



Liquids 2025, 5(4), 28; https://doi.org/10.3390/liquids5040028 (registering DOI) - 26 Oct 2025
Abstract
The rheological properties of aqueous solutions of wormlike micelles (WLMs) of cationic surfactant erucyl bis(hydroxyethyl)methylammonium chloride (EHAC) in the presence of hydrotropic salt sodium salicylate (NaSal) and inorganic salt sodium chloride (NaCl) have been studied. The conditions for maximum zero-shear viscosity at fixed
[...] Read more.
The rheological properties of aqueous solutions of wormlike micelles (WLMs) of cationic surfactant erucyl bis(hydroxyethyl)methylammonium chloride (EHAC) in the presence of hydrotropic salt sodium salicylate (NaSal) and inorganic salt sodium chloride (NaCl) have been studied. The conditions for maximum zero-shear viscosity at fixed surfactant concentration were investigated. It has been shown that charged WLMs in the presence of NaSal have higher viscosities than well-screened micelles in the presence of NaCl. This is because the adsorption of hydrophobic salicylate ions onto the micelles increases their length more significantly than the presence of a large amount of sodium ions in the solution. It was discovered that the effect of temperature on the rheological properties depends on both the type of salt used and the salt/surfactant molar ratio. An unusual increase in zero-shear viscosity and elastic modulus was observed at a NaSal concentration that corresponds to the maximum zero-shear viscosity when the WLMs are linear, charged, and “unbreakable”. These results expand the possibilities of using hydrotropic salts to create stable, highly viscous systems in various fields, and opening up new horizons for applications in oil production, cosmetics, and household chemicals.
Full article
(This article belongs to the Section Chemical Physics of Liquids)
►
Show Figures
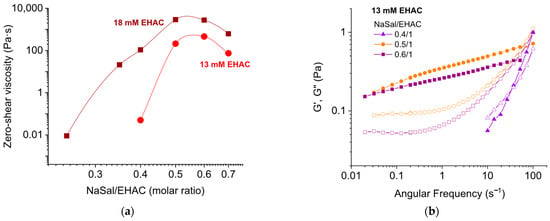
Figure 1





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}