Meat consumption, health, and the environment. Science 2018, 361, eaam5324. [Google Scholar] [CrossRef]
- Pelletier, N.; Tyedmers, P. Forecasting potential global environmental costs of livestock production 2000–2050. Proc. Natl. Acad. Sci. USA 2010, 107, 18371–18374. [Google Scholar] [CrossRef]
- Paz, H.A.; Hales, K.E.; Wells, J.E.; Kuehn, L.A.; Freetly, H.C.; Berry, E.D.; Flythe, M.D.; Spangler, M.L.; Fernando, S.C. Rumen bacterial community structure impacts feed efficiency in beef cattle. J. Anim. Sci. 2018, 96, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Mottet, A.; de Haan, C.; Falcucci, A.; Tempio, G.; Opio, C.; Gerber, P. Livestock: On our plates or eating at our table? A new analysis of the feed/food debate. Glob. Food Secur. 2017, 14, 1–8. [Google Scholar] [CrossRef]
- de Vries, M.; de Boer, I.J. Comparing environmental impacts for livestock products: A review of life cycle assessments. Livest. Sci. 2010, 128, 1–11. [Google Scholar] [CrossRef]
- Rotz, C.A.; Asem-Hiablie, S.; Place, S.; Thoma, G. Environmental footprints of beef cattle production in the United States. Agric. Syst. 2019, 169, 1–13. [Google Scholar] [CrossRef]
- Wagner, J.J.; Archibeque, S.L.; Feuz, D.M. The modern feedlot for finishing cattle. Annu. Rev. Anim. Biosci. 2014, 2, 535–554. [Google Scholar] [CrossRef]
- Kahn, L.; Cottle, D. Beef Cattle Production and Trade; Csiro Publishing: Clayton, Australia, 2014. [Google Scholar]
- Coffey, D.; Dawson, K.; Ferket, P.; Connolly, A. Review of the feed industry from a historical perspective and implications for its future. J. Appl. Anim. Nutr. 2016, 4, e3. [Google Scholar] [CrossRef]
- Ojo, A.O.; Mulim, H.A.; Campos, G.S.; Junqueira, V.S.; Lemenager, R.P.; Schoonmaker, J.P.; Oliveira, H.R. Exploring Feed Efficiency in Beef Cattle: From Data Collection to Genetic and Nutritional Modeling. Animals 2024, 14, 3633. [Google Scholar] [CrossRef]
- Herd, R.; Oddy, V.; Richardson, E. Biological basis for variation in residual feed intake in beef cattle. 1. Review of potential mechanisms. Aust. J. Exp. Agric. 2004, 44, 423–430. [Google Scholar] [CrossRef]
- Fitzsimons, C.; McGee, M.; Keogh, K.; Waters, S.M.; Kenny, D.A. Molecular physiology of feed efficiency in beef cattle. In Biology of Domestic Animals; CRC Press: Boca Raton, FL, USA, 2017; pp. 122–165. [Google Scholar]
- Kenny, D.; Fitzsimons, C.; Waters, S.; McGee, M. Invited review: Improving feed efficiency of beef cattle–the current state of the art and future challenges. Animal 2018, 12, 1815–1826. [Google Scholar] [CrossRef]
- Nkrumah, J.; Okine, E.; Mathison, G.; Schmid, K.; Li, C.; Basarab, J.; Price, M.; Wang, Z.; Moore, S. Relationships of feedlot feed efficiency, performance, and feeding behavior with metabolic rate, methane production, and energy partitioning in beef cattle. J. Anim. Sci. 2006, 84, 145–153. [Google Scholar] [CrossRef]
- Herd, R.; Arthur, P. Physiological basis for residual feed intake. J. Anim. Sci. 2009, 87, E64–E71. [Google Scholar] [CrossRef] [PubMed]
- Moloney, A.P.; McGee, M. Factors influencing the growth of meat animals. In Lawrie’ s Meat Science; Elsevier: Amsterdam, The Netherlands, 2017; pp. 19–47. [Google Scholar]
- Zhou, M.; Hernandez-Sanabria, E. Characterization of variation in rumen methanogenic communities under different dietary and host feed efficiency conditions, as determined by PCR-denaturing gradient gel electrophoresis analysis. Appl. Environ. Microbiol. 2010, 76, 3776–3786. [Google Scholar] [CrossRef]
- Petri, R.; Schwaiger, T.; Penner, G.; Beauchemin, K.; Forster, R.; McKinnon, J.; McAllister, T. Changes in the rumen epimural bacterial diversity of beef cattle as affected by diet and induced ruminal acidosis. Appl. Environ. Microbiol. 2013, 79, 3744–3755. [Google Scholar] [CrossRef] [PubMed]
- Fernando, S.C.; Purvis, H.; Najar, F.; Sukharnikov, L.; Krehbiel, C.; Nagaraja, T.; Roe, B.; DeSilva, U. Rumen microbial population dynamics during adaptation to a high-grain diet. Appl. Environ. Microbiol. 2010, 76, 7482–7490. [Google Scholar] [CrossRef]
- Inda, M.E.; Broset, E.; Lu, T.K.; de la Fuente-Nunez, C. Emerging Frontiers in Microbiome Engineering. Trends Immunol. 2019, 40, 952–973. [Google Scholar] [CrossRef]
- Sheth, R.U.; Cabral, V.; Chen, S.P.; Wang, H.H. Manipulating bacterial communities by in situ microbiome engineering. Trends Genet. 2016, 32, 189–200. [Google Scholar] [CrossRef]
- Clemmons, B.A.; Voy, B.H.; Myer, P.R. Altering the gut microbiome of cattle: Considerations of host-microbiome interactions for persistent microbiome manipulation. Microb. Ecol. 2019, 77, 523–536. [Google Scholar] [CrossRef]
- DePeters, E.; George, L. Rumen transfaunation. Immunol. Lett. 2014, 162, 69–76. [Google Scholar] [CrossRef]
- McAllister, T.A.; Beauchemin, K.A.; Alazzeh, A.Y.; Baah, J.; Teather, R.M.; Stanford, K. Review: The use of direct fed microbials to mitigate pathogens and enhance production in cattle. Can. J. Anim. Sci. 2011, 91, 193–211. [Google Scholar] [CrossRef]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Collaborators, G.R.C.; Abecia, L.; Angarita, E.; Aravena, P.; Arenas, G.N. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Webb, M.; Ghimire, S.; Blair, A.; Olson, K.; Fenske, G.J.; Fonder, A.T.; Christopher-Hennings, J.; Brake, D.; Scaria, J. Metagenomic characterization of the effect of feed additives on the gut microbiome and antibiotic resistome of feedlot cattle. Sci. Rep. 2017, 7, 12257. [Google Scholar] [CrossRef]
- Callaway, T.; Edrington, T.; Anderson, R.; Harvey, R.; Genovese, K.; Kennedy, C.; Venn, D.; Nisbet, D. Probiotics, prebiotics and competitive exclusion for prophylaxis against bacterial disease. Anim. Health Res. Rev. 2008, 9, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Gill, H.S.; Shu, Q.; Leng, R.A. Immunization with Streptococcus bovis protects against lactic acidosis in sheep. Vaccine 2000, 18, 2541–2548. [Google Scholar] [CrossRef] [PubMed]
- Wedlock, D.; Janssen, P.; Leahy, S.; Shu, D.; Buddle, B. Progress in the development of vaccines against rumen methanogens. Animal 2013, 7, 244–252. [Google Scholar] [CrossRef]
- Nagaraja, T.; Miller, G. Rumen Microbial Changes in Ionophore Antiobiotic-Treated Steers with Experimenally Induced Acidosis. Asian-Australas. J. Anim. Sci. 1989, 2, 465–468. [Google Scholar] [CrossRef]
- Jo, S.J.; Kwon, J.; Kim, S.G.; Lee, S.J. The Biotechnological Application of Bacteriophages: What to Do and Where to Go in the Middle of the Post-Antibiotic Era. Microorganisms 2023, 11, 2311. [Google Scholar] [CrossRef]
- D’Herelle, F. On an invisible microbe antagonistic toward dysenteric bacilli: Brief note by Mr. F. D’Herelle, presented by Mr. Roux. 1917. Res. Microbiol. 2007, 158, 553–554. [Google Scholar] [CrossRef]
- Schwarz, C.; Mathieu, J.; Laverde Gomez, J.A.; Yu, P.; Alvarez, P.J. Renaissance for Phage-Based Bacterial Control. Environ. Sci. Technol. 2021, 56, 4691–4701. [Google Scholar] [CrossRef]
- Schwarz, C.; Mathieu, J.; Laverde Gomez, J.; Miller, M.R.; Tikhonova, M.; Hamor, C.; Alvarez, P.J. Isolation and Characterization of Six Novel Fusobacterium necrophorum Phages. PHAGE 2024, 5, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, C. Ruminal Fusobacterium and Their Phages-Fundamentals and Applications; University of California: Berkeley, CA, USA, 2023. [Google Scholar]
- Russell, J.B.; Rychlik, J.L. Factors that alter rumen microbial ecology. Science 2001, 292, 1119–1122. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.-J.; McAllister, T.; Popp, J.; Hristov, A.; Mir, Z.; Shin, H. A review of bloat in feedlot cattle. J. Anim. Sci. 1998, 76, 299–308. [Google Scholar] [CrossRef]
- Goodrich, R.D.; Garrett, J.E.; Gast, D.R.; Kirick, M.A.; Larson, D.A.; Meiske, J.C. Influence of Monensin on the Performance of Cattle. J. Anim. Sci. 1984, 58, 1484–1498. [Google Scholar] [CrossRef]
- Marques, R.d.S.; Cooke, R.F. Effects of Ionophores on Ruminal Function of Beef Cattle. Animals 2021, 11, 2871. [Google Scholar] [CrossRef]
- Gilbert, R.A.; Klieve, A.V. Ruminal Viruses (Bacteriophages, Archaeaphages). In Rumen Microbiology: From Evolution to Revolution; Puniya, A.K., Singh, R., Kamra, D.N., Eds.; Springer: New Delhi, India, 2015; pp. 121–141. [Google Scholar]
- Tarakanov, B.V. Biology of lysogenic strains of Streptococcus bovis and virulent mutants of their temperate phages. Mikrobiologiia 1996, 65, 656–662. [Google Scholar] [PubMed]
- Klieve, A.V.; Heck, G.L.; Prance, M.A.; Shu, Q. Genetic homogeneity and phage susceptibility of ruminal strains of Streptococcus bovis isolated in Australia. Lett. Appl. Microbiol. 1999, 29, 108–112. [Google Scholar] [CrossRef]
- Iverson, W.G.; Millis, N.F. Characterization of Streptococcus bovis bacteriophages. Can. J. Microbiol. 1976, 22, 847–852. [Google Scholar] [CrossRef]
- Štyriak, I.; Španová, A.; Montagová, H.; Kmet, V. Isolation and characterization of a new ruminal bacteriophage lytic to Streptococcus bovis. Curr. Microbiol. 1994, 28, 355–358. [Google Scholar] [CrossRef]
- Gilbert, R.A.; Kelly, W.J.; Altermann, E.; Leahy, S.C.; Minchin, C.; Ouwerkerk, D.; Klieve, A.V. Toward Understanding Phage:Host Interactions in the Rumen; Complete Genome Sequences of Lytic Phages Infecting Rumen Bacteria. Front. Microbiol. 2017, 8, 2340. [Google Scholar] [CrossRef]
- Park, S.Y.; Kwon, H.; Kim, S.G.; Park, S.C.; Kim, J.H.; Seo, S. Characterization of two lytic bacteriophages, infecting Streptococcus bovis/equinus complex (SBSEC) from Korean ruminant. Sci. Rep. 2023, 13, 9110. [Google Scholar] [CrossRef]
- Köhne, M.; Hüsch, R.; Tönissen, A.; Schmidt, M.; Müsken, M.; Böttcher, D.; Hirnet, J.; Plötz, M.; Kittler, S.; Sieme, H. Isolation and characterization of bacteriophages specific to Streptococcus equi subspecies zooepidemicus and evaluation of efficacy ex vivo. Front. Microbiol. 2024, 15, 1448958. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.A.; Townsend, E.M.; Crew, K.S.; Hitch, T.C.A.; Friedersdorff, J.C.A.; Creevey, C.J.; Pope, P.B.; Ouwerkerk, D.; Jameson, E. Rumen Virus Populations: Technological Advances Enhancing Current Understanding. Front. Microbiol. 2020, 11, 450. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, Z.S.; Hales, K.E.; Nagaraja, T.G.; Lawrence, T.E.; Tennant, T.C.; Amachawadi, R.G.; Carroll, J.A.; Burdick Sanchez, N.C.; Galyean, M.L.; Davis, E.; et al. Validation of an experimental model to induce liver abscesses in Holstein steers using an acidotic diet challenge and intraruminal bacterial inoculation. Appl. Anim. Sci. 2024, 40, 398–413. [Google Scholar] [CrossRef]
- Schwarz, C.; Mathieu, J.; Gomez, J.L.; Miller, M.R.; Tikhonova, M.; Nagaraja, T.G.; Alvarez, P.J. Unexpected finding of Fusobacterium varium as the dominant Fusobacterium species in cattle rumen: Potential implications for liver abscess etiology and interventions. J. Anim. Sci. 2023, 101, skad130. [Google Scholar] [CrossRef]
- Russell, J.B.; Robinson, P.H. Compositions and characteristics of strains of Streptococcus bovis. J. Dairy Sci. 1984, 67, 1525–1531. [Google Scholar] [CrossRef]
- Martin, S.A.; Russell, J.B. Transport and phosphorylation of disaccharides by the ruminal bacterium Streptococcus bovis. Appl. Environ. Microbiol. 1987, 53, 2388–2393. [Google Scholar] [CrossRef]
- Heuer, H.; Krsek, M.; Baker, P.; Smalla, K.; Wellington, E.M. Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl. Environ. Microbiol. 1997, 63, 3233–3241. [Google Scholar] [CrossRef]
- Adams, M.H. Bacteriophages; Inter-Science Publishers: New York, NY, USA, 1959. [Google Scholar]
- Protocol: Phage DNA Extraction-Traditional; Center for Phage Technology, Texas A&M University: College Station, TX, USA, 2018.
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Shen, W.; Sipos, B.; Zhao, L. SeqKit2: A Swiss army knife for sequence and alignment processing. iMeta 2024, 3, e191. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 2017, 3, e000132. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Walenz, B.P.; Rhie, A.; Vollger, M.R.; Logsdon, G.A.; Grothe, R.; Miga, K.H.; Eichler, E.E.; Phillippy, A.M.; Koren, S. HiCanu: Accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 2020, 30, 1291–1305. [Google Scholar] [CrossRef]
- Bouras, G.; Nepal, R.; Houtak, G.; Psaltis, A.J.; Wormald, P.-J.; Vreugde, S. Pharokka: A fast scalable bacteriophage annotation tool. Bioinformatics 2022, 39, btac776. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.-y.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.H. clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Lee, I.; Ouk Kim, Y.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Göker, M.; García-Blázquez, G.; Voglmayr, H.; Tellería, M.T.; Martín, M.P. Molecular taxonomy of phytopathogenic fungi: A case study in Peronospora. PLoS ONE 2009, 4, e6319. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Göker, M. Highly parallelized inference of large genome-based phylogenies. Concurr. Comput. Pract. Exp. 2014, 26, 1715–1729. [Google Scholar] [CrossRef]
- Tiwari, R.; Artiushin, S.; Timoney, J.F. P9, a temperate bacteriophage of Streptococcus equi. Int. Congr. Ser. 2006, 1289, 165–168. [Google Scholar] [CrossRef]
- Nelson, D.; Schuch, R.; Zhu, S.; Tscherne, D.M.; Fischetti, V.A. Genomic Sequence of C1 the First Streptococcal Phage. J. Bacteriol. 2003, 185, 3325–3332. [Google Scholar] [CrossRef] [PubMed]
- Sabri, M.; Häuser, R.; Ouellette, M.; Liu, J.; Dehbi, M.; Moeck, G.; García, E.; Titz, B.; Uetz, P.; Moineau, S. Genome annotation and intraviral interactome for the Streptococcus pneumoniae virulent phage Dp-1. J. Bacteriol. 2011, 193, 551–562. [Google Scholar] [CrossRef]
- Philippe, C.; Levesque, S.; Dion, M.; Tremblay, D.; Horvath, P.; Lüth, N.; Cambillau, C.; Franz, C.; Neve, H.; Fremaux, C. Genomic and morphological characterization of a novel genus of phages infecting Streptococcus thermophilus. Appl. Environ. Microbiol. 2020, 86, e00227-20. [Google Scholar] [CrossRef]
- Rezaei Javan, R.; Ramos-Sevillano, E.; Akter, A.; Brown, J.; Brueggemann, A.B. Prophages and satellite prophages are widespread in Streptococcus and may play a role in pneumococcal pathogenesis. Nat. Commun. 2019, 10, 4852. [Google Scholar] [CrossRef]
- Blazanin, M.; Vasen, E.; Jolis, C.V.; An, W.; Turner, P.E. Theoretical validation of growth curves for quantifying phage-bacteria interactions. bioRxiv 2023. [Google Scholar] [CrossRef]
- Rajnovic, D.; Muñoz-Berbel, X.; Mas, J. Fast phage detection and quantification: An optical density-based approach. PLoS ONE 2019, 14, e0216292. [Google Scholar] [CrossRef]
- West, R.M. Best practice in statistics: Use the Welch t-test when testing the difference between two groups. Ann. Clin. Biochem. 2021, 58, 267–269. [Google Scholar] [CrossRef]
- Garneau, J.R.; Depardieu, F.; Fortier, L.-C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- Shang, J.; Tang, X.; Sun, Y. PhaTYP: Predicting the lifestyle for bacteriophages using BERT. Brief. Bioinform. 2022, 24, bbac487. [Google Scholar] [CrossRef]
- Shang, J.; Peng, C.; Liao, H.; Tang, X.; Sun, Y. PhaBOX: A web server for identifying and characterizing phage contigs in metagenomic data. Bioinform. Adv. 2023, 3, vbad101. [Google Scholar] [CrossRef] [PubMed]
- Tynecki, P.; Guziński, A.; Kazimierczak, J.; Jadczuk, M.; Dastych, J.; Onisko, A. PhageAI—Bacteriophage Life Cycle Recognition with Machine Learning and Natural Language Processing. bioRxiv 2020. [Google Scholar] [CrossRef]
- Altamirano, F.L.G.; Barr, J.J. Screening for Lysogen Activity in Therapeutically Relevant Bacteriophages. Bio-Protocol 2021, 11, e3997. [Google Scholar] [CrossRef]
- Golding, I.; Coleman, S.; Nguyen, T.; Yao, T. Decision Making by Temperate Phages. Encycl. Virol. 2019, 1, 5. [Google Scholar]
- Luque, A.; Silveira, C.B. Quantification of Lysogeny Caused by Phage Coinfections in Microbial Communities from Biophysical Principles. mSystems 2020, 5, e00353-20. [Google Scholar] [CrossRef]
- Levine, M. Effect of mitomycin C on interactions between temperate phages and bacteria. Virology 1961, 13, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Borin, J.M.; Avrani, S.; Barrick, J.E.; Petrie, K.L.; Meyer, J.R. Coevolutionary phage training leads to greater bacterial suppression and delays the evolution of phage resistance. Proc. Natl. Acad. Sci. USA 2021, 118, e2104592118. [Google Scholar] [CrossRef]
- Meyer, J.R.; Dobias, D.T.; Weitz, J.S.; Barrick, J.E.; Quick, R.T.; Lenski, R.E. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science 2012, 335, 428–432. [Google Scholar] [CrossRef]
- Ngiam, L.; Weynberg, K.; Guo, J. Evolutionary and co-evolutionary phage training approaches enhance bacterial suppression and delay the emergence of phage resistance. ISME Commun. 2024, 4, ycae082. [Google Scholar] [CrossRef] [PubMed]
- Borges, A.L. How to train your bacteriophage. Proc. Natl. Acad. Sci. USA 2021, 118, e2109434118. [Google Scholar] [CrossRef]
- Gao, X.Y.; Zhi, X.Y.; Li, H.W.; Klenk, H.P.; Li, W.J. Comparative genomics of the bacterial genus Streptococcus illuminates evolutionary implications of species groups. PLoS ONE 2014, 9, e101229. [Google Scholar] [CrossRef] [PubMed]
- Klieve, A.V.; Swain, R.A.; Nolan, J. Bacteriophages in the rumen: Types present, population size and implications for the efficiency of feed utilisation. In Proceedings of the Australian Society of Animal Production, Brisbane, QLD, Australia, 8–12 July 1996; pp. 92–94. [Google Scholar]
- Sun, R.; Yu, P.; Zuo, P.; Villagrán, D.; Mathieu, J.; Alvarez, P.J.J. Biofilm Control in Flow-Through Systems Using Polyvalent Phages Delivered by Peptide-Modified M13 Coliphages with Enhanced Polysaccharide Affinity. Environ. Sci. Technol. 2022, 56, 17177–17187. [Google Scholar] [CrossRef]
- Yu, P.; Mathieu, J.; Yang, Y.; Alvarez, P.J.J. Suppression of Enteric Bacteria by Bacteriophages: Importance of Phage Polyvalence in the Presence of Soil Bacteria. Environ. Sci. Technol. 2017, 51, 5270–5278. [Google Scholar] [CrossRef]
- Klieve, A.V.; Bauchop, T. Phage resistance and altered growth habit in a strain of Streptococcus bovis. FEMS Microbiol. Lett. 1991, 80, 155–159. [Google Scholar] [CrossRef]
- Štyriak, I.; Španová, A.; Žitňan, R. Partial characterization of two ruminal bacteriophages with similar restriction patterns and different capsids morphology. Arch. Anim. Breed. 2005, 48, 572–579. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Assembling the perfect bacterial genome using Oxford Nanopore and Illumina sequencing. PLoS Comput. Biol. 2023, 19, e1010905. [Google Scholar] [CrossRef]
- Hernandez, S.I.; Berezin, C.-T.; Miller, K.M.; Peccoud, S.J.; Peccoud, J. Hybrid sequencing facilitates robust de novo plasmid assembly. bioRxiv 2024. [Google Scholar] [CrossRef]
- Peters, T.L.; Schow, J.; Spencer, E.; Leuven, J.T.V.; Wichman, H.; Miller, C. Directed evolution of bacteriophages: Thwarted by prolific prophage. Appl. Environ. Microbiol. 2024, 90, e00884-24. [Google Scholar] [CrossRef]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Philippe, C.; Chaïb, A.; Jaomanjaka, F.; Claisse, O.; Lucas, P.M.; Samot, J.; Cambillau, C.; Le Marrec, C. Characterization of the First Virulent Phage Infecting Oenococcus oeni, the Queen of the Cellars. Front. Microbiol. 2021, 11, 596541. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, D.; Martínez, B.; Rodríguez, A.; García, P. Genomic characterization of two Staphylococcus epidermidis bacteriophages with anti-biofilm potential. BMC Genom. 2012, 13, 228. [Google Scholar] [CrossRef]
- Cumby, N.; Davidson, A.R.; Maxwell, K.L. The moron comes of age. Bacteriophage 2012, 2, 225–228. [Google Scholar] [CrossRef]
- Farlow, J.; Bolkvadze, D.; Leshkasheli, L.; Kusradze, I.; Kotorashvili, A.; Kotaria, N.; Balarjishvili, N.; Kvachadze, L.; Nikolich, M.; Kutateladze, M. Genomic characterization of three novel Basilisk-like phages infecting Bacillus anthracis. BMC Genom. 2018, 19, 685. [Google Scholar] [CrossRef]
- Wallace, B.A.; Varona, N.S.; Hesketh-Best, P.J.; Stiffler, A.K.; Silveira, C.B. Globally distributed bacteriophage genomes reveal mechanisms of tripartite phage–bacteria–coral interactions. ISME J. 2024, 18, wrae132. [Google Scholar] [CrossRef]
- Kieft, K.; Zhou, Z.; Anderson, R.E.; Buchan, A.; Campbell, B.J.; Hallam, S.J.; Hess, M.; Sullivan, M.B.; Walsh, D.A.; Roux, S.; et al. Ecology of inorganic sulfur auxiliary metabolism in widespread bacteriophages. Nat. Commun. 2021, 12, 3503. [Google Scholar] [CrossRef]
- Yang, H.; Ma, Y.; Wang, Y.; Yang, H.; Shen, W.; Chen, X. Transcription regulation mechanisms of bacteriophages: Recent advances and future prospects. Bioengineered 2014, 5, 300–304. [Google Scholar] [CrossRef]
- Aravind, L.; Makarova, K.S.; Koonin, E.V. SURVEY AND SUMMARY: Holliday junction resolvases and related nucleases: Identification of new families, phyletic distribution and evolutionary trajectories. Nucleic Acids Res. 2000, 28, 3417–3432. [Google Scholar] [CrossRef] [PubMed]
- Neamah, M.M.; Mir-Sanchis, I.; López-Sanz, M.; Acosta, S.; Baquedano, I.; Haag, A.F.; Marina, A.; Ayora, S.; Penadés, J.R. Sak and Sak4 recombinases are required for bacteriophage replication in Staphylococcus aureus. Nucleic Acids Res. 2017, 45, 6507–6519. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.; Amarir-Bouhram, J.; Faure, G.; Petit, M.-A.; Guerois, R. Detection of novel recombinases in bacteriophage genomes unveils Rad52, Rad51 and Gp2.5 remote homologs. Nucleic Acids Res. 2010, 38, 3952–3962. [Google Scholar] [CrossRef] [PubMed]
- Hutinet, G.; Besle, A.; Son, O.; McGovern, S.; Guerois, R.; Petit, M.-A.; Ochsenbein, F.; Lecointe, F. Sak4 of Phage HK620 Is a RecA Remote Homolog With Single-Strand Annealing Activity Stimulated by Its Cognate SSB Protein. Front. Microbiol. 2018, 9, 743. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, D.; Huang, Y.; Zhu, X.; Rui, M.; Wan, T.; Zheng, X.; Shen, Y.; Chen, X.; Ma, K.; et al. Structural and functional characterization of deep-sea thermophilic bacteriophage GVE2 HNH endonuclease. Sci. Rep. 2017, 7, 42542. [Google Scholar] [CrossRef]
- Wilson, G.W.; Edgell, D.R. Phage T4 mobE promotes trans homing of the defunct homing endonuclease I-TevIII. Nucleic Acids Res. 2009, 37, 7110–7123. [Google Scholar] [CrossRef]
- Moodley, S.; Maxwell, K.L.; Kanelis, V. The protein gp74 from the bacteriophage HK97 functions as a HNH endonuclease. Protein Sci. 2012, 21, 809–818. [Google Scholar] [CrossRef]
- Bignell, C.; Thomas, C.M. The bacterial ParA-ParB partitioning proteins. J. Biotechnol. 2001, 91, 1–34. [Google Scholar] [CrossRef]
- Ludtke, D.N.; Eichorn, B.G.; Austin, S.J. Plasmid-partition functions of the P7 prophage. J. Mol. Biol. 1989, 209, 393–406. [Google Scholar] [CrossRef]
- Andrews, T.; Hoyer, J.S.; Ficken, K.; Fey, P.D.; Duffy, S.; Boyd, J.M. A Transducing Bacteriophage Infecting Staphylococcus epidermidis Contributes to the Expansion of a Novel Siphovirus Genus and Implies the Genus Is Inappropriate for Phage Therapy. Msphere 2023, 8, e00524-22. [Google Scholar] [CrossRef]
- Łobocka, M.B.; Rose, D.J.; Plunkett, G.; Rusin, M.; Samojedny, A.; Lehnherr, H.; Yarmolinsky, M.B.; Blattner, F.R. Genome of Bacteriophage P1. J. Bacteriol. 2004, 186, 7032–7068. [Google Scholar] [CrossRef] [PubMed]
- Lehnherr, H.; Maguin, E.; Jafri, S.; Yarmolinsky, M.B. Plasmid addiction genes of bacteriophage P1: Doc, which causes cell death on curing of prophage, and phd, which prevents host death when prophage is retained. J. Mol. Biol. 1993, 233, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Bull, J.J.; Vegge, C.S.; Schmerer, M.; Chaudhry, W.N.; Levin, B.R. Phenotypic resistance and the dynamics of bacterial escape from phage control. PLoS ONE 2014, 9, e94690. [Google Scholar] [CrossRef] [PubMed]
- Egido, J.E.; Costa, A.R.; Aparicio-Maldonado, C.; Haas, P.-J.; Brouns, S.J.J. Mechanisms and clinical importance of bacteriophage resistance. FEMS Microbiol. Rev. 2021, 46, fuab048. [Google Scholar] [CrossRef]
- Habusha, M.; Tzipilevich, E.; Fiyaksel, O.; Ben-Yehuda, S. A mutant bacteriophage evolved to infect resistant bacteria gained a broader host range. Mol. Microbiol. 2019, 111, 1463–1475. [Google Scholar] [CrossRef]
- Favor, A.H.; Llanos, C.D.; Youngblut, M.D.; Bardales, J.A. Optimizing bacteriophage engineering through an accelerated evolution platform. Sci. Rep. 2020, 10, 13981. [Google Scholar] [CrossRef]
- Laanto, E.; Mäkelä, K.; Hoikkala, V.; Ravantti, J.J.; Sundberg, L.-R. Adapting a phage to combat phage resistance. Antibiotics 2020, 9, 291. [Google Scholar] [CrossRef]
- Peters, T.L.; Song, Y.; Bryan, D.W.; Hudson, L.K.; Denes, T.G. Mutant and recombinant phages selected from in vitro coevolution conditions overcome phage-resistant Listeria monocytogenes. Appl. Environ. Microbiol. 2020, 86, e02138-20. [Google Scholar] [CrossRef]
- Paterson, S.; Vogwill, T.; Buckling, A.; Benmayor, R.; Spiers, A.J.; Thomson, N.R.; Quail, M.; Smith, F.; Walker, D.; Libberton, B.; et al. Antagonistic coevolution accelerates molecular evolution. Nature 2010, 464, 275–278. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolation Source | Cluster | Phage | Production Host | Genome (bp) | GC Content (%) | Predicted CDS | CDS Hypothetical |
|---|---|---|---|---|---|---|---|
| Fecal | 1 | CSJC | OC2C | 33,868 | 37.16 | 58 | 32 |
| Fecal | 1 | Mushu | B+ | 36,293 | 37.1 | 67 | 34 |
| Fecal | 1 | Taco | OC1D | 35,445 | 37.16 | 66 | 37 |
| Rumen | 2 | Pika | MEM36 | 40,558 | 39.5 | 68 | 39 |
| Rumen | 2 | 36L | MEM36 | 40,696 | 39.41 | 68 | 39 |
| Rumen | 2 | B-single | C5D10 | 40,564 | 39.5 | 67 | 38 |
| Rumen | 2 | PYS40 | MEM35 | 40,560 | 39.49 | 68 | 39 |
| Rumen | 2 | PY1 | MEM7 | 40,576 | 39.49 | 72 | 41 |
| Rumen | 2 | PY2 | MEM7 | 40,589 | 39.49 | 71 | 38 |
| Rumen | 2 | PY3 | MEM7 | 40,590 | 39.5 | 67 | 38 |
| Rumen | 2 | PY4 | MEM7 | 40,595 | 39.5 | 72 | 40 |
| Rumen | 2 | PY7 | MEM35 | 40,588 | 39.5 | 70 | 39 |
| Rumen | 2 | PY9 | MEM7 | 40,592 | 39.49 | 72 | 37 |
| Fecal | - | Vroast | MP2-7 | 32,968 | 40.6 | 58 | 31 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laverde Gomez, J.; Schwarz, C.; Tikhonova, M.; Hamor, C.; Tao, Y.J.; Alvarez, P.J.J.; Mathieu, J. Isolation and Optimization of Phages Infecting Members of the Streptococcus bovis/Streptococcus equinus Complex. Appl. Microbiol. 2025, 5, 28. https://doi.org/10.3390/applmicrobiol5010028
Laverde Gomez J, Schwarz C, Tikhonova M, Hamor C, Tao YJ, Alvarez PJJ, Mathieu J. Isolation and Optimization of Phages Infecting Members of the Streptococcus bovis/Streptococcus equinus Complex. Applied Microbiology. 2025; 5(1):28. https://doi.org/10.3390/applmicrobiol5010028
Chicago/Turabian StyleLaverde Gomez, Jenny, Cory Schwarz, Marina Tikhonova, Clark Hamor, Yizhi J. Tao, Pedro J. J. Alvarez, and Jacques Mathieu. 2025. "Isolation and Optimization of Phages Infecting Members of the Streptococcus bovis/Streptococcus equinus Complex" Applied Microbiology 5, no. 1: 28. https://doi.org/10.3390/applmicrobiol5010028
APA StyleLaverde Gomez, J., Schwarz, C., Tikhonova, M., Hamor, C., Tao, Y. J., Alvarez, P. J. J., & Mathieu, J. (2025). Isolation and Optimization of Phages Infecting Members of the Streptococcus bovis/Streptococcus equinus Complex. Applied Microbiology, 5(1), 28. https://doi.org/10.3390/applmicrobiol5010028