Abstract
The structure of lead(II) is not well known in aqueous solution. The Hartree–Fock and second order Møller–Plesset levels of theory using the CEP, LANL2, and SDD effective core potentials in combination with their associated basis sets, or with the 6-31G* and 6-31+G* basis sets were used to calculate the energies, structures, and vibrational frequencies of Pb2+(H2O)n, n = 0–9, 18. The lead–oxygen distances and totally symmetric stretching frequency of the aqualead(II) ions from different levels of theory were compared with each other, and with solution measurements where available. The calculations support a hemidirected hexacoordinate structure.
1. Introduction
Although the structure of many metal ions in solution is known, some remain elusive [1]. Many are known to be toxic to man, however this is dependent on oxidation state and speciation, which often depends on pH and the presence of counterions that solubilize the metal by complex formation. While computational chemistry can assist in supporting and rationalizing proposed speciation models, one drawback is that there are typically few all-electron basis sets that can be used. For elements of high atomic number, relativistic effects can play an important role. Effective core potentials replace the explicit description of core electrons by a potential, and are paired with basis sets describing the outermost electrons. In a previous work, we benchmarked some common effective core potentials for the aqua complexes of the heavy metals mercury(II) and thallium(III), both of which have valence electron configuration 5d10 [2]. We extend our work now to benchmark these common effective core potentials for the aqua complexes of lead(II), which has valence electron configuration 6s25d10, with the aim of predicting Pb-O distances and vibrational frequencies. The presence of the 6s2 subshell will be shown to have a pronounced effect on the structures compared to those metals (Tl3+, Hg2+) without it. Aqualead(II) is predicted to be hemidirected at lower coordination numbers (the ligands are not symmetrically distributed about the central ion) whereas aquamercury(II) and thallium(III) are holodirected (the ligands are symmetrically distributed about the central ion).
It appears as though the lead(II) ion has not been characterized as an aqua ion in aqueous solution, although it is believed to be the predominant form of lead for pH < 6 [3]. Lead has a propensity to oligomerize at or near neutral pH. One perchlorate salt that has been structurally characterized at a hydrolysis ratio of 1.33 is Pb6(μ4-O)(μ3-OH)6(ClO4)4∙H2O [4]. The structure may be viewed as three-face shared lead tetrahedra, with tetrahedrally-coordinated oxygen in the central tetrahedral and hydroxyl groups sitting on the faces of the distal tetrahedral. A second polymorph exists [5]. At a hydrolysis ratio [OH−]/[Pb2+] of 1.00, the solution X-ray structure is consistent with the formation of a Pb4(OH)44+ ion [6]. The Pb-O distances were around 2.6 Å. The Pb4(OH)44+ ion was characterized by single crystal X-ray diffraction in the compounds [Pb4(OH)4]3(CO3)(ClO4)10∙6H2O [7] and [Pb4(OH)4](ClO4)4∙2H2O [8]. In the mixed carbonate-perchlorate, formed by the introduction of adventitious carbon dioxide, the Pb-O distances varied from 2.293(17)–2.488(16) Å [7]. In the compound without carbonate ion, the Pb-O distances range from 2.38(2)–2.54(2) Å [8].
In the gas-phase, it was reported that tin(II), lead(II), and mercury(II) underwent a facile proton transfer reaction and that the only species observed in the mass spectra are the deprotonated MOH+(H2O)n-1 ions, not the M(H2O)n2+ ions, i.e., these ions have anomalously high acidity in the gas phase as well as the solution phase. An ab initio study was carried out to rationalize this behavior, with a focus on the pathways to deprotonation [9]. However, in the same year, the existence of lead(II) monohydrate was reported in the mass spectrometer from the reaction of the lead(II) acetonitrile monosolvate with water, albeit in low yield [10]. It can be abundantly produced by ligand exchange with nitrogen-solvated lead(II) [11]. Electrospray ionization mass spectrometry (ESI-MS) was used to produce [Pb(H2O)n]2+, n = 8–47, however water loss eventually resulted in [Pb(OH)m]+ and [H(H2O)k]+, m + k = n < 8, k = 3, 4. For n = 8–10, both [Pb(OH)m]+ and [H(H2O)k]+ were observed, showing that Pb(H2O)n]2+, n = 8–10 were metastable [12].
Computationally, lead(II) has been studied by a few research groups. A Hartree–Fock and four-component density functional study using an all-electron basis set has been carried out on [Pb(H2O)]2+ and [Pb(OH)]+ [13]. The monohydrate possessed C2v symmetry. Scalar calculations were also carried out using the 6-31+G** basis set on oxygen and hydrogen and a variety of pseudopotentials. Tetraaqualead(II) has also been studied and shown to possess a hemidirected structure [14]. In addition, several molecular dynamics studies have been carried out. Hofer and Rode suggested that the coordination number of lead in aqueous solution is nine, based on HF-QM/MM/MD methods, with a maximum in the Pb-O radial distribution of 2.60 Å (shoulder 2.65 Å) [15]. The dynamical information was given in a follow-up publication, which suggested a Pb-O stretching frequency of 217 cm−1 [16]. However, another research group using CPMD calculations suggested that lead(II) was actually heptacoordinated, with an average Pb-O distance of 2.70 Å and a very wide distribution [17]. Wander and Clark studied Pb(H2O)n2+, n = 0–9 using both the large-core (78 core electrons for Pb) B3LYP/LANL2DZ and the small core (60 core electrons for Pb) B3LYP/aug-cc-pVDZ(-pp) levels of theory and concluded thermodynamically with single point PCM calculations that 6–8 inner-shell water molecules were possible. They noted that the use of the B3LYP/LANL2DZ method gave unstable higher coordination complexes (n = 5–9) [18]. In conjunction with their ESI-MS measurements, Stace and coworkers calculated the structures of aqualead(II) complexes, n = 8, 10–12 using B3LYP/LANL2DZ and found that the 4-coordinate structure was preferred [12]. Bhattacharjee et al. examined the hydration of lead(II) using QMCF-MD, based on the cc-pVDZ-pp basis set and effective core potential on lead and DZP on oxygen and hydrogen. They found a most probable coordination number of eight, with a significant population of seven- and nine-coordination as well. The mean Pb-O distance was 2.72 Å, and the power spectrum gave a Pb-O stretching frequency with band maximum at 196 cm−1. [19]. Lei and Pan studied [Pb(H2O)n]2+, n = 1–7, using B3LYP, BLYP, and MP2 with the aug-cc-pVDZ-pp, LANL2DZ, and SDD effective core potential and basis set on lead, with aug-cc-pVDZ, 6-311+G(d,p), 6-31G(d), and LANL2DZ basis sets on oxygen and hydrogen, and extended n to 17 at the B3LYP/aug-cc-pVDZ-pp level. They also examined the structures [PbOH(H2O)n]+, n = 1, 3, 7, formed from a single proton transfer [20]. Wang et al. studied the structures of [Pb(H2O)n]2+, n = 1–9, using the PBE functional with a plane wave basis set and supersoft pseudopotentials. They found stable conformations for n = 6–8, but not for 9, and the hemi- to holodirected transition occurred at n = 6. They found a hydration number of 6.3 [21]. Leon-Pimentel et al. performed Born–Oppenheimer molecular dynamics (BOMD) simulations using the aug-cc-pVDZ-pp basis set and effective core potential on lead and both the 6-311G and aug-cc-pVDZ basis sets on oxygen and hydrogen. They concluded that lead(II) was hemidirected and 4-coordinate. They also examined proton-transfer events [22]. Kuznetsov carried out density functional calculations on [Pb(H2O)n]2+, n = 6–9, using the B3LYP, B3LYP-D3, ω-B97X, ω-B97XD, M06L and TPSSH functionals and the 6-311++G(d,p) and aug-cc-pVDZ basis sets on O and H and the aug-cc-pVDZ-pp basis set and effective core potential on lead. They found that for n = 6, 7 a hemidirected species was formed, however for n = 8,9 a holodirected species was found. All were comparable in energy [23]. Leon-Pimentel et al. again studied the hydration of lead(II), but with a polarizable mobile charge densities in harmonic oscillators (MCDHO) force field in a classical MD simulation. In addition, MP2, SCS-MP2, B3LYP, M06-2X, ω-B97X, PBE, and CCSD levels of theory were employed with the aug-cc-pVDZ and aug-cc-pVTZ for H and O (-pp for Pb) [24]. In a study of the interaction of lead(II) with biological media, Tolbatov and Marrone studied [Pb(H2O)n]2+, n = 1–9, using the ω-B97X functional and the LANL08(d) basis set on lead and either 6-311++G** or 6-31+G(d) for H and O and concluded that both n = 4 and n = 6 were particularly stable [25].
2. Materials and Methods
Calculations were performed using Gaussian 98 [26]. This program version was the first that allows an analytical frequency calculation of molecules, in which core electrons are described by effective core potentials (ECPs), and thus many variants of these were tried. Although these effective core potentials only include scalar relativistic effects, the effect of spin-orbit coupling is small (3 kJ/mol, <0.001 Å) [27]. The MP2 calculations use the frozen core approximation. A stepping-stone approach was used for geometry optimization, in which the geometries at the levels HF/CEP-4G, HF/CEP-31G*, HF/CEP-121G*, HF/LANL2MB, HF/LANL2DZ, and HF/SDD were sequentially optimized. For minimum energy structures, the MP2/CEP-31G* and MP2/CEP-121G* calculations were also performed. Calculations were also carried out using the 6-31G* and 6-31+G* basis sets on the atoms of the water molecules (5d) with an effective core potential and basis set on the metal ion (denoted as ECP+6-31G* or 6-31+G*). For shorthand, we denote the mixed basis sets as follows: CEP-121G* on Pb, 6-31G* on O,H, as basis set A; LANL2DZ on Pb, 6-31G* on O, H, as basis set B; SDD on Pb, 6-31G* on O,H, as basis set C; and the corresponding basis sets with diffuse functions are indicated by adding a “+” to the basis set name. Default optimization specifications were normally used. After each level, where possible, a frequency calculation was performed at the same level and the resulting Hessian used in the following optimization. Z-matrix coordinates constrained to the appropriate symmetry were used as required to speed up the optimizations. Since frequency calculations are done at each level, any problems with the Z-matrix coordinates would manifest themselves by giving imaginary frequencies corresponding to modes orthogonal to the spanned Z-matrix space. The Hessian was evaluated at the first geometry (opt = CalcFC) for the first level in a series in order to aid geometry convergence. We note that, for the heavy elements only, the three different CEP basis sets are equivalent (CEP-121G*) but differ for the oxygen and hydrogen atoms. The choice of core electrons defining the pseudopotential is constant ([Xe]5d104f14). In some cases, Gaussian 03 [28] and Gaussian 16 [29] was used to correct errors and omissions.
In many cases to follow, the symmetry of the minimum-energy complexes was the same as those previously found for bismuth [30]. To confirm these results, starting with high symmetry structures, systematic desymmetrization along the various irreducible representations was carried out [31,32] using Gaussian 16.
The authors have elected not to apply an implicit solvation model. One of the authors [33] has experience that demonstrates that, at least for the metal-oxygen stretching and deformation modes of [M(H2O)n+], the use of post-Hartree–Fock treatments (e.g., MP2) leads to no improvement in the vibrational frequencies relative to Hartree–Fock methods. In addition, scaling by the usual multiplicative factors (less than one) for specific theoretical levels usually makes the agreement worse as the stretching vibrational frequencies are already underestimated. We demonstrated for numerous systems that the calculated vibrational frequency can be brought into nearly perfect agreement with experiment if an explicit model of the full second-hydration sphere is used. For bromozinc complexes [ZnBrm(H2O)n](2-m)+, m = 0–4, n = 0–6−m, inclusion of thermal and entropy term changed the relative energies by only a few kJ/mol, although inclusion of solvation effects using the CPCM model tends to stabilize more compact structures with higher coordination numbers relative to those with more water molecules in the second hydration shell (by ~20 kJ/mol per second-shell water molecule) [33].
3. Results
3.1. A Survey of Structures
Lead(II), like bismuth(III) [21] might be expected to show deviations from the ideal symmetry point groups listed above, due to the potentially stereochemically active inert 6s2 subshell. The point group symmetry for the mono- through octaaquabismuth(III) was found to be C2v, C2, C3, C2, C2v, C3, C2, and S8, respectively. Lead(II) follows the same pattern (see Figure 1; Tables S1 and S2 of SM), except that the heptaaqualead(II) has no symmetry (C1). In addition lead(II) can form an enneahydrate of C3 symmetry at selected levels of theory. The diaqualead(II) species ascends in symmetry to a planar C2v structure at HF/LANL2MB. The tetraaqualead(II) forms a Cs structure at HF/LANL2MB. The pentaaqualead(II) forms a Cs structure at HF/A, HF/B, and HF/C levels. At some levels, the heptaaqualead(II) ion gives structures such as [6 + 1], [5 + 2], or [4 + 3]. If a heptacoordinate structure [7 + 0] exists, it is usually more stable than the corresponding [(7 − m) + m] structures (i.e., 7 − m water molecules in the first hydration shell and m water molecules in the second hydration shell). In some cases the octaaqualead(II) prefers a lower coordination number (as in [4 + 4], [4 + 3 + 1], [5 + 3]) which suggests that the octacoordinate structure is not particularly stable. The enneaaqualead(II) structure can exist in one of two C3 forms, the intermittently stable [9 + 0] and the more stable [6 + 3]. The octadecaaqualead(II) structure [6 + 12] was also examined, and in accordance with previous results the structure with symmetry T was chosen [2]. To our surprise, the structure was actually stable at some levels of theory (see Table 1), indicating that the hemidirected and holodirected structures for hexaaqualead(II) could be interchanged by inclusion of a second hydration sphere. In accordance with earlier calculations [2], the effect of the second hydration sphere is to shorten slightly the Pb-O distance and raise the symmetric stretching vibrational frequency by 20–30 cm−1.
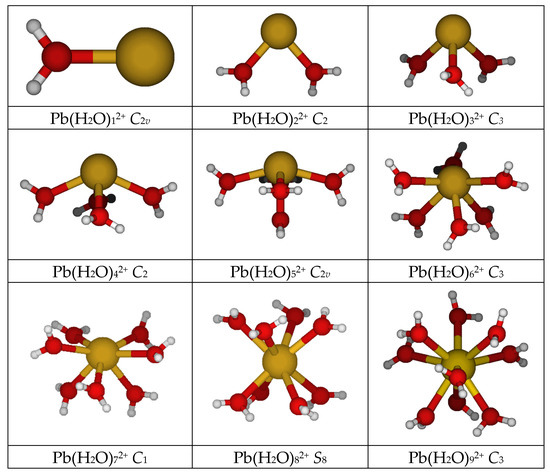
Figure 1.
The minimum energy structures of aqualead(II).

Table 1.
Totally symmetric stretching frequency (cm−1) and Pb-O distance(s) (Å) of the aqualead(II) cation. n/c = not calculated, * = imaginary frequency.
The results of the systematic desymmetrization procedure [31] for aqualead(II) follow (please see video in SM). The differences in energy reported below are exaggerated for the minimal basis sets:
- The bent C2 symmetry for diaqualead(II) is confirmed, with a Cs structure slightly higher and the linear D2d structure approximately 40 kJ/mol higher in energy. The related [1 + 1] and PbOH+ … H3O+ structures are 50–100 kJ/mol higher in energy. The use of either the SDD or LANL2DZ effective core potential on lead gives numbers on the lower end of this range. In some cases, the [1 + 1] structure does not exist, reverting to the PbOH+ … H3O+ (all MP2; HF/LANL2DZ) or [2 + 0] structure (HF/CEP-121G*);
- The pyramidal C3 symmetry for triaqualead(II) is confirmed, with the two C3v structures 10–20 kJ/mol higher in energy, and with the two D3h and one D3 planar structures approximately 40–60 kJ/mol higher in energy. The [2 + 1] C2v (HF/CEP-4G Cs) structure is 25–90 kJ/mol higher in energy, with the SDD or LANL2DZ effective core potential on lead giving numbers on the lower end of this range;
- The C2 see-saw structure for tetraaqualead(II) is confirmed at all levels except HF/LANL2MB (Cs), with the C2v #3 structure only slightly higher in energy. The distorted tetrahedral D2d #1, D2d #2, D2, and S4 structures are 20–40 kJ/mol higher in energy. The C2v #1 and 2 see-saw structures are approximately 10–20 kJ/mol higher in energy. The [3 + 1] Cs #2/C1 #1 structure tends to be about 25 kJ/mol higher in energy than the most stable [4 + 0] structure when using the CEP effective core potential/basis set combination, but is competitive in energy with the others (and even slightly lower for HF/LANL2DZ and HF/SDD), which may be a result of the lack of polarization functions in the basis set;
- The C2v #3/Cs square pyramidal structure is confirmed for pentaaqualead(II), with the other C2v structures (#1, #2, #4) being within 30 kJ/mol. The most stable [4 + 1] structure is either competitive in energy (Pb: SDD, LANL2DZ) or 10–25 kJ/mol less stable than the [5 + 0] structures;
- The C3 distorted octahedral structure is confirmed for hexaaqualead(II), with the undistorted Th structure being 2–30 kJ/mol higher in energy. For the CEP methods, the structure is competitive in energy with the [5 + 1] Cs structure, whereas for the LANL2DZ and SDD methods, the [5 + 1] is more stable by 10–25 kJ/mol;
- There are 16 possible C2v structures for heptaaqualead(II) spanning a range of 60 kJ/mol, with #1 and #16 being the most stable. In some cases, at some levels, these optimize to [6 + 1] or [5 + 2] structures. The C1 [7 + 0], [6 + 1], and [5 + 2] structures are competitive in energy. A stable [7 + 0] structure appears to be nonexistent at HF/LANL2MB, HF/LANL2DZ, HF/SDD, and HF/B. In some cases, the C2v [5 + 2] structures revert to [7 + 0] structures. Twelve C2v [5 + 2] structures and four C2v [6 + 1] structures were also located and desymmetrized. At least one stable structure was located for each by desymmetrization, except for the [6 + 1] structures at HF/LANL2MB, HF/LANL2DZ, and HF/SDD, which reverted to a [5 + 2] structure;
- For octaaqualead(II), the square prism D4h #1 and #2 and square antiprism D4d #1 and #2 have multiple imaginary frequencies, however the antiprism is much more stable. In some cases, stable S8 and D4 structures can be derived from these, and usually S8 is slightly more stable. Other possibilities include C4h, D2d, D2h, and C4v, although these are higher in energy. In some cases, a stable C4 structure forms from an unstable S8 (MP2/CEP-121G*, MP2/A), D4 (HF/SDD), or C4h structure (MP2/A+). Other C4, D2, S4, C2h, and C2v structures from desymmetrization are either not stable, or ascend in symmetry to give the S8 and D4 structure. Stable S4 structures exist at some of the MP2 levels. In all cases, the C1 #1 or #2 [7 + 1] structure is more stable than the S8, S4, or C4 [8 + 0] structures;
- For enneaaqualead(II), the tricapped trigonal prisms D3h #1–4 have multiple imaginary frequencies. Desymmetrization along A2′, A1”, and A2” modes led to C3h, D3, and C3v structures, respectively. These structures are unstable. The D3 #4 structure is often found, however the D3 #3 structure reverts to [6 + 3] in some cases. The C3h #3 and #4 coalesce to the corresponding #1 and #2 structures. In most cases, the C3v structures become [6 + 3]. To our surprise, the C3 #1–#3 structures formed from desymmetrization of D3 #4 (along A2), C3h #1, and #2 (along A”), respectively, were stable [9 + 0] structures if the split valence CEP basis sets were employed, although they usually reverted to stable [6 + 3] structures when using the LANL2DZ or SDD basis sets on lead. Desymmetrization along the E’ mode of the D3h structures gives C2v structures, whereas desymmetrization along the E” mode could give either a C2 or Cs structure. None of the C2v structures were stable, and many reverted to a [8 + 1], [7 + 2], [5 + 4], [5 + 2 + 2], or [4 + 5] structures. Desymmetrization of these along the A2 mode occasionally gave a stable C2 structure, whereas along the B1 or B2 modes gave either unstable Cs structures, ascension in symmetry to give C3h #1 or #2, or decoordination to give [8 + 1], [7 + 2], [6 + 3], [6 + 2 + 1], [5 + 4], [5 + 3 + 1], [4 + 5], or [4 + 3 + 2] structures.
The systematic desymmetrization procedure employed here allows us to efficiently find many stationary points, in addition to local minima, that may be difficult or impossible to find by other means commonly employed, such as turning off symmetry or employing classical molecular dynamics to generate candidate structures. These can tend to favor the lowest energy structures at the level of theory used to generate them.
3.2. The Pb-O Distance
The average Pb-O distance as a function of coordination number is plotted for all of the levels studied here. Examination of Figure 2 shows some clear trends. The Pb-O distance always lengthens upon an increase in the coordination number. There are some gaps present, in particular for n = 7, 9 where a directly bound structure does not exist, as indicated in the previous section. The Pb-O distance using the minimal basis HF/LANL2MB are shorter than the other levels at the same hydration number by 0.1–0.3 Å. The Pb-O distance using the SDD basis set/pseudopotential on Pb (HF/SDD, HF/C, HF/C+) tend to be the longest (2.35–2.80 Å), whereas the Pb-O distance using the LANL2DZ basis set/pseudopotential on Pb (HF/LANL2DZ, HF/B, HF/B+) tends to fall in the middle, especially at higher coordination numbers (5–8). With the exception of the HF/LANL2MB calculations, the Pb-O distances using the CEP basis set/pseudopotential cluster together as the shortest distances. The effect of basis set/pseudopotential combination seems to be more important here than the presence or absence of electron correlation (MP2/CEP-31G*, MP2/CEP-121G*, MP2/A, and MP2/A+).
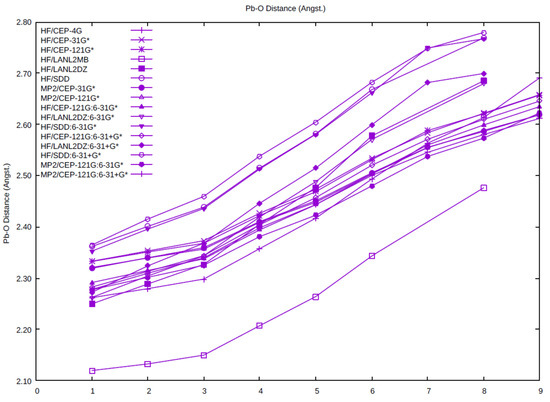
Figure 2.
The dependence of the average Pb-O distance on the coordination number and level of theory.
Although the average Pb-O distance shows some clear trends, there is much variation in the individual Pb-O distances where they are not related by symmetry. The stereochemically active electron pair (6s2) is typically on the principal axis of symmetry of the hydrated ion and acts similarly to an additional ligand, forming hemidirected complexes. If we label this as E, then the trend seen is that the Pb-O distances corresponding to the smaller E-Pb-O angles tend to be much longer than those with the larger E-Pb-O angles (see, for example the Pb-O distances in C3-[Pb(H2O)6]2+, Table 1). For n = 7, the bond is so weak that the water prefers to detach from the Pb2+ and hydrogen-bond to form structures such as [5 + 2]. Indeed, in some cases, it is difficult to distinguish whether a bond exists at all!
3.3. The Pb-O Vibrational Frequency
The Pb-O symmetric stretching vibrational frequency as a function of coordination number is plotted for all of the levels (Figure 3). As the symmetric stretching frequency is inversely related to the distance, the vibrational frequencies decrease as the coordination number increases. The highest frequencies occur for the HF/LANL2MB level, as expected, as these distances are the shortest. The next highest correspond to the HF/CEP-4G level, in which a minimal basis set for oxygen and hydrogen is used. The smallest frequencies correspond to the HF/SDD, HF/C, and HF/C+, which all contain the SDD basis set/pseudopotential on lead, followed closely by the HF/LANL2DZ, HF/B, and HF/B+, which all contain the LANL2DZ basis set/pseudopotential on lead. Most of the levels containing the CEP-121G basis set/pseudopotential on lead cluster together in the middle.
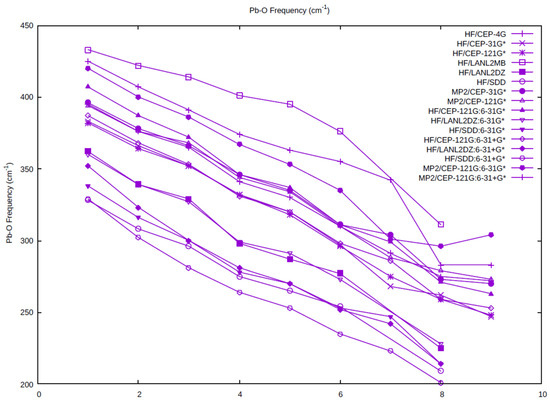
Figure 3.
The dependence of the totally symmetric Pb-O frequency on the coordination number and level of theory.
4. Discussion
We may compare our calculations to some of those previously conducted. The B3LYP/LANL2DZ calculations of Wander and Clark [18] gave unstable coordination for coordination numbers 5–9, which is in line with our HF/LANL2DZ calculations, suggesting that this feature is due to the basis set and effective core potential, not to correlation energy. Although the distances reported in Table 2 of Ref. [19] appear accurate using the same level of theory, we were not able to reproduce the binding energies for n = 1 presented in Table 1 of Bhattacharjee et al. [19] The HF, MP2, and CCSD binding energies are rather close to each other, yet different than the B3LYP results, and it is unclear why [35]. One possibility that could account for this is that the HF energy results were used to calculate the binding energies at the optimized MP2 and CCSD geometries. The results of Lei and Pan seem to suggest that the lowest energy structures of aqualead(II) complexes have tetracoordination, although with low-lying higher-coordination structures close in energy [20]. Molecular dynamics studies give varying numbers for the hydration number of lead (e.g., 8.1 [19], 6.3 [21], 4 [22]). Clearly the result has a strong dependence on the potential (level of theory) used, which is a main point we are trying to make. Although Leon-Pimentel et al. [22] carefully calibrated the aug-cc-pVDZ basis set with the 6-311G basis set for their MD simulations, finding a systematic difference of about 50 kJ/mol for the average binding energy, the lack of polarization functions on the latter, and their reliance on the structures in Ref. [20] is cause for concern.
Due to the lack of experimental structural data on aqualead(II) complexes, it is difficult to state with any certainty what the structure and vibrational frequencies of the aqualead(II) ion actually are. The thermodynamics of our calculations (i.e., comparisons between [n + 0] and [(n − m) + m] structures) suggest that a coordination number n of between four and seven is most likely. This in turn, suggests an average Pb-O distance in the range 2.38–2.75 Å (Figure 2) and a totally symmetric stretching frequency in the range between 230–370 cm−1 (Figure 3). Some recent measurements from the group of Persson [35] on single crystals of the related compounds [Pb2(H2O)2(ClO4)4]n, [Pb(H2O)(CF3SO3)2]n, [Pb(dma)6](ClO4)2, and [Pb(dmpu)6](ClO4)2 (dma = dimethylacetamide, dmpu = N,N’-dimethylpropyleneurea) gave relevant Pb-O distances of 2.338(24), 2.434(40) (Pb-OH2); 2.473(26), 2.459(28) (Pb-OH2); 2.519(3) (Pb-O = C); and 2.520(6) Å (Pb-O = C), respectively, which are consistent with the lower end of this range. In the same paper, Extended X-ray Absorption Fine Structure (EXAFS) measurements on 1.0 and 1.9 mol L−1 solutions of lead(II) perchlorate could be well fitted to structures with mean Pb-O distance of 2.541(6) and 2.543(6) Å. The large Debye–Waller factors indicate a significant spread about the mean values. These same solutions, examined using large angle X-ray scattering (LAXS), gave values of 2.527(7) and 2.536(7) Å. Persson and coworkers concluded that lead was hexacoordinate hemidirected in aqueous perchlorate solutions, which is consistent with our hemidirected Pb(H2O)62+ C3 structure. For similarly solvated lead ions, the Pb-O distance was determined to be in the range 2.48–2.56 Å for the hexacoordinated solvates of dimethylsulfoxide (dmso), tetramethylurea (tmu), dimethylformamide (dmf), dma, and dmpu, using EXAFS and LAXS. Some of the structures are holodirected, whereas others are hemidirected. As the data for aqualead(II) indicates a hexaaqua hemidirected structure akin to our C3 form, our calculations can be narrowed to the Pb-O range 2.47–2.54 Å (corresponding to the middle group), which would also narrow the vibrational frequency to the range 300–350 cm−1. A second hydration sphere would add 20–30 cm−1 to these values (Table 1). The experiments suggest that calculations using the SDD and LANL2DZ basis set/pseudopotential combinations may overestimate the Pb-O distance and underestimate the totally symmetric vibrational frequency compared to the CEP-121G* basis set/pseudopotential.
5. Conclusions
The common CEP, LANL2DZ, and SDD pseudopotentials are paired with various basis sets to study the hydrated lead(II) ion. Calculations with minimal basis sets perform poorly. The lead-oxygen distances and totally symmetric stretching frequency with the other levels divide themselves into three groups (CEP, LANL2DZ, SDD) depending on the basis set/pseudopotential on lead. It appears as though the lead SDD and LANL2DZ give Pb-O distances that are too large compared to the experiment and tend to favor lower coordination numbers. However, the structures computed are consistent with the more recent experimental result of a six-coordinate hemidirected aqua complex. The careful use of symmetry can be used to guide the search for new structures.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/liquids2010004/s1, Table S1: Total Energies of Aqualead(II) Species; Table S2: Selected optimized structures (MP2/B, B = CEP-121G on Pb, 6-31+G* on O, H); SM_Desymmetrization.mp4 (video describing the desymmetrization procedure, 20 min); Translation_of_ActaChimSin_Wang_2013_71_1307.pdf.
Author Contributions
Conceptualization, C.C.P.; methodology, C.C.P.; investigation, C.C.P. and C.M.G.; resources, C.C.P.; data curation, C.C.P..; writing—original draft preparation, C.C.P.; writing—review and editing, C.C.P.; visualization, C.C.P.; supervision, C.C.P.; project administration, C.C.P.; funding acquisition, C.C.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Sciences and Engineering Research Council of Canada.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and Supplementary Materials.
Acknowledgments
The authors thank the Department of Astronomy and Physics, Saint Mary’s University (AP-SMU), for providing access to computing facilities, in particular, to Cygnus, a 10-processor Sun server, purchased with assistance from the Canada Foundation for Innovation, Sun Microsystems, the Atlantic Canada Opportunities Agency, and SMU. CCP also thanks ACEnet and Compute Canada for providing access to computers and Gaussian 03/16. CCP acknowledges the former financial support of NSERC. CMG acknowledges the support of the Department of Economic Development (Government of Nova Scotia), the Office of the Dean of Science, SMU, and the Cooperative Education program (SMU—Work Term 2, Fall 2001 and Work Term 3, Winter 2002).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References and Notes
- Richens, D.T. The Chemistry of Aqua Ions; Wiley: Chichester, UK, 1997. [Google Scholar]
- Pye, C.C.; Gunasekara, C.M. An Ab Initio Investigation of the Hydration of Thallium(III) and Mercury(II). J. Solut. Chem. 2020, 49, 1419–1429. [Google Scholar] [CrossRef]
- Baes, C.F., Jr.; Mesmer, R.E. The Hydrolysis of Cations; Wiley: New York, NY, USA, 1976. [Google Scholar]
- Spiro, T.G.; Templeton, D.H.; Zalkin, A. The Crystal Structure of a Hexanuclear Basic Lead(II) Perchlorate Hydrate: Pb6O(OH)6(ClO4)4∙H2O. Inorg. Chem. 1968, 8, 856–861. [Google Scholar] [CrossRef]
- Olin, A.; Soderquist, R. The Crystal Structure of β-[Pb6O(OH)6] (ClO4)4∙H2O. Acta Chem. Scand. 1972, 26, 3505–3514. [Google Scholar] [CrossRef]
- Johansson, G.; Olin, A. On the Structures of the Dominating Hydrolysis Products of Lead(II) in Solution. Acta Chem. Scand. 1968, 22, 3197–3201. [Google Scholar] [CrossRef]
- Hong, S.-H.; Olin, A. On the Crystal Structure of [Pb4(OH)4]3(CO3)(ClO4)10∙6H2O. Acta Chem. Scand. 1973, 27, 2309–2320. [Google Scholar] [CrossRef]
- Hong, S.-H.; Olin, A. The Crystal Structure of [Pb4(OH)4](ClO4)4∙2H2O. Acta Chem. Scand. A 1974, 28, 233–238. [Google Scholar] [CrossRef]
- Cox, H.; Stace, A.J. Molecular View of the Anomalous Acidities of Sn2+, Pb2+, and Hg2+. J. Am. Chem. Soc. 2004, 126, 3939–3947. [Google Scholar] [CrossRef]
- Shi, T.; Orlova, G.; Guo, J.; Bohme, D.K.; Hopkinson, A.C.; Siu, K.W.M. Evidence of Doubly Charged Lead Monohydrate: Experimental Evidence and Theoretical Examination. J. Am. Chem. Soc. 2004, 126, 7975–7980. [Google Scholar] [CrossRef]
- Shi, T.; Zhao, J.; Hopkinson, A.C.; Siu, K.W.M. Formation of Abundant [Pb(H2O)]2+ by Ligand-Exchange Reaction between [Pb(N2)n]2+ (n = 1–3) and H2O. J. Phys. Chem. B 2005, 109, 10590–10593. [Google Scholar] [CrossRef]
- McQuinn, K.; Hof, F.; McIndoe, J.S.; Chen, X.; Wu, G.; Stace, A.J. Evidence of asymmetric cation solvation from the instability of [Pb(H2O)n]2+ complexes. Chem. Commun. 2009, 27, 4088–4090. [Google Scholar] [CrossRef]
- Gourlaouen, C.; Piquemal, J.-P.; Parisel, O. [Pb(H2O)]2+ and [Pb(OH)]+: Four-component density functional theory calculations, correlated scalar relativistic constrained-space orbital variation energy decompositions, and topological analysis. J. Chem. Phys. 2006, 124, 174311. [Google Scholar] [CrossRef] [PubMed]
- Arfa, M.; Olier, R.; Privat, M. Ab Initio Calculations on the Electronic Structure of the Divalent Lead-Water Complex. J. Phys. Chem. A 2008, 112, 6004–6008. [Google Scholar] [CrossRef] [PubMed]
- Hofer, T.S.; Rode, B.M. The solvation structure of Pb(II) in dilute aqueous solution: An ab initio quantum mechanical/molecular mechanical molecular dynamics approach. J. Chem. Phys. 2004, 121, 6406–6411. [Google Scholar] [CrossRef] [PubMed]
- Hofer, T.S.; Randolf, B.R.; Rode, B.M. The dynamics of the solvation of Pb(II) in aqueous solution obtained by an ab initio QM/MM MD approach. Chem. Phys. 2006, 323, 473–478. [Google Scholar] [CrossRef]
- Gourlaouen, C.; Gerard, H.; Parisel, O. Exploring the Hydration of Pb2+: Ab Initio Studies and First-Principles Molecular Dynamics. Chem. Eur. J. 2006, 12, 5024–5032. [Google Scholar] [CrossRef]
- Wander, M.C.F.; Clark, A.E. Hydration Properties of Aqueous Pb(II) Ion. Inorg. Chem. 2008, 47, 8233–8241. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Hofer, T.S.; Pribil, A.B.; Randolf, B.R.; Lim, L.H.V.; Lichtenberger, A.F.; Rode, B.M. Revisiting the Hydration of Pb(II): A QMCF MD Approach. J. Phys. Chem. B 2009, 113, 13007–13013. [Google Scholar] [CrossRef]
- Lei, X.L.; Pan, B.C. The Geometries and Proton Transfer of Hydrated Divalent Lead Ion Clusters [Pb(H2O)n]2+ (n = 1–17). J. Theor. Comput. Chem. 2012, 11, 1149–1164. [Google Scholar] [CrossRef]
- Wang, J.; Xia, S.; Yu, L. Hydration Structure of Pb(II) from Density Functional Theory Studies and First-Principles Molecular Dynamics. Acta Chim. Sin. 2013, 71, 1307–1312. (In Chinese)An English translation for ease of understanding is provided in the SM [Google Scholar] [CrossRef]
- Leon-Pimentel, C.I.; Amaro-Estrada, J.I.; Saint-Martin, H.; Ramirez-Solis, A. Born-Oppenheimer molecular dynamics studies of Pb(II) micro hydrated gas phase clusters. J. Chem. Phys. 2017, 146, 084307. [Google Scholar] [CrossRef]
- Kuznetsov, A.M.; Masliy, A.N.; Korshin, G.V. Quantum-chemical simulations of the hydration of Pb(II) ion: Structure, hydration energies, and pKa1 value. J. Mol. Model. 2018, 24, 193. [Google Scholar] [CrossRef] [PubMed]
- Leon-Pimentel, C.I.; Martinez-Jimenez, M.; Saint-Martin, H. Study of the Elusive Hydration of Pb2+ from the Gas Phase to the Liquid Aqueous Solution: Modeling the Hemidirected Solvation with a Polarizable MCDHO Force-Field. J. Phys. Chem. B 2019, 123, 9155–9166. [Google Scholar] [CrossRef] [PubMed]
- Tolbatov, I.; Marrone, A. Molecular dynamics simulation of the Pb(II) coordination in biological media via cationic dummy atom models. Theor. Chem. Acc. 2021, 140, 20. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A., Jr.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian 98, Revision A.9; Gaussian, Inc.: Pittsburgh, PA, USA, 1998. [Google Scholar]
- We thank an anonymous reviewer for carrying out some scalar relativistic (SR) and spin-orbit (SO) ZORA/BP86/DZP-small core Gibbs free energy of binding calculations using the Amsterdam Density Functional (ADF) package to confirm this: n = 1, −202.27 kJ/mol (SR), −205.13 kJ/mol; n = 2, −353.12 kJ/mol (SR), −356.05 kJ/mol (SO). The bond distances are: n = 1, 2.352 Å (SR and SO); n = 2, 2.394, 2.397 Å (both SR and SO). These compare favorably with our HF/C calculations.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision D.02; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Pye, C.C.; Gunasekara, C.M.; Rudolph, W.W. An ab initio investigation of bismuth hydration. Can. J. Chem. 2007, 85, 945–950. [Google Scholar] [CrossRef]
- Pye, C.C.; Whynot, D.C.M.; Corbeil, C.R.; Mercer, D.J. Desymmetrization in geometry optimization: Application to an ab initio study of copper(I) hydration. Pure Appl. Chem. 2020, 92, 1643–1654. [Google Scholar] [CrossRef]
- Pye, C.C.; Gilbert, C.R. An ab initio investigation of the second hydration shell of metal cations. Comput. Appl. Chem. 2020, 208, 395–397. [Google Scholar]
- Pye, C.C.; Black, S.M.; Rudolph, W.W. An Ab Initio Investigation of Zinc Bromo Complexes. J. Sol. Chem. 2011, 40, 1932–1954, and references 7-16 therein. [Google Scholar] [CrossRef]
- Persson, I.; Lyczko, K.; Lundberg, D.; Eriksson, L.; Placzek, A. Coordination Chemistry Study of Hydrated and Solvated Lead(II) Ions in Solution and Solid State. Inorg. Chem. 2011, 50, 1058–1072. [Google Scholar] [CrossRef]
- Rode, B.M. (University of Innsbruck, Innsbruck, Austria). Personal communication, 2 February 2022.



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).