
Evolution of SARS-CoV-2 Strains in Senegal: From a Wild Wuhan Strain to the Omicron Variant
 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Study Subject
2.2. Genome Sequencing and Analysis
2.2.1. RNA Extraction
2.2.2. Reverse Transcriptase-Polymerase Chain Reaction
2.3. Data Analysis
3. Results
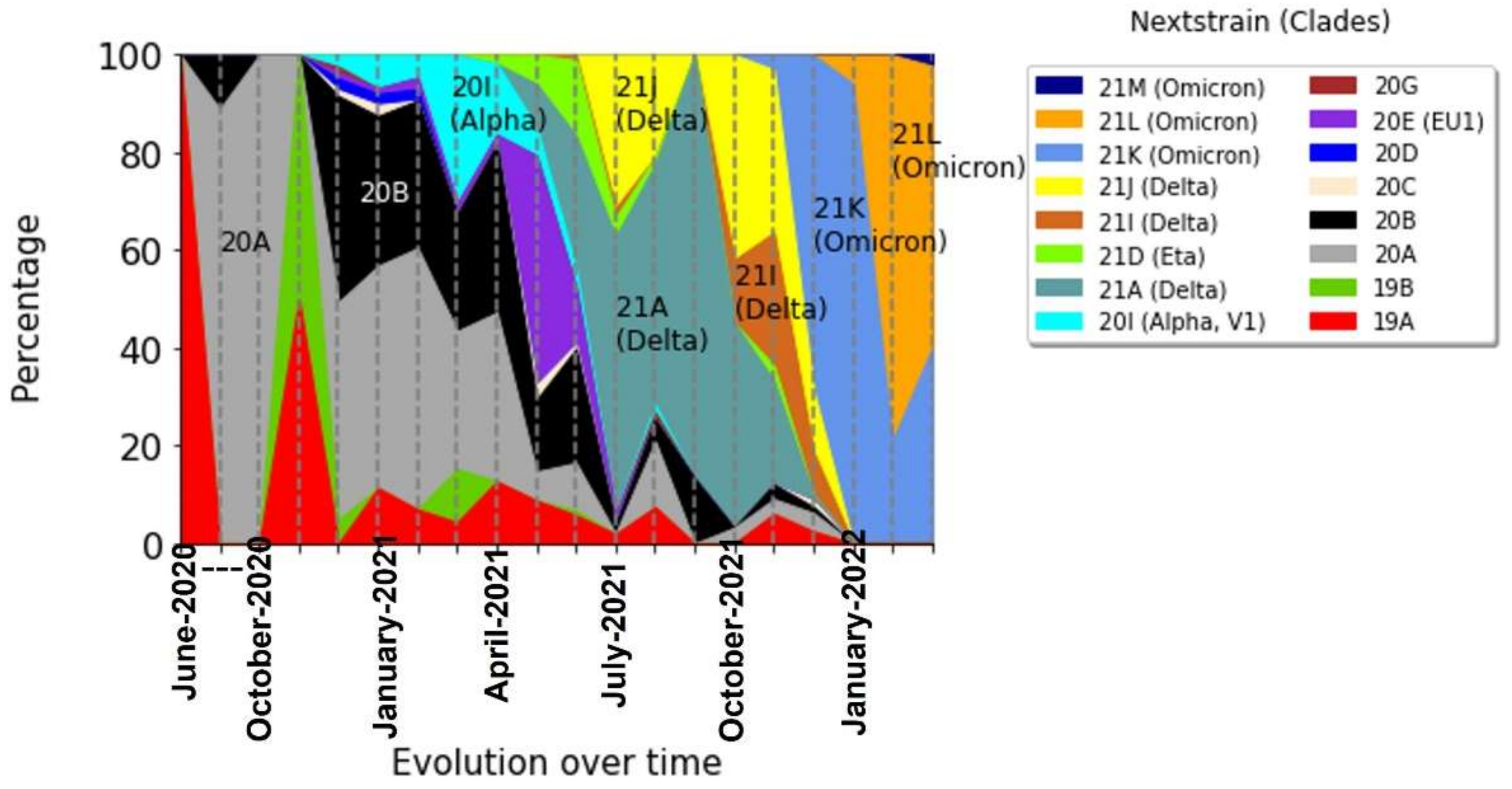
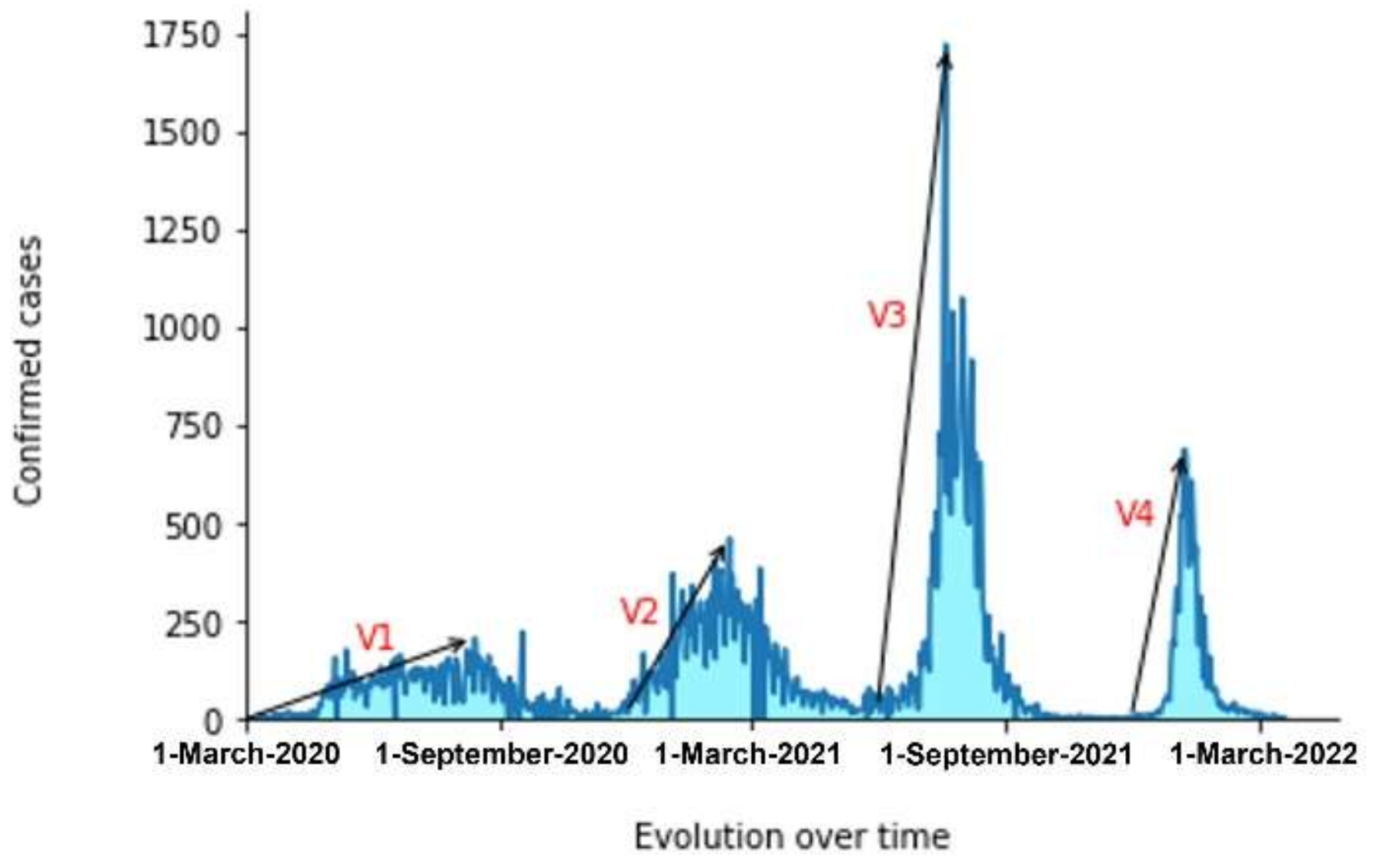
3.1. The Evolution of the Different Variants over Time
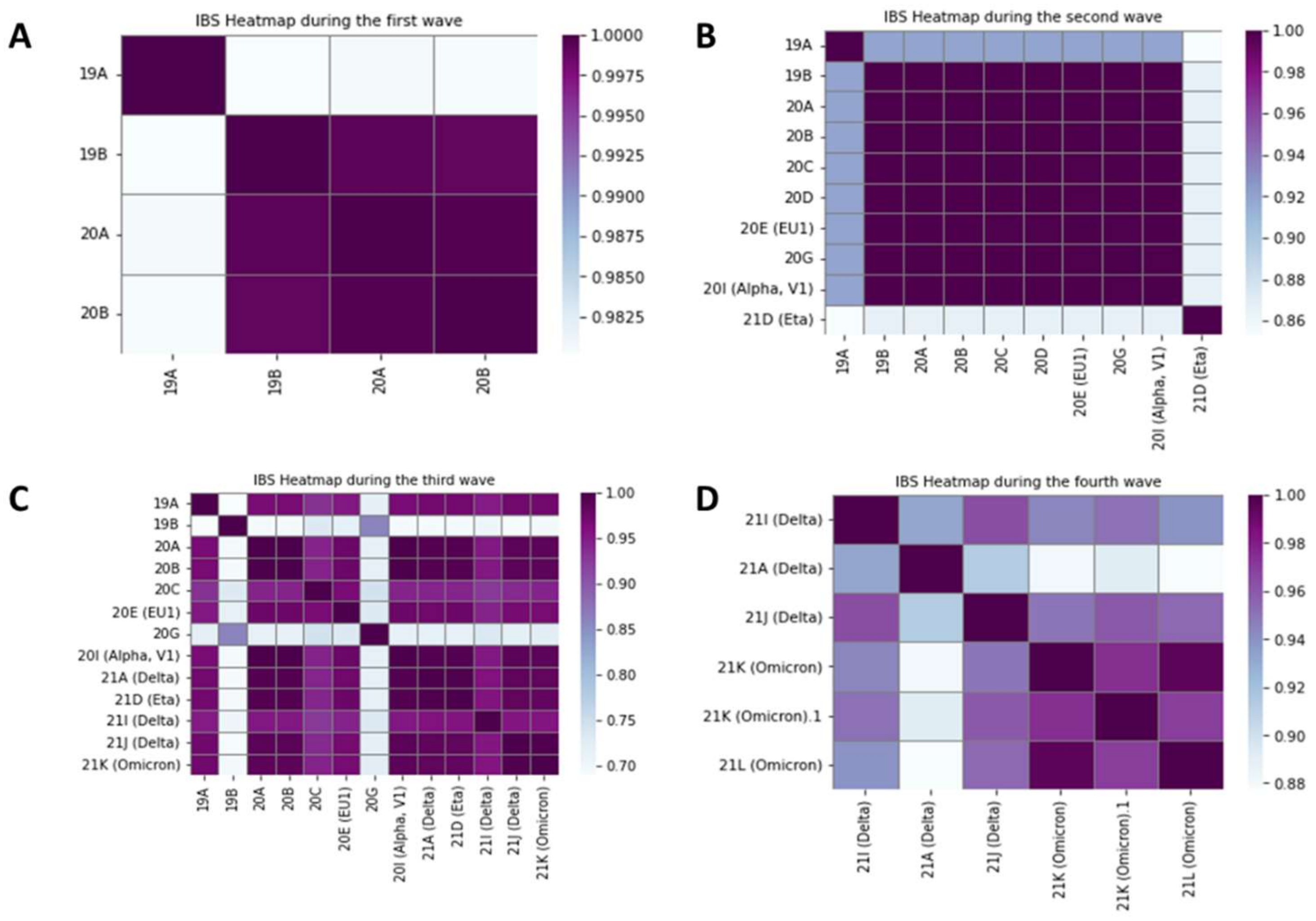
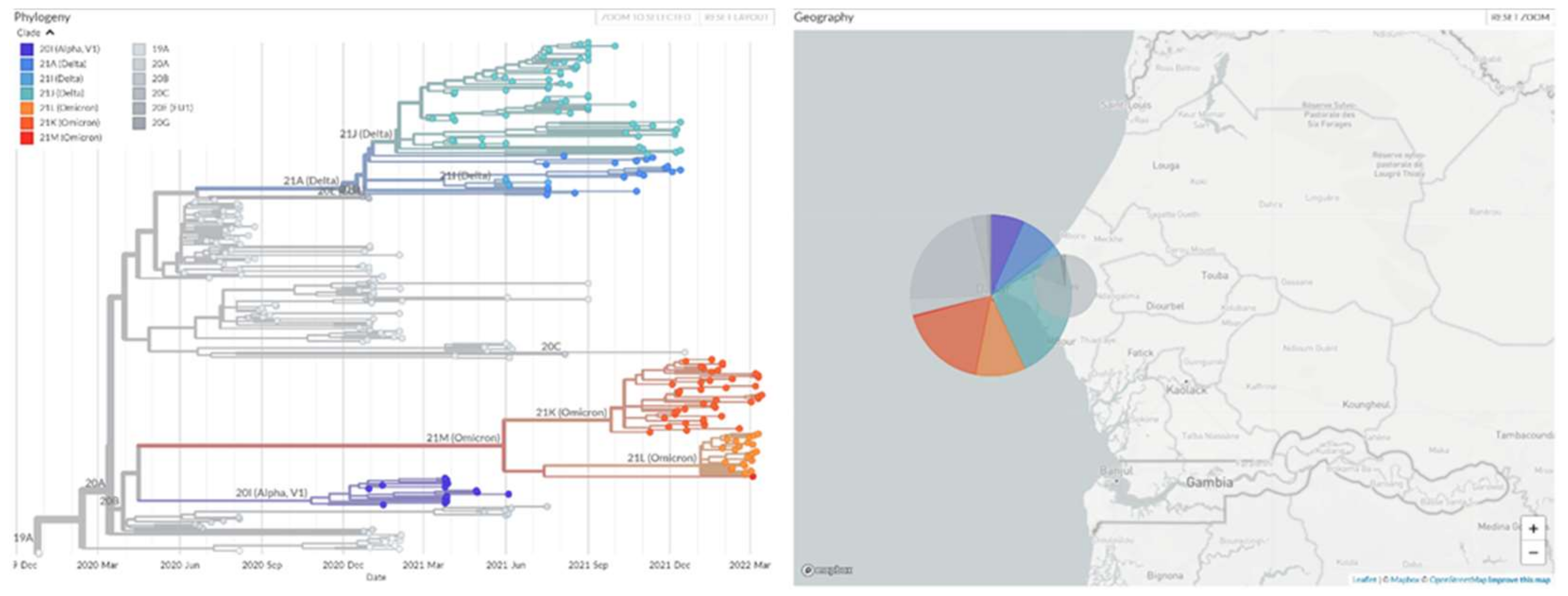
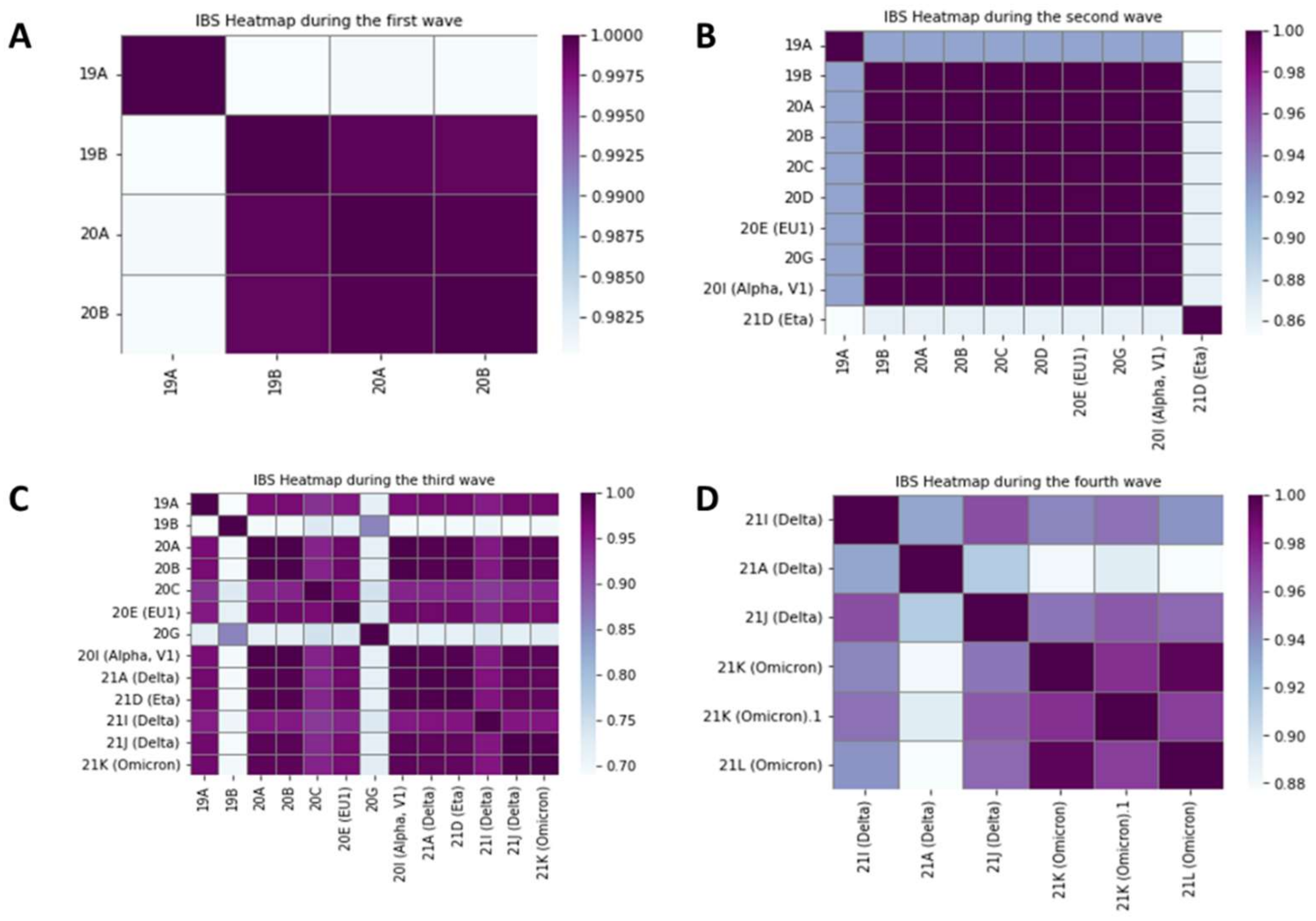
3.2. Relationships between Variants on Each Wave
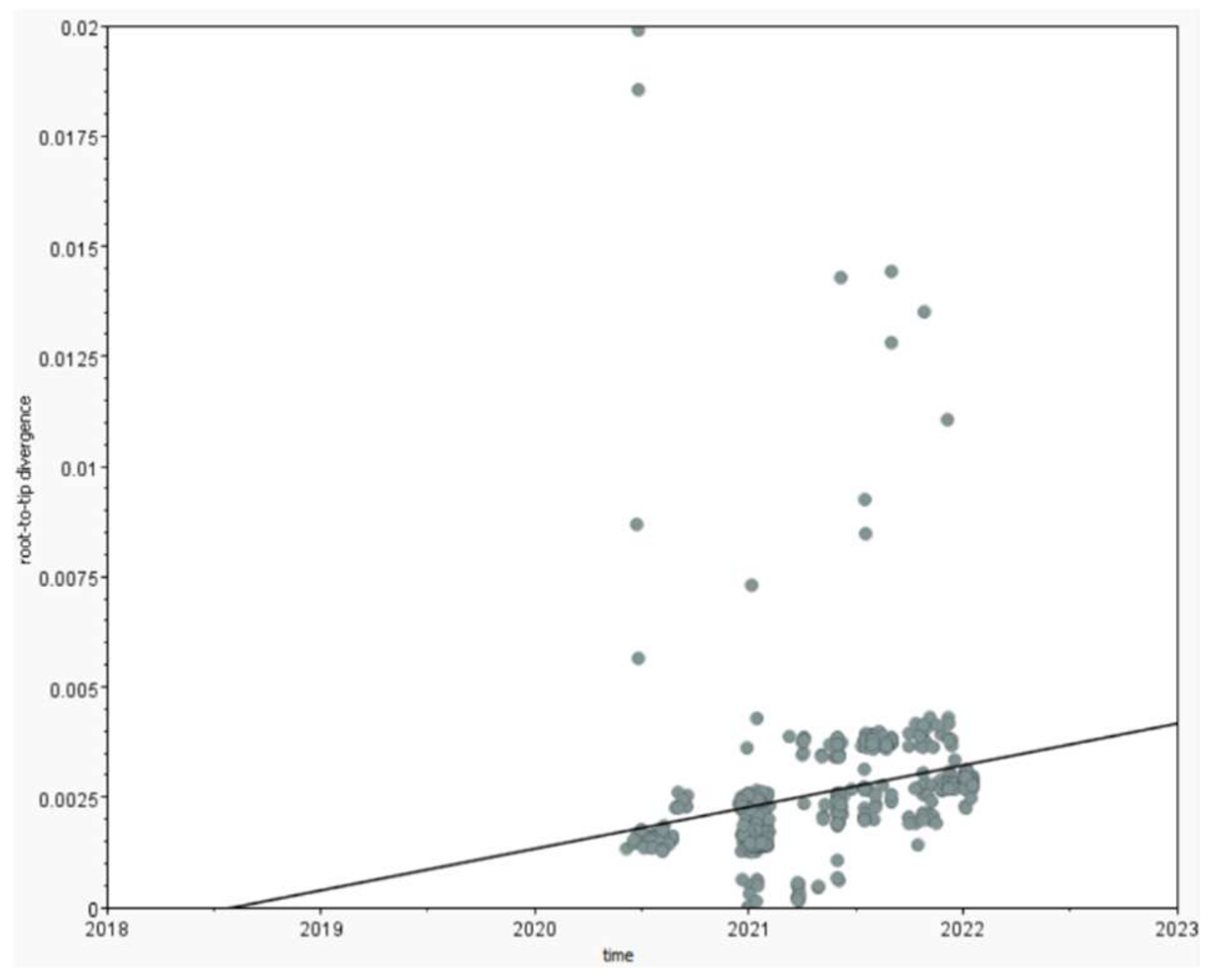
3.3. The Pandemic’s Speed of Spread
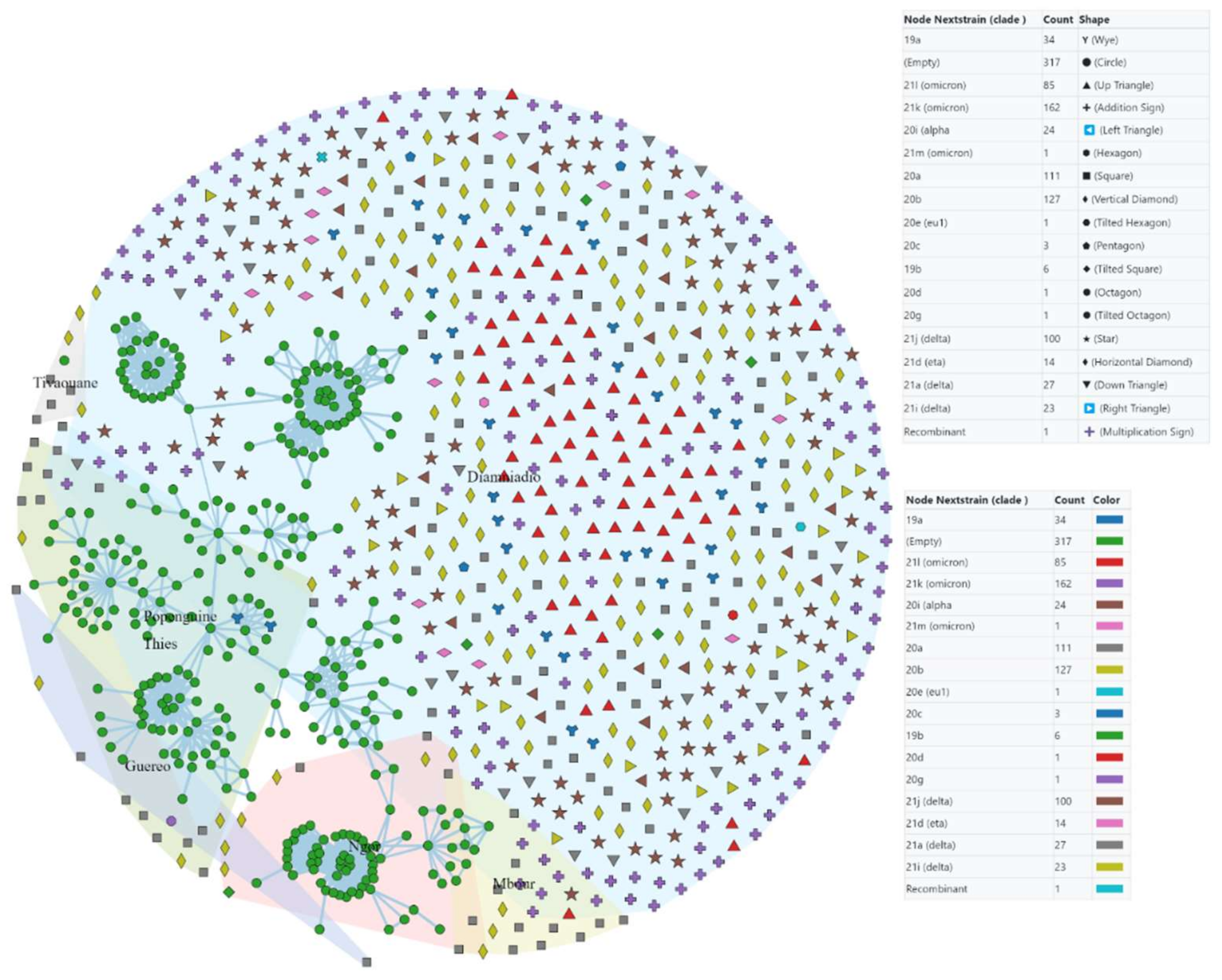
3.4. Origin and Transmission Network of the Pandemic
4. Discussion
4.1. The Evolution of Phylodynamic Variants and Their Transmissibility
4.2. The Pandemic’s Origin, Speed of Spread, and Transmission Network
5. Conclusions
- Continue to monitor the genome for new mutations in SARS-CoV-2 that could cause new outbreaks.
- Expand this research to the whole country to better understand the progression of the disease.
- Prepare for new outbreaks.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- WHO|Novel Coronavirus—China. WHO n.d. Available online: http://www.who.int/csr/don/12-january-2020-novel-coronavirus-china/en/ (accessed on 25 January 2022).
- WHO Director-General’s Remarks at the Media Briefing on 2019-nCoV on 11 February 2020 n.d. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-remarks-at-the-media-briefing-on-2019-ncov-on-11-february-2020 (accessed on 25 January 2022).
- Coronavirus-West-Africa—Sahel and West Africa Club Secretariat n.d. Available online: https://www.oecd.org/swac/coronavirus-west-africa/ (accessed on 25 January 2022).
- Senegal Reports First COVID-19 Case. WHO|Regional Office for Africa n.d. Available online: https://www.afro.who.int/news/senegal-reports-first-COVID-19-case (accessed on 25 January 2022).
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef] [PubMed]
- Aleem, A.; Akbar Samad, A.B.; Slenker, A.K. Emerging Variants of SARS-CoV-2 and Novel Therapeutics Against Coronavirus (COVID-19); StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Padane, A.; Kanteh, A.; Leye, N.; Mboup, A.; Manneh, J.; Mbow, M.; Diaw, P.; Ndiaye, B.; Lo, G.; Lo, C.; et al. First detection of SARS-CoV-2 variant B.1.1.7 in Senegal. N. Microbes N. Infect. 2021, 41, 100877. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Deng, A.; Li, K.; Hu, Y.; Li, Z.; Xiong, Q.; Liu, Z.; Guo, Q.; Zou, L.; Zhang, H.; et al. Viral infection and transmission in a large well-traced outbreak caused by the Delta SARS-CoV-2 variant. medRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, J.P.; Samal, K.C. World on Alert: WHO Designated South African New COVID Strain (Omicron/B.1.1.529) as a Variant of Concern. Biot. Res. Today 2021, 3, 1086–1088. [Google Scholar]
- The Transmission Modes and Sources of COVID-19: A Systematic Review—PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7484735/ (accessed on 28 March 2022).
- Xu, T.; Chen, C.; Zhu, Z.; Cui, M.; Chen, C.; Dai, H.; Xue, Y. Clinical features and dynamics of viral load in imported and non-imported patients with COVID-19. Int. J. Infect. Dis. 2020, 94, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Xiao, Y.; Kang, L.; Ma, W.; Shi, L.; Zhang, L.; Zhou, Z.; Yang, J.; Zhong, J.; Yang, D.; et al. Genomic diversity of SARS-CoV-2 in Coronavirus Disease 2019 patients. Clin. Infect. Dis. 2020, 71, 713–720, published online ahead of print. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coronavirus: Circulation Des Variants Du SARS-CoV-2. Available online: https://www.santepubliquefrance.fr/dossiers/coronavirus-COVID-19/coronavirus-circulation-des-variants-du-sars-cov-2 (accessed on 25 January 2022).
- Otto, S.P.; Day, T.; Arino, J.; Colijn, C.; Dushoff, J.; Li, M.; Mechai, S.; Van Domselaar, G.; Wu, J.; Earn, D.J.; et al. The origins and potential future of SARS-CoV-2 variants of concern in the evolving COVID-19 pandemic. Curr. Biol. 2021, 31, R918–R929. [Google Scholar] [CrossRef] [PubMed]
- Bertholom, C. Évolution Génétique du Sars-CoV-2 et ses Conséquences. Option/Bio 2021, 32, 22–23. [Google Scholar] [CrossRef]
- Kannan, S.R.; Spratt, A.N.; Cohen, A.R.; Naqvi, S.H.; Chand, H.S.; Quinn, T.P.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Evolutionary analysis of the Delta and Delta Plus variants of the SARS-CoV-2 viruses. J. Autoimmun. 2021, 124, 102715. [Google Scholar] [CrossRef] [PubMed]
- COVID-19: Qu’est-Ce Que Le Variant Omicron? ONU France, 13 Janvier 2022. Available online: https://unric.org/fr/COVID-19-quest-ce-que-le-variant-omicron/ (accessed on 25 January 2022).
- Amir, I.J.; Lebar, Z.; Yahyaoui, G.; Mahmoud, M. Covid-19: Virologie, épidémiologie et diagnostic biologique. Option/Bio 2020, 31, 15–20. [Google Scholar] [CrossRef]
- Alizon, S.; Sofonea, M.T. SARS-CoV-2 contagious evolution: Acontagious theory, immunity and trade-offs. J. Evol. Biol. Juill. 2021, 34, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, J.; Holmes, E.C. The phylogenomics of evolving virus virulence. Nat. Rev. Genet. 2018, 19, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Meyerowitz, E.A.; Richterman, A.; Gandhi, R.T.; Sax, P.E. Transmission of SARS-CoV-2: A Review of Viral, Host, and Environmental Factors. Ann. Intern. Med. 2021, 174, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Le Sénégal Déclare Le Premier Cas De COVID-19, OMS|Bureau Régional Pour l’Afrique. Available online: https://www.afro.who.int/fr/news/le-senegal-declare-le-premier-cas-de-COVID-19 (accessed on 25 January 2022).
- Pham, T.Q.; Hoang, N.-A.; Quach, H.-L.; Nguyen, K.C.; Colquhoun, S.; Lambert, S.; Duong, L.H.; Tran, Q.D.; Ha, D.A.; Phung, D.C.; et al. Timeliness of contact tracing among flight passengers during the COVID-19 epidemic in Vietnam. BMC Infect. Dis. 2021, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lotfi, M.; Hamblin, M.R.; Rezaei, N. COVID-19: Transmission, prevention, and potential therapeutic opportunities. Clin. Chim. Acta 2020, 508, 254–266. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gueye, K.; Padane, A.; Diédhiou, C.K.; Ndiour, S.; Diagne, N.D.; Mboup, A.; Mbow, M.; Lo, C.I.; Leye, N.; Ndoye, A.S.; et al. Evolution of SARS-CoV-2 Strains in Senegal: From a Wild Wuhan Strain to the Omicron Variant. COVID 2022, 2, 1116-1124. https://doi.org/10.3390/covid2080082
Gueye K, Padane A, Diédhiou CK, Ndiour S, Diagne ND, Mboup A, Mbow M, Lo CI, Leye N, Ndoye AS, et al. Evolution of SARS-CoV-2 Strains in Senegal: From a Wild Wuhan Strain to the Omicron Variant. COVID. 2022; 2(8):1116-1124. https://doi.org/10.3390/covid2080082
Chicago/Turabian StyleGueye, Khadim, Abdou Padane, Cyrille Kouligueul Diédhiou, Samba Ndiour, Ndéye Diabou Diagne, Aminata Mboup, Moustapha Mbow, Cheikh Ibrahima Lo, Nafissatou Leye, Aissatou Sow Ndoye, and et al. 2022. "Evolution of SARS-CoV-2 Strains in Senegal: From a Wild Wuhan Strain to the Omicron Variant" COVID 2, no. 8: 1116-1124. https://doi.org/10.3390/covid2080082